
Preparation, Performance and Challenges of Catalyst Layer for Proton Exchange Membrane Fuel Cell

 , and
, and 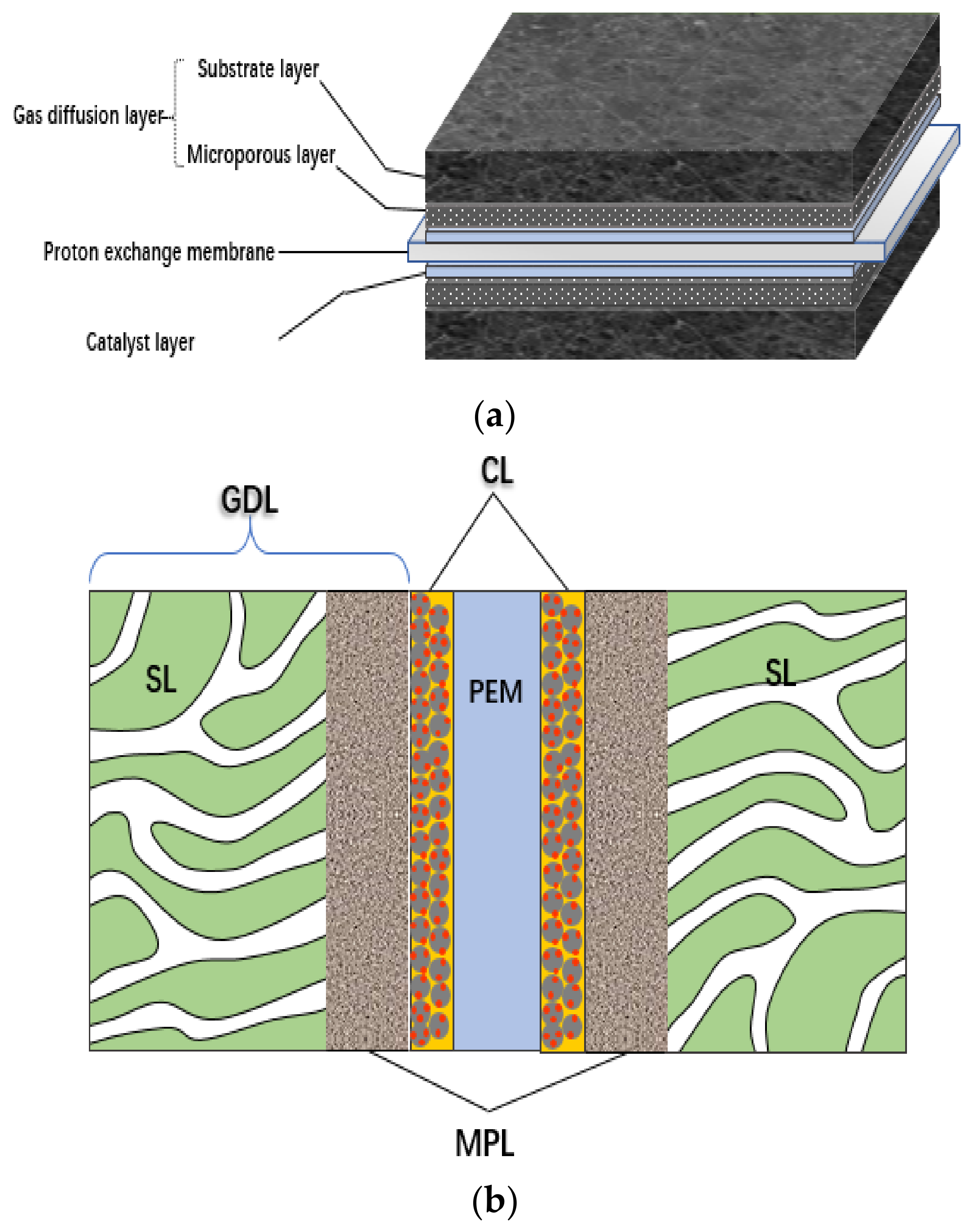{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
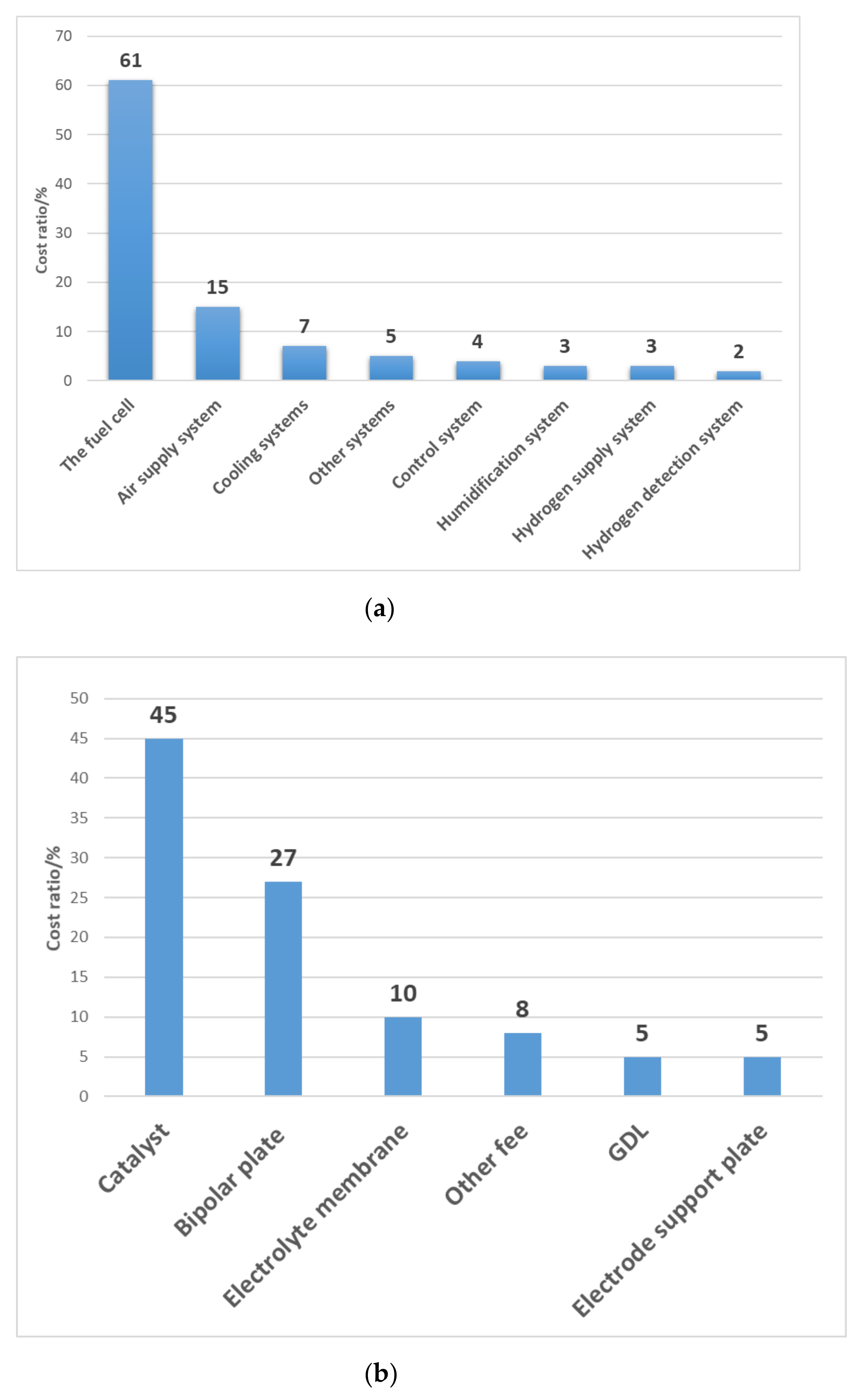
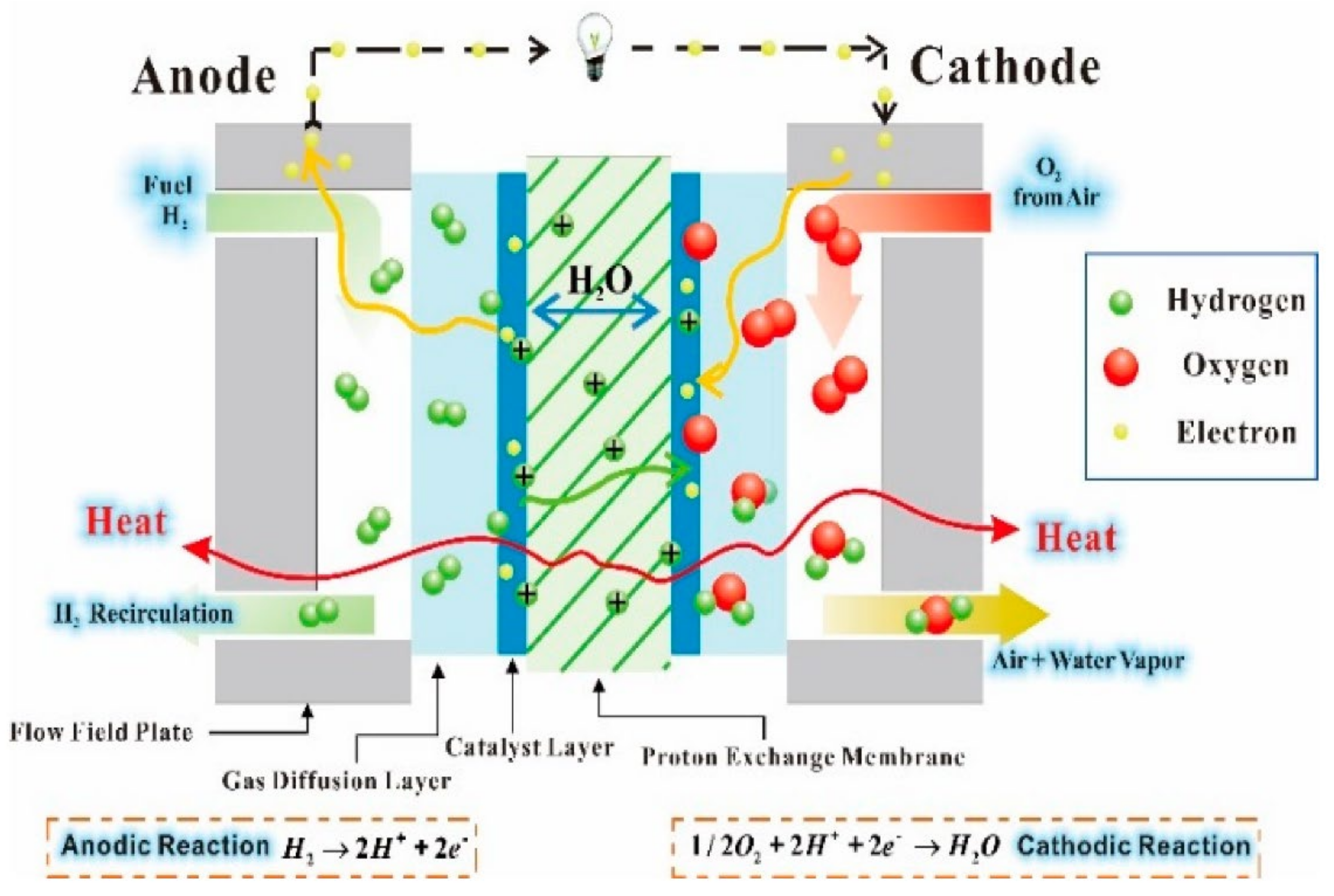
:1. Introduction
2. Composition and Structure of CL
2.1. Composition Materials of CL and Their Functions
- (1)
- Proton conductivity. The higher the proton conductivity, the smaller the proton conductivity resistance of PEM, which makes the overall internal resistance of MEA decrease, and the current density increase. Because protons transports in the form of hydronium ions, the proton conductivity is usually related to the water content and temperature of PEM, so it is often necessary to test at a specific temperature and humidity when evaluating their proton conductivity [63,64].
