

Red Blood Cell Membrane Cholesterol May Be a Key Regulator of Sickle Cell Disease Microvascular Complications
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Role of Membrane Cholesterol and Lipids in RBC Physiology
2. Cholesterol Movements between RBCs and Plasma Lipoproteins
- a.
- RBC cholesterol movement via plasma lipoproteins and ApoA1
- b.
- Lecithin Cholesterol Acyl Transferase (LCAT) and Scavenger Receptor B1 (SRB1) deficiencies
3. Low Plasma HDL and High RBC Cholesterol Concentration in SCD and Thalassemia
4. Potential Interventions to Regulate RBC Membrane Cholesterol: HDL and ApoA1 Mimetics
5. Evidence for a Role of HDL and HDL Mimetics on RBC Deformability and Physiology
5.1. Methods
5.2. Results
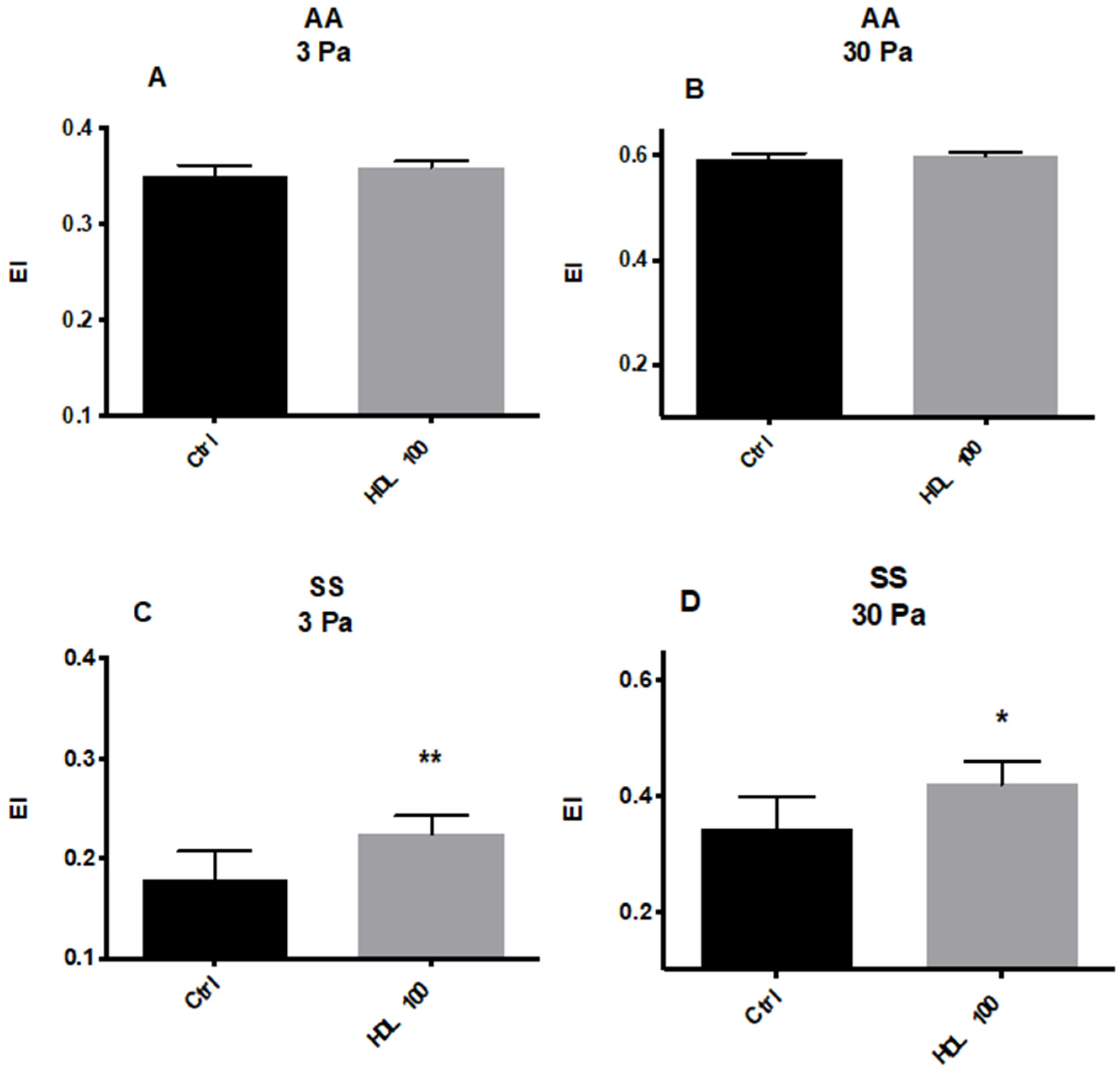
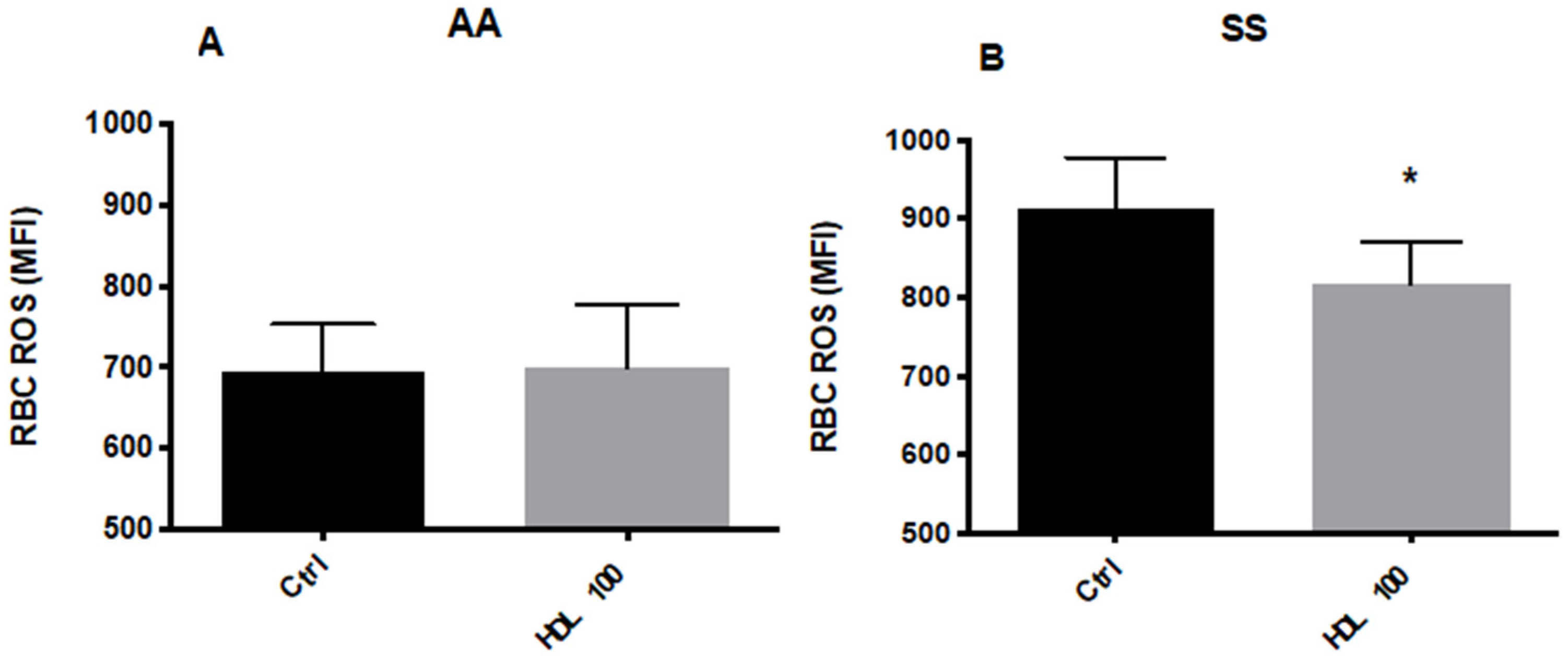
5.2.1. Effects of HDL on RBC Deformability and ROS Levels
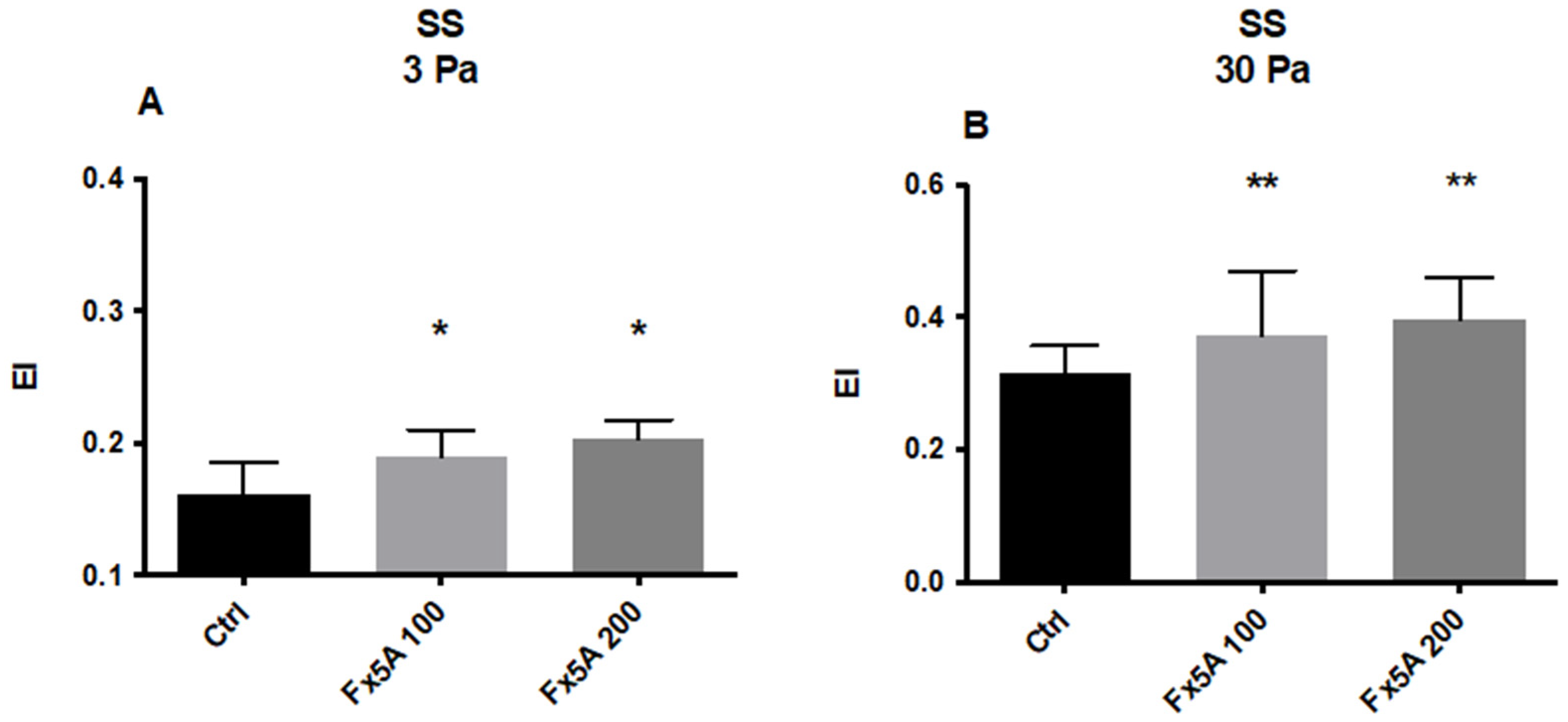
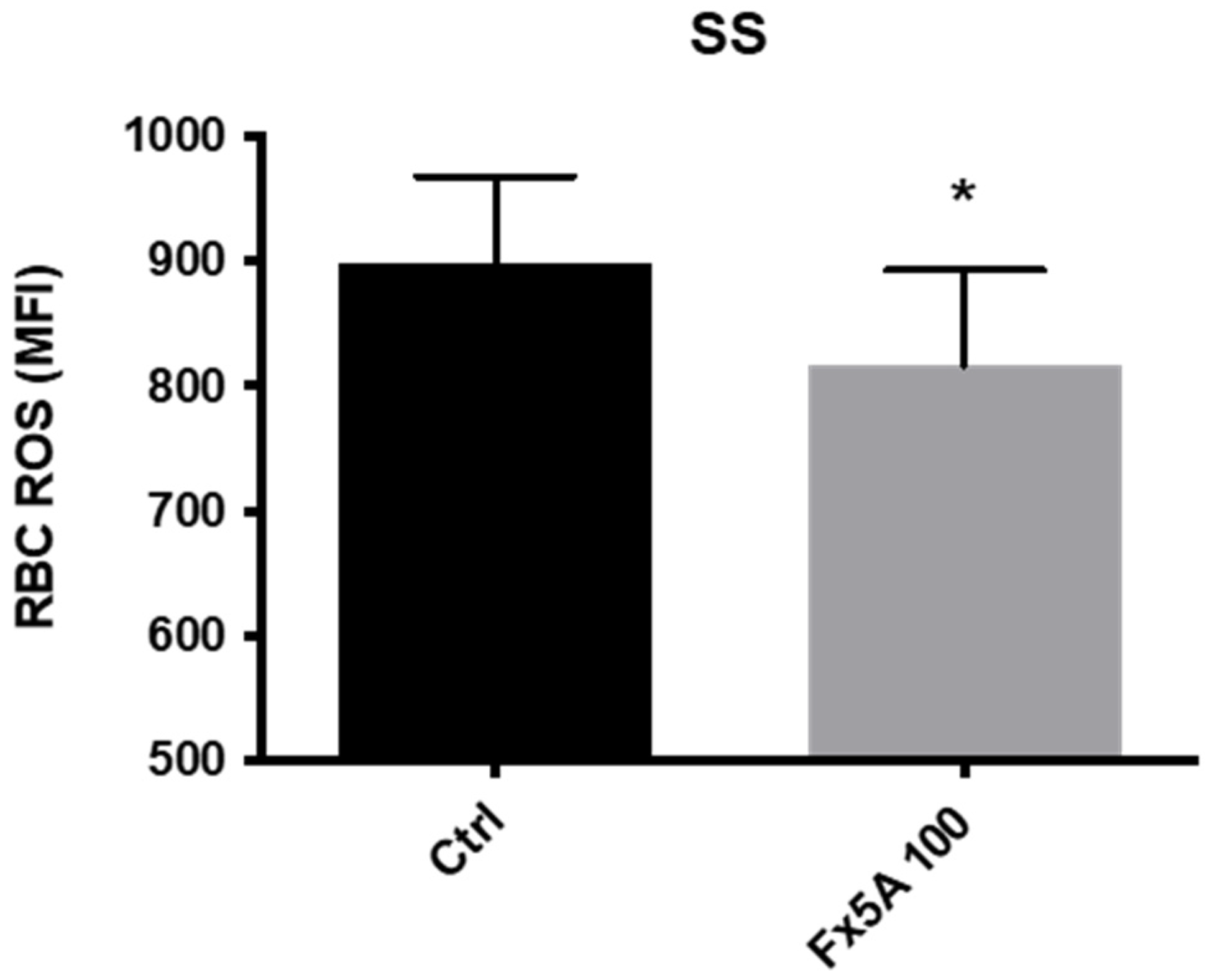
5.2.2. Effects of Fx5A on RBC Deformability and ROS Levels
6. Are Learnings from Sickle Cell Disease Applicable to Type 2 Diabetes and Cardiovascular Diseases?
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High Cholesterol/Low Cholesterol: Effects in Biological Membranes: A Review. Cell Biochem. Biophys. 2017, 75, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Al-Samir, S.; Itel, F.; Hegermann, J.; Gros, G.; Tsiavaliaris, G.; Endeward, V. O2 permeability of lipid bilayers is low, but increases with membrane cholesterol. Cell. Mol. Life Sci. 2021, 78, 7649–7662. [Google Scholar] [CrossRef] [PubMed]
- Itel, F.; Al-Samir, S.; Oberg, F.; Chami, M.; Kumar, M.; Supuran, C.T.; Deen, P.M.; Meier, W.; Hedfalk, K.; Gros, G.; et al. CO2 permeability of cell membranes is regulated by membrane cholesterol and protein gas channels. FASEB J. 2012, 26, 5182–5191. [Google Scholar] [CrossRef]
- Subczynski, W.K.; Widomska, J.; Feix, J.B. Physical properties of lipid bilayers from EPR spin labeling and their influence on chemical reactions in a membrane environment. Free Radic. Biol. Med. 2009, 46, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Miersch, S.; Espey, M.G.; Chaube, R.; Akarca, A.; Tweten, R.; Ananvoranich, S.; Mutus, B. Plasma membrane cholesterol content affects nitric oxide diffusion dynamics and signaling. J. Biol. Chem. 2008, 283, 18513–18521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchwald, H.; O’Dea, T.J.; Menchaca, H.J.; Michalek, V.N.; Rohde, T.D. Effect of plasma cholesterol on red blood cell oxygen transport. Clin. Exp. Pharmacol. Physiol. 2000, 27, 951–955. [Google Scholar] [CrossRef] [Green Version]
- Buchwald, H.; Menchaca, H.J.; Michalek, V.N.; Rohde, T.D.; Hunninghake, D.B.; O’Dea, T.J. Plasma cholesterol: An influencing factor in red blood cell oxygen release and cellular oxygen availability. J. Am. Coll. Surg. 2000, 191, 490–497. [Google Scholar] [CrossRef]