- (2)
- Gas permeation rate. One of the most important functions of PEM is to separate the reactions on both sides of MEA, so it is necessary to have a low gas permeability to prevent the fuel from mixing with the oxides, otherwise it will lead to local overheating, which greatly affects the efficiency and lifetime of fuel cell.
- (3)
- Dry-wet conversion characteristics. PEM water content not only affects the proton conductivity but also its mass and volume, and the humidity and temperature of the electrode are not constant during operation. The ideal PEM requires higher water content at lower humidity or higher temperature, to prevent the PEM from drying and affecting its performance. However, when the water content of PEM is too high, its mass and volume will increase accordingly. The change of mass is usually expressed by the water absorption rate, and the change of volume is expressed by the swelling rate. The smaller the swelling rate, the better because in the process of CL fabrication, excessive swelling rate will affect the degree of binding between CL and PEM, which may lead to interfacial delamination in severe cases and increase the proton conduction resistance, thus affecting the MEA performance.
- (4)
- Stability. The PEM for fuel cell requires good chemical stability, thermal stability, and mechanical stability. Sufficient stability ensures the durability of PEM and thus prolong its lifetime. PEM operates under acidic conditions (take PEMFCs for example), and its temperature varies during the operating. Under such operating conditions, it must have good acid resistance. Thermal stability ensures that the PEM does not degrade at the working temperature. Higher mechanical strength prevents PEM damage during assembly.
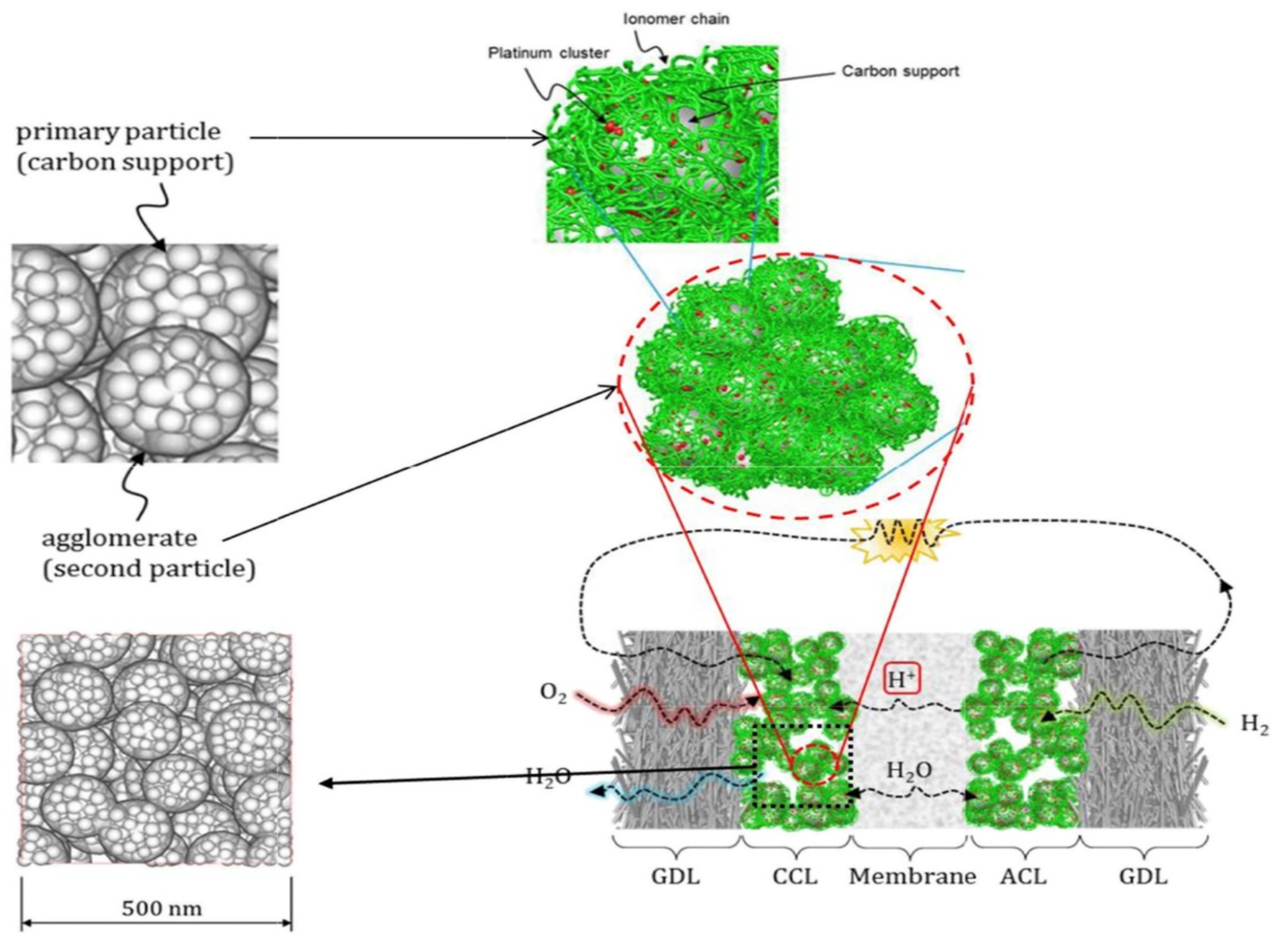
2.2. Structural Characteristics of CL
2.3. Interface of CL
2.3.1. Interface of PEM|CL
2.3.2. Interface of CL|MPL
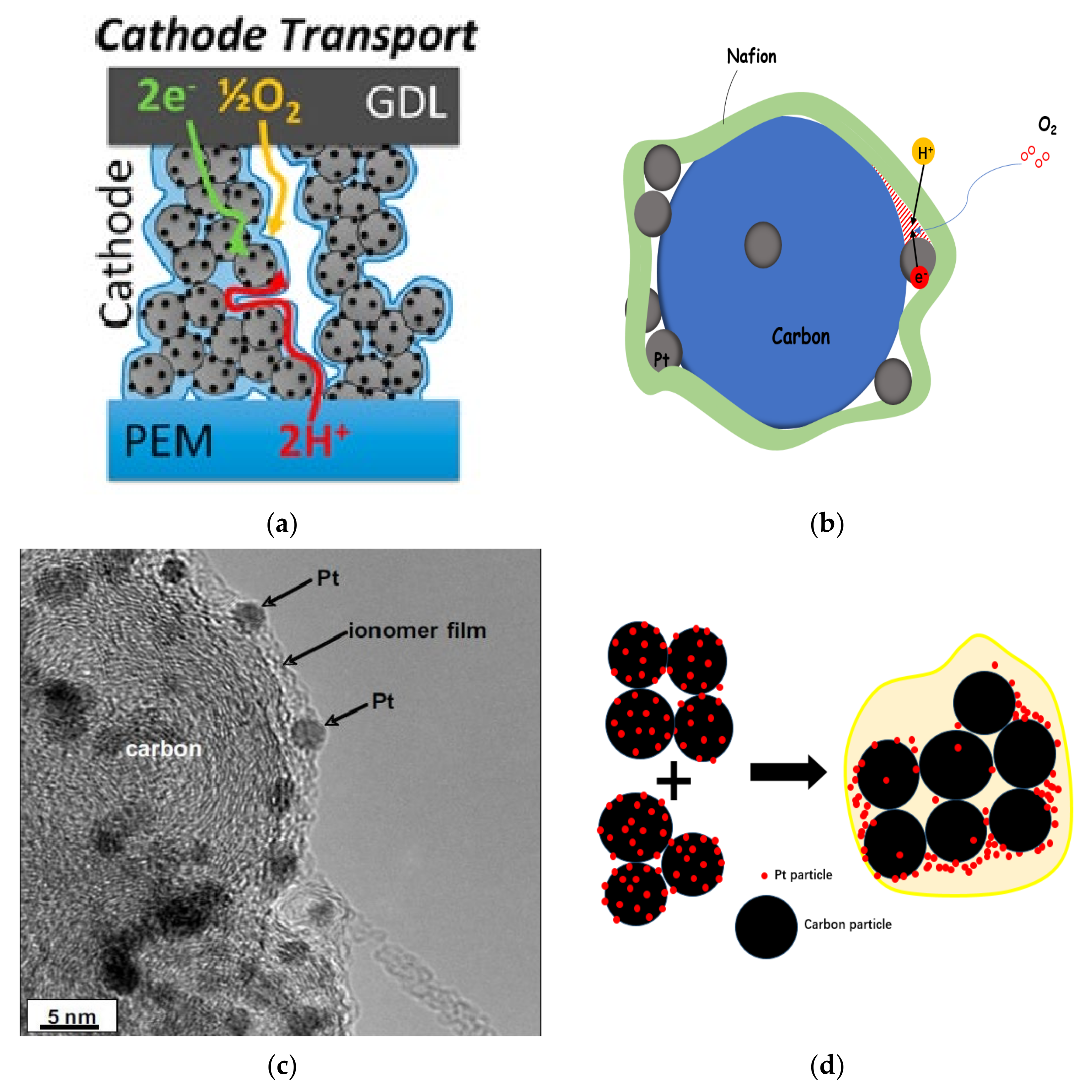
2.4. Transmission, Failure and Optimization in CL
- (1)
- The porous structure of carbon support allows Pt particles to enter, but the ionomer cannot reach the corresponding site, and Pt particles lack proton transfer channel and reduce their activity. In general, micropores smaller than 20 nm prevent the entry of ionomer.
- (2)
- When Pt particles enter the pores of carbon support, the ionomer may block the entrance of pores, increase the diffusion resistance of reaction gas, and reduce the catalytic efficiency of Pt particles.
- (3)
- If the ionomer is too thick, since the gas need be transferred to the three-phase boundary through the ionomer, the gas concentration will gradually decrease from the outside to the inside, forming in a concentration gradient, resulting in the reduction of catalytic efficiency of Pt particles.
- (4)
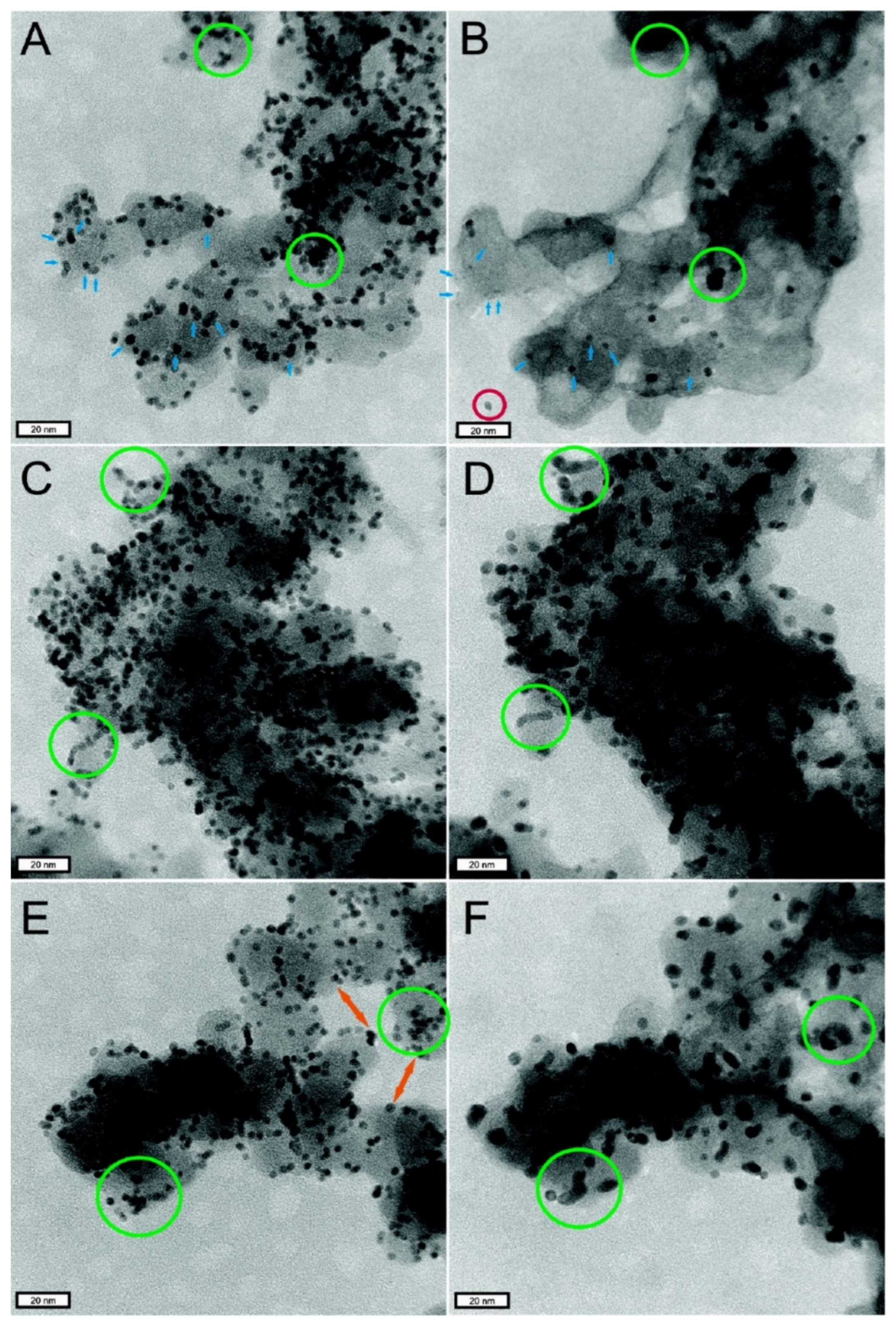
- During the preparation of catalyst and slurry or the coating of CL, the catalyst particles themselves will agglomerate. All of these will lead to a decrease in the utilization rate of Pt particles in CL (as shown in Figure 4d).