- Cooper, R.A. Influence of increased membrane cholesterol on membrane fluidity and cell function in human red blood cells. J. Supramol. Struct. 1978, 8, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Sagawa, S.; Shiraki, K. Changes of osmotic fragility of red blood cells due to repletion or depletion of cholesterol in human and rat red cells in vitro. J. Nutr. Sci. Vitaminol. 1980, 26, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Kanakaraj, P.; Singh, M. Influence of hypercholesterolemia on morphological and rheological characteristics of erythrocytes. Atherosclerosis 1989, 76, 209–218. [Google Scholar] [CrossRef]
- Chabanel, A.; Flamm, M.; Sung, K.L.; Lee, M.M.; Schachter, D.; Chien, S. Influence of cholesterol content on red cell membrane viscoelasticity and fluidity. Biophys. J. 1983, 44, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Brun, J.F.; Varlet-Marie, E.; Myzia, J.; Raynaud de Mauverger, E.; Pretorius, E. Metabolic Influences Modulating Erythrocyte Deformability and Eryptosis. Metabolites 2022, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.W.; Stewart, C.M.; Carpenter, M.P.; Stewart, T.P. Effects of cholesterol on lipid organization in human erythrocyte membrane. J. Cell Biol. 1980, 85, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, B.D.; Smith, P.; McGill, B.; Marshall, S.; Trick, J.L.; Chumakov, A.P.; Winlove, C.P.; Konovalov, O.V.; Lorenz, C.D.; Petrov, P.G. The Effects of Cholesterol Oxidation on Erythrocyte Plasma Membranes: A Monolayer Study. Membranes 2022, 12, 828. [Google Scholar] [CrossRef] [PubMed]
- Jewell, S.A.; Petrov, P.G.; Winlove, C.P. The effect of oxidative stress on the membrane dipole potential of human red blood cells. Biochim. Biophys. Acta 2013, 1828, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, J.P.; Winlove, C.P.; Petrov, P.G. Effect of hydroperoxides on red blood cell membrane mechanical properties. Biophys. J. 2011, 101, 1921–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulig, W.; Olzynska, A.; Jurkiewicz, P.; Kantola, A.M.; Komulainen, S.; Manna, M.; Pourmousa, M.; Vazdar, M.; Cwiklik, L.; Rog, T.; et al. Cholesterol under oxidative stress-How lipid membranes sense oxidation as cholesterol is being replaced by oxysterols. Free Radic. Biol. Med. 2015, 84, 30–41. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Chapman, M.J.; Fazio, S.; Hussain, M.M.; Kontush, A.; Krauss, R.M.; Otvos, J.D.; Remaley, A.T.; Schaefer, E.J. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 2011, 57, 392–410. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, H.; Yokoyama, S. Lecithin:cholesterol acyltransferase reaction on cellular lipid released by free apolipoprotein-mediated efflux. Biochemistry 1995, 34, 4385–4392. [Google Scholar] [CrossRef]