2.5. Failure Mechanism of CL
2.5.1. Failure of Pt Particles in CL
2.5.2. Crack of CL
2.5.3. Flooding of CL
2.5.4. Degradation of CL
Degradation of Ionomer
Corrosion of Carbon Support
Degradation of Catalyst
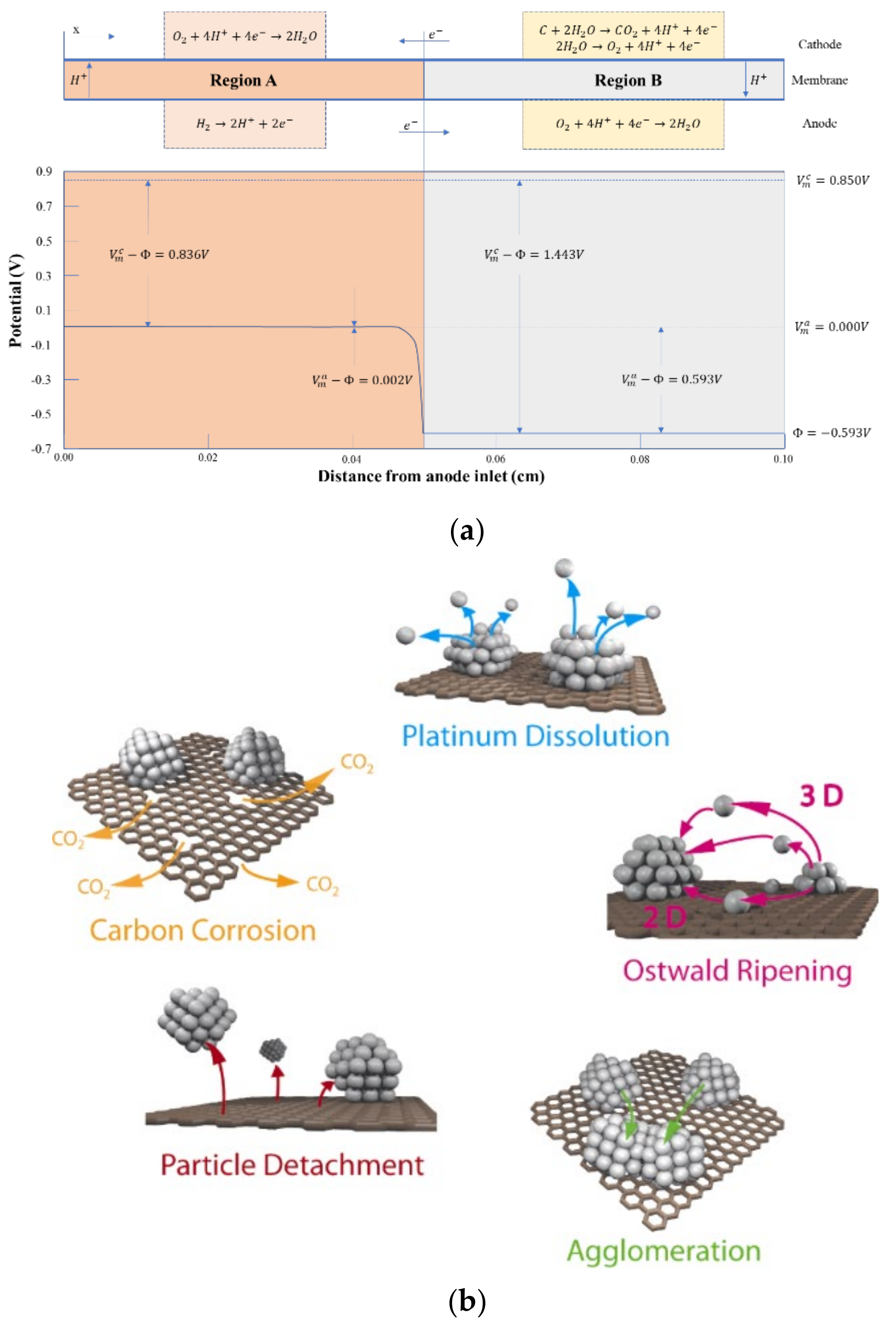
- (1)
- Dissolution and redeposition of Pt particles [135] (Ostwald ripening): Within the fuel cell environment, Pt nanoparticles tend to dissolve into ionic form at high potential and re-precipitate on the surface of large particles at low potential, resulting in the continuous reduction of small particles and the continuous growth of large particles, or the dissolved Pt ions will diffuse into PEM and be reduced to Pt particles in the presence of hydrogen. The dissolution process of Pt particles can be described as follows:
- (2)
- (3)
- Corrosion of carbon support [130,140]: It is consistent with the degradation mechanism of carbon materials in CL described in Section Corrosion of Carbon Support. Under high potential conditions, the carbon support of catalyst is oxidizes and corrodes easily, resulting in the loss of catalytic activity of Pt nanoparticles due to the lack of electron transport channel. At the same time, the electrons released from the carbon corrosion reaction will further accelerate the dissolution and precipitation of Pt particles.