- Li, Q.; Komaba, A.; Yokoyama, S. Cholesterol is poorly available for free apolipoprotein-mediated cellular lipid efflux from smooth muscle cells. Biochemistry 1993, 32, 4597–4603. [Google Scholar] [CrossRef]
- Schwartz, C.C.; Berman, M.; Vlahcevic, Z.R.; Swell, L. Multicompartmental analysis of cholesterol metabolism in man. Quantitative kinetic evaluation of precursor sources and turnover of high density lipoprotein cholesterol esters. J. Clin. Investig. 1982, 70, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.; Voogt, J.; Davidson, M.; Glass, A.; Killion, S.; Decaris, J.; Mohammed, H.; Minehira, K.; Boban, D.; Murphy, E.; et al. Measurement of reverse cholesterol transport pathways in humans: In vivo rates of free cholesterol efflux, esterification, and excretion. J. Am. Heart Assoc. 2012, 1, e001826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, K.T.; Berisha, S.Z.; Ritchey, B.M.; Santore, J.; Smith, J.D. Red blood cells play a role in reverse cholesterol transport. Arter. Thromb. Vasc. Biol. 2012, 32, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Glomset, J.A. The plasma lecithins:cholesterol acyltransferase reaction. J. Lipid Res. 1968, 9, 155–167. [Google Scholar] [CrossRef]
- Lai, S.J.; Ohkawa, R.; Horiuchi, Y.; Kubota, T.; Tozuka, M. Red blood cells participate in reverse cholesterol transport by mediating cholesterol efflux of high-density lipoprotein and apolipoprotein A-I from THP-1 macrophages. Biol. Chem. 2019, 400, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- Lange, Y.; Steck, T.L. Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog. Lipid Res. 2008, 47, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, R.S.; Ingham, S.A.; Kozlitina, J.; Gay, A.; Cohen, J.C.; Radhakrishnan, A.; Hobbs, H.H. Variability of cholesterol accessibility in human red blood cells measured using a bacterial cholesterol-binding toxin. Elife 2017, 6, e23355. [Google Scholar] [CrossRef]
- Quarfordt, S.H.; Hilderman, H.L. Quantitation of the in vitro free cholesterol exchange of human red cells and lipoproteins. J. Lipid Res. 1970, 11, 528–535. [Google Scholar] [CrossRef]
- Dahlan, W.; Richelle, M.; Kulapongse, S.; Rossle, C.; Deckelbaum, R.J.; Carpentier, Y.A. Modification of erythrocyte membrane lipid composition induced by a single intravenous infusion of phospholipid-triacylglycerol emulsions in man. Clin. Nutr. 1992, 11, 255–261. [Google Scholar] [CrossRef]
- Suda, T.; Akamatsu, A.; Nakaya, Y.; Masuda, Y.; Desaki, J. Alterations in erythrocyte membrane lipid and its fragility in a patient with familial lecithin:cholesterol acyltrasferase (LCAT) deficiency. J. Med. Investig. 2002, 49, 147–155. [Google Scholar]
- Gillett, M.P.; Obineche, E.N.; El-Rokhaimi, M.; Lakhani, M.S.; Abdulle, A.; Sulaiman, M. Lecithin: Cholesterol acyltransfer, dyslipoproteinaemia and membrane lipids in uraemia. J. Nephrol. 2001, 14, 472–480. [Google Scholar] [PubMed]
- Meurs, I.; Hoekstra, M.; van Wanrooij, E.J.; Hildebrand, R.B.; Kuiper, J.; Kuipers, F.; Hardeman, M.R.; Van Berkel, T.J.; Van Eck, M. HDL cholesterol levels are an important factor for determining the lifespan of erythrocytes. Exp. Hematol. 2005, 33, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.J.; Jia, S.S.; Man, Q.Q.; Song, S.; Li, Y.Q.; Song, P.K.; Zhao, W.H.; Zhang, J. Dietary Cholesterol in the Elderly Chinese Population: An Analysis of CNHS 2010-2012. Nutrients 2017, 9, 934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, R.; van den Hurk, K.; Schoonmade, L.J.; de Kort, W.; Brug, J.; Lakerveld, J. Urban-rural differences in the association between blood lipids and characteristics of the built environment: A systematic review and meta-analysis. BMJ Glob. Health 2019, 4, e001017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.J. Comparative analysis of mammalian plasma lipoproteins. Methods Enzymol. 1986, 128, 70–143. [Google Scholar] [PubMed]
- Zorca, S.; Freeman, L.; Hildesheim, M.; Allen, D.; Remaley, A.T.; Taylor, J.G.; Kato, G.J. Lipid levels in sickle-cell disease associated with haemolytic severity, vascular dysfunction and pulmonary hypertension. Br. J. Haematol. 2010, 149, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Seixas, M.O.; Rocha, L.C.; Carvalho, M.B.; Menezes, J.F.; Lyra, I.M.; Nascimento, V.M.; Couto, R.D.; Atta, A.M.; Reis, M.G.; Goncalves, M.S. Levels of high-density lipoprotein cholesterol (HDL-C) among children with steady-state sickle cell disease. Lipids Health Dis. 2010, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, R.S.; Arriaga, M.B.; Terse-Ramos, R.; Ferreira, T.A.; Machado, V.R.; Rissatto-Lago, M.R.; Silveira-Mattos, P.S.; Boa-Sorte, N.; Ladeia, A.M.T.; Andrade, B.B. Higher values of triglycerides:HDL-cholesterol ratio hallmark disease severity in children and adolescents with sickle cell anemia. Braz. J. Med. Biol. Res. 2019, 52, e8833. [Google Scholar] [CrossRef]
- Lalanne-Mistrih, M.L.; Connes, P.; Lamarre, Y.; Lemonne, N.; Hardy-Dessources, M.D.; Tarer, V.; Etienne-Julan, M.; Mougenel, D.; Tressieres, B.; Romana, M. Lipid profiles in French West Indies sickle cell disease cohorts, and their general population. Lipids Health Dis. 2018, 17, 38. [Google Scholar] [CrossRef] [Green Version]
- Yuditskaya, S.; Tumblin, A.; Hoehn, G.T.; Wang, G.; Drake, S.K.; Xu, X.; Ying, S.; Chi, A.H.; Remaley, A.T.; Shen, R.F.; et al. Proteomic identification of altered apolipoprotein patterns in pulmonary hypertension and vasculopathy of sickle cell disease. Blood 2009, 113, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Westerman, M.P.; Pierce, L.E.; Jensen, W.N. Erythrocyte and Plasma Lipids in Sickle Cell Anemia. Blood 1964, 23, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Homan, R.; Esmaeil, N.; Mendelsohn, L.; Kato, G.J. A fluorescence method to detect and quantitate sterol esterification by lecithin:cholesterol acyltransferase. Anal. Biochem. 