2.6. Structural Optimization of CL
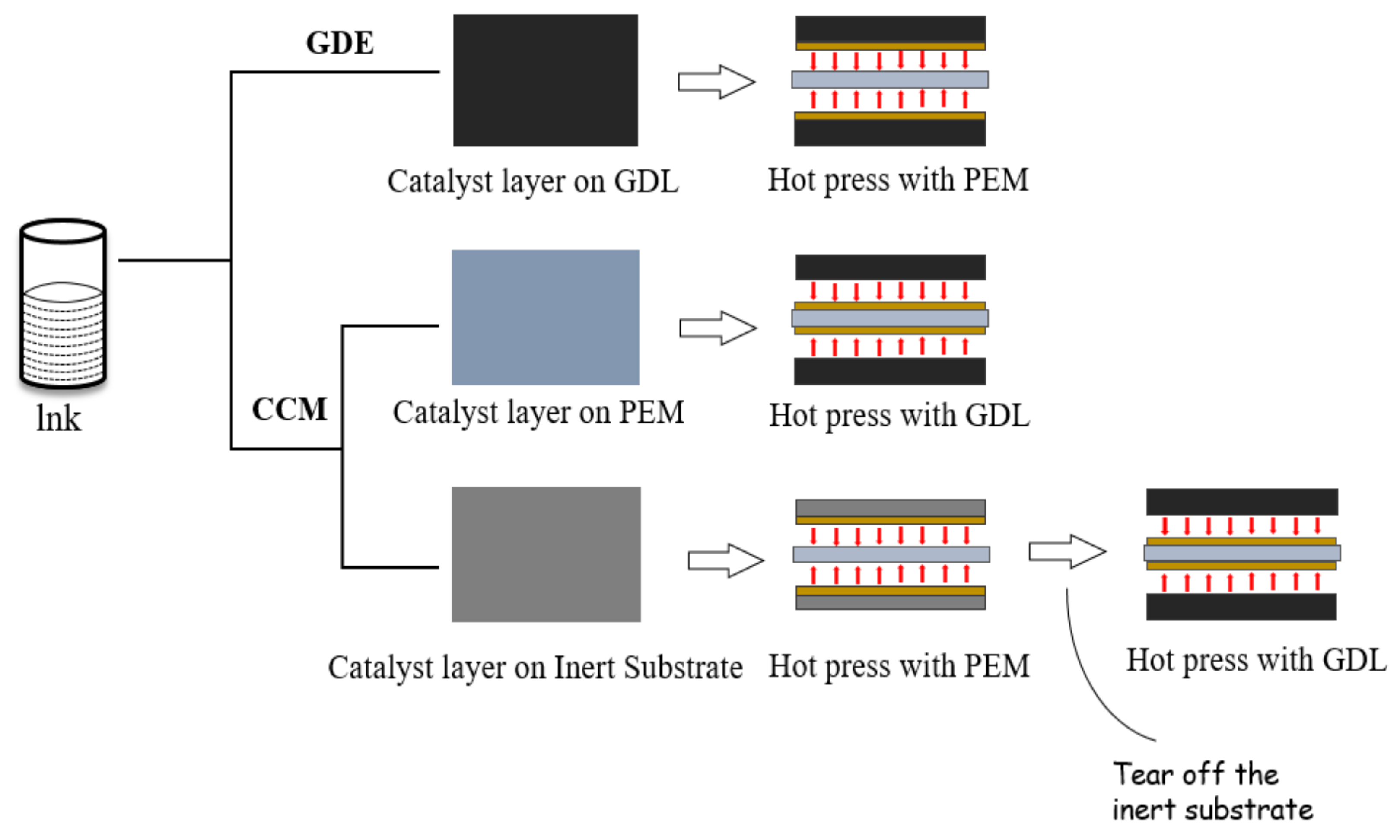
3. Preparation of CL
3.1. Coating Process of CL
- (1)
- Spraying method [175]
3.2. Drying Process of CL
- (1)
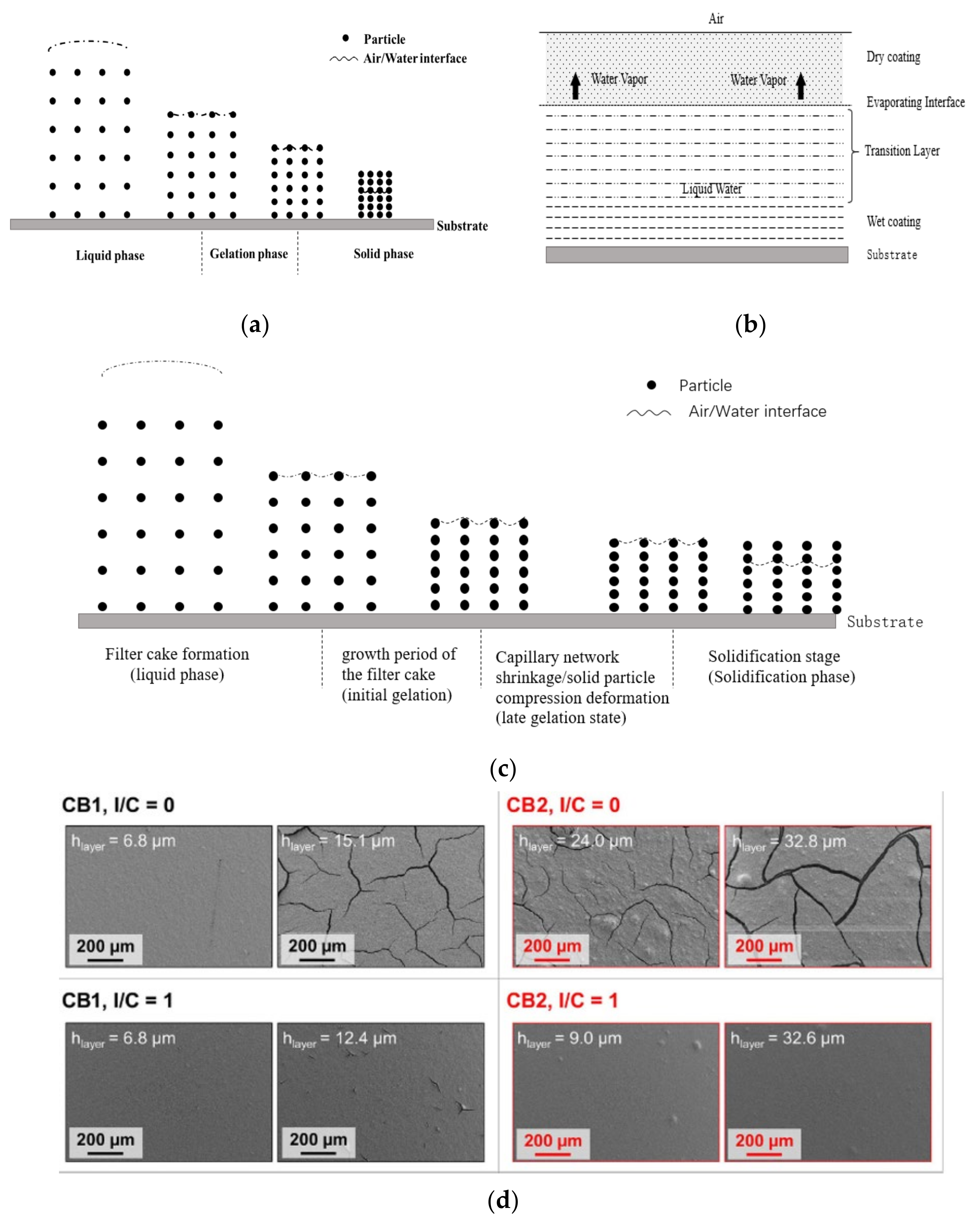
- After the catalyst slurry coats on PEM, the wet coating surface is covered by a water-like film. The coating has the characteristics of stable suspension, such as particles being able to move under the action of thermal movement and diffusion of liquid molecules, and the wet coating can flow under the action of external forces. Liquid evaporation mainly occurs at the interface between the wet coating surface and the air. The volume of the evaporating solvent is equivalent to the change of volume in the wet coating. This process is called the liquid phase.