2013, 441, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meleard, P.; Gerbeaud, C.; Pott, T.; Fernandez-Puente, L.; Bivas, I.; Mitov, M.D.; Dufourcq, J.; Bothorel, P. Bending elasticities of model membranes: Influences of temperature and sterol content. Biophys. J. 1997, 72, 2616–2629. [Google Scholar] [CrossRef] [Green Version]
- Ashar, S.; Sultan, S.; Irfan, S.M.; Sheeraz, A. Serum fasting lipid profile in children and adolescents with beta-thalassaemia major in southern Pakistan. Malays. J. Pathol. 2015, 37, 233–238. [Google Scholar] [PubMed]
- Hamdy, M.M.; Mosallam, D.S.; Jamal, A.M.; Rabie, W.A. Selenium and Vitamin E as antioxidants in chronic hemolytic anemia: Are they deficient? A case-control study in a group of Egyptian children. J. Adv. Res. 2015, 6, 1071–1077. [Google Scholar] [CrossRef]
- Wang, X.M.L.; Freeman, L.; Vaisman, B.; Remaley, A.; Kato, G.J. Stimulation of Nitric Oxide Synthase Activity By Plasma Apolipoproteins: A Biomarker of Endothelial Function in Adults with Sickle Cell Disease. Blood 2014, 124, 4015. [Google Scholar] [CrossRef]
- Sasaki, J.; Waterman, M.R.; Cottam, G.L. Decreased apolipoprotein A-I and B content in plasma of individuals with sickle cell anemia. Clin. Chem. 1986, 32 Pt 1, 226–227. [Google Scholar] [CrossRef] [PubMed]
- Tumblin, A.; Tailor, A.; Hoehn, G.T.; Mack, A.K.; Mendelsohn, L.; Freeman, L.; Xu, X.; Remaley, A.T.; Munson, P.J.; Suffredini, A.F.; et al. Apolipoprotein A-I and serum amyloid A plasma levels are biomarkers of acute painful episodes in patients with sickle cell disease. Haematologica 2010, 95, 1467–1472. [Google Scholar] [CrossRef] [Green Version]
- Ou, J.; Ou, Z.; Jones, D.W.; Holzhauer, S.; Hatoum, O.A.; Ackerman, A.W.; Weihrauch, D.W.; Gutterman, D.D.; Guice, K.; Oldham, K.T.; et al. L-4F, an apolipoprotein A-1 mimetic, dramatically improves vasodilation in hypercholesterolemia and sickle cell disease. Circulation 2003, 107, 2337–2341. [Google Scholar] [CrossRef] [Green Version]
- Niesor, E.J.; Benghozi, R. Potential Signal Transduction Regulation by HDL of the beta2-Adrenergic Receptor Pathway. Implications in Selected Pathological Situations. Arch. Med. Res. 2015, 46, 361–371. [Google Scholar] [CrossRef]
- Colin, Y.; Rahuel, C.; Wautier, M.P.; El Nemer, W.; Filipe, A.; Cartron, J.P.; Le Van Kim, C.; Wautier, J.L. Red cell and endothelial Lu/BCAM beyond sickle cell disease. Transfus. Clin. Biol. 2008, 15, 402–405. [Google Scholar] [CrossRef] [PubMed]
- El Nemer, W.; Gauthier, E.; Wautier, M.P.; Rahuel, C.; Gane, P.; Galacteros, F.; Wautier, J.L.; Cartron, J.P.; Colin, Y.; Le Van Kim, C. Role of Lu/BCAM in abnormal adhesion of sickle red blood cells to vascular endothelium. Transfus. Clin. Biol. 2008, 15, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Zennadi, R.; Moeller, B.J.; Whalen, E.J.; Batchvarova, M.; Xu, K.; Shan, S.; Delahunty, M.; Dewhirst, M.W.; Telen, M.J. Epinephrine-induced activation of LW-mediated sickle cell adhesion and vaso-occlusion in vivo. Blood 2007, 110, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Jackson, T.; Elliott, L.E.; De Castro, L.M.; Jonassaint, J.; Ashley-Koch, A.; Telen, M.J. beta(2)-Adrenergic receptor and adenylate cyclase gene polymorphisms affect sickle red cell adhesion. Br. J. Haematol. 2008, 141, 105–108. [Google Scholar] [CrossRef]
- Uehara, Y.; Chiesa, G.; Saku, K. High-Density Lipoprotein-Targeted Therapy and Apolipoprotein A-I Mimetic Peptides. Circ. J. 2015, 79, 2523–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamburek, R.D.; Bakker-Arkema, R.; Auerbach, B.J.; Krause, B.R.; Homan, R.; Amar, M.J.; Freeman, L.A.; Remaley, A.T. Familial lecithin:cholesterol acyltransferase deficiency: First-in-human treatment with enzyme replacement. J. Clin. Lipidol. 2016, 10, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Gunawardane, R.N.; Fordstrom, P.; Piper, D.E.; Masterman, S.; Siu, S.; Liu, D.; Brown, M.; Lu, M.; Tang, J.; Zhang, R.; et al. Agonistic Human Antibodies Binding to Lecithin-Cholesterol Acyltransferase Modulate High Density Lipoprotein Metabolism. J. Biol. Chem. 2016, 291, 2799–2811. [Google Scholar] [CrossRef] [Green Version]
- Sethi, A.A.; Stonik, J.A.; Thomas, F.; Demosky, S.J.; Amar, M.; Neufeld, E.; Brewer, H.B.; Davidson, W.S.; D’Souza, W.; Sviridov, D.; et al. Asymmetry in the lipid affinity of bihelical amphipathic peptides. A structural determinant for the specificity of ABCA1-dependent cholesterol efflux by peptides. J. Biol. Chem. 2008, 283, 32273–32282. [Google Scholar] [CrossRef] [Green Version]
- Connes, P.; Alexy, T.; Detterich, J.; Romana, M.; Hardy-Dessources, M.D.; Ballas, S.K. The role of blood rheology in sickle cell disease. Blood Rev. 2016, 30, 111–118. [Google Scholar] [CrossRef]
- Boisson, C.; Nader, E.; Renoux, C.; Gauthier, A.; Poutrel, S.; Bertrand, Y.; Stauffer, E.; Virot, E.; Hot, A.; Fort, R.; et al. Shear-Stress-Gradient and Oxygen-Gradient Ektacytometry in Sickle Cell Patients at Steady State and during Vaso-Occlusive Crises. Cells 2022, 11, 585. [Google Scholar] [CrossRef]
- Hierso, R.; Waltz, X.; Mora, P.; Romana, M.; Lemonne, N.; Connes, P.; Hardy-Dessources, M.D. Effects of oxidative stress on red blood cell rheology in sickle cell patients. Br. J. Haematol. 2014, 166, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.B.; Delbosc, S.; Ho-Tin-Noe, B.; Leseche, G.; Nicoletti, A.; Meilhac, O.; Martin-Ventura, J.L. From intraplaque haemorrhages to plaque vulnerability: Biological consequences of intraplaque haemorrhages. J. Cardiovasc. Med. (Hagerstown) 2012, 13, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Delbosc, S.; Bayles, R.G.; Laschet, J.; Ollivier, V.; Ho-Tin-Noe, B.; Touat, Z.; Deschildre, C.; Morvan, M.; Louedec, L.; Gouya, L.; et al. Erythrocyte Efferocytosis by the Arterial Wall Promotes Oxidation in Early-Stage Atheroma in Humans. Front. Cardiovasc. Med. 2017, 4, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamos, T.D.; Rosenson, R.S. Low high density lipoprotein levels are associated with an elevated blood viscosity. Atherosclerosis 1999, 146, 161–165. [Google Scholar] [CrossRef]
- Barnes, H.J. Blood rheology in diabetes mellitus. Acta Med. Port. 1986, 7, S36–S39. [Google Scholar]
- Namazi, G.; Pourfarzam, M.; Jamshidi Rad, S.; Movahedian Attar, A.; Sarrafzadegan, N.; Sadeghi, M.; Asa, P. Association of the total cholesterol content of erythrocyte membranes with the severity of disease in stable coronary artery disease. Cholesterol 2014, 2014, 821686. [Google Scholar] [CrossRef] [Green Version]
- McMillan, D.E. Development of vascular complications in diabetes. Vasc. Med. 1997, 2, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Cignarelli, M.; Damato, A.; Cospite, M.R.; Guastamacchia, E.; Nardelli, G.M.; Giorgino, R. Erythrocyte cholesterol and red blood cells deformability in diabetes mellitus. Boll. Soc. Ital. Biol Sper. 1982, 58, 1115–1118. [Google Scholar]
- Tsukada, K.; Sekizuka, E.; Oshio, C.; Minamitani, H. Direct measurement of erythrocyte deformability in diabetes mellitus with a transparent microchannel capillary model and high-speed video camera system. Microvasc. Res. 2001, 61, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, H.; Kim, K.; Sohn, Y.; Jang, S.; Park, Y. Refractive index tomograms and dynamic membrane fluctuations of red blood cells from patients with diabetes mellitus. Sci. Rep. 2017, 7, 1039. [Google Scholar] [CrossRef]
- Zeng, N.F.; Mancuso, J.E.; Zivkovic, A.M.; Smilowitz, J.T.; Ristenpart, W.D. Red Blood Cells from Individuals with Abdominal Obesity or Metabolic Abnormalities Exhibit Less Deformability upon Entering a Constriction. PLoS ONE 2016, 11, e0156070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.R.; Macdougall, D.A.; Tyser, R.; Pugh, S.D.; Calaghan, S.C.; Harvey, R.D. Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 500–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshamwala, S.M.; Patil, N.D. Biochemical changes in erythrocyte membrane in uncontrolled type 2 diabetes mellitus. Indian J. Biochem. Biophys. 2005, 42, 250–253. [Google Scholar] [PubMed]
- Li, H.; Papageorgiou, D.P.; Chang, H.Y.; Lu, L.; Yang, J.; Deng, Y. Synergistic Integration of Laboratory and Numerical Approaches in Studies of the Biomechanics of Diseased Red Blood Cells. Biosensors 2018, 8, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tziakas, D.N.; Kaski, J.C.; Chalikias, G.K.; Romero, C.; Fredericks, S.; Tentes, I.K.; Kortsaris, A.X.; Hatseras, D.I.; Holt, D.W. Total cholesterol content of erythrocyte membranes is increased in patients with acute coronary syndrome: A new marker of clinical instability? J. Am. Coll. Cardiol. 2007, 49, 2081–2089. [Google Scholar] [CrossRef] [Green Version]
- Tziakas, D.N.; Chalikias, G.K.; Stakos, D.; Boudoulas, H. The role of red blood cells in the progression and instability of atherosclerotic plaque. Int. J. Cardiol. 2010, 142, 2–7. [Google Scholar] [CrossRef]
- Tziakas, D.N.; Chalikias, G.K.; Stakos, D.; Tentes, I.K.; Papazoglou, D.; Thomaidi, A.; Grapsa, A.; Gioka, G.; Kaski, J.C.; Boudoulas, H. Independent and additive predictive value of total cholesterol content of erythrocyte membranes with regard to coronary artery disease clinical presentation. Int. J. Cardiol. 2011, 150, 22–27. [Google Scholar] [CrossRef]
- Nunes, J.M.; Pretorius, E. Red blood cell membrane cholesterol in type 2 diabetes mellitus. Thromb. Res. 2019, 178, 91–98. [Google Scholar] [CrossRef]
- Kostara, C.E.; Tsiafoulis, C.G.; Bairaktari, E.T.; Tsimihodimos, V. Altered RBC membrane lipidome: A possible etiopathogenic link for the microvascular impairment in Type 2 diabetes. J. Diabetes Complicat. 2021, 35, 107998. [Google Scholar] [CrossRef]
- Skinner, S.; Pialoux, V.; Fromy, B.; Sigaudo-Roussel, D.; Connes, P. Sickle-cell trait and diagnosis of type 2 diabetes. Lancet Diabetes Endocrinol. 2018, 6, 840–843. [Google Scholar] [CrossRef]
- Skinner, S.C.; Diaw, M.; Pialoux, V.; Mbaye, M.N.; Mury, P.; Lopez, P.; Bousquet, D.; Gueye, F.; Diedhiou, D.; Joly, P.; et al. Increased Prevalence of Type 2 Diabetes-Related Complications in Combined Type 2 Diabetes and Sickle Cell Trait. Diabetes Care 2018, 41, 2595–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlaki, M.; Kourkouli, A.; Chalikias, G.; Kikas, P.; Tziakas, D. Cholesterol content of erythrocyte membranes and elusive target. Thromb. Res. 2020, 185, 32. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niesor, E.J.; Nader, E.; Perez, A.; Lamour, F.; Benghozi, R.; Remaley, A.; Thein, S.L.; Connes, P. Red Blood Cell Membrane Cholesterol May Be a Key Regulator of Sickle Cell Disease Microvascular Complications. Membranes 2022, 12, 1134. https://doi.org/10.3390/membranes12111134
Niesor EJ, Nader E, Perez A, Lamour F, Benghozi R, Remaley A, Thein SL, Connes P. Red Blood Cell Membrane Cholesterol May Be a Key Regulator of Sickle Cell Disease Microvascular Complications. Membranes. 2022; 12(11):1134. https://doi.org/10.3390/membranes12111134
Chicago/Turabian StyleNiesor, Eric J., Elie Nader, Anne Perez, François Lamour, Renée Benghozi, Alan Remaley, Swee Lay Thein, and Philippe Connes. 2022. "Red Blood Cell Membrane Cholesterol May Be a Key Regulator of Sickle Cell Disease Microvascular Complications" Membranes 12, no. 11: 1134. https://doi.org/10.3390/membranes12111134