- (2)
- The duration of the liquid phase relates to many factors, such as the slurry solid content, the temperature, humidity and so on. As the solvent continues to evaporate, the water-like film on the wet coating surface disappears. A three-dimensional capillary network forms between the carbon-based Pt catalyst and the Nafion binder within the coating, which could still contract and deform although it can greatly restrict the free movement of particles. In this process, the evaporation of solvent still occurs on the coating surface, but it is different from the position of the liquid phase, at the interface between the meniscus surface formed by the solvent in the capillary and the air. Since CL has formed the capillary structure, the solvent can be transferred to the evaporation surface (the interface between the meniscus surface of the capillary and the air) by capillary force to maintain the solvent evaporation process. Due to the continuous evaporation, the volume of the coating further shrinks and the gap between the solid particles (catalyst particles and Nafion particles) reduces gradually, resulting in gradual increase of capillary force. When the solid content reaches a certain degree, the solid particles compress and deform under the action of capillarity until they no longer change. This process is called the gelation phase.
- (3)
- Finally, there is the solid phase. When the solid particles of the coating are no longer deformed, the amount of solvent in the coating is insufficient to continue to transfer to the coating surface through the capillary tube for evaporation, and the evaporation interface reduces from the outside to the inside gradually, causing air to occupy the void above the evaporation interface. This process determines the spatial distribution and the pore size distribution of the catalyst, Nafion and other compositions within CL.
- (1)
- Filter cake formation (liquid phase). The stage from the slurry coats on substrate, to continuous water-like film disappears on the wet coating surface.
- (2)
- The growth period of the filter cake (initial gelation). The stage from the continuous water-like film disappears, and the free water disappears in the wet coating. Free water does not contain the water confined to capillaries formed by particles.
- (3)
- Capillary network shrinkage/solid particle compression deformation (late gelation state). This stage begins with the evaporation of water in the capillaries, until the coating is no longer in compressed deformation. In the second stage, the loss of free water and the shrinking of coating volume lead to the decrease of particle spacing, and the increase of capillary force. The evaporation interface stays on the meniscus surface of the coating surface. With the increase of capillary force, the compressible particles in the coating deform gradually until the particle spacing cannot be reduced. This stage relates to the content of compressible material and its elastic modulus within the coating.
- (4)
- Solidification stage (solidification phase). In the final stage, the coating structure has been formed and no longer is compressed or deformed with the increase of capillary force. The evaporation interface moves down gradually until it makes contact with the substrate, which is the same as the last stage of Lepoutre’s classic coating solidification theory.
4. Conclusions and Prospects
4.1. Conclusions
4.2. Prospects and Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arcot, M.P.; Zheng, K.; McGrory, J.; Fowler, M.W.; Pritzker, M.D. Investigation of catalyst layer defects in catalyst-coated membrane for PEMFC application: Non-destructive method. Int. J. Energy Res. 2018, 42, 3615–3632. [Google Scholar] [CrossRef]
- Nanadegani, F.S.; Lay, E.N.; Sunden, B. Effects of an MPL on water and thermal management in a PEMFC. Int. J. Energy Res. 2019, 43, 274–296. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, N.K.; Chatterjee, A.; Harris, T.A.L. Effect of cell compression on the performance of a non-hot-pressed MEA for PEMFC. Int. J. Energy Res. 2019, 44, 370–387. [Google Scholar] [CrossRef]
- Sassin, M.B.; Garsany, Y.; Gould, B.D.; Swider-Lyons, K. Impact of Compressive Stress on MEA Pore Structure and Its Consequence on PEMFC Performance. J. Electrochem. Soc. 2016, 163, F808–F815. [Google Scholar] [CrossRef]
- Fu, X.G.; Gao, R.; Jiang, G.P.; Li, M.; Li, S.; Luo, D.; Hu, Y.F.; Yuan, Q.X.; Huang, W.X.; Zhu, N.; et al. Evolution of atomic-scale dispersion of FeNx in hierarchically porous 3D air electrode to boost the interfacial electrocatalysis of oxygen reduction in PEMFC. Nano Energy 2021, 83, 105734. [Google Scholar] [CrossRef]
- Dyantyi, N.; Parsons, A.; Sita, C.; Pasupathi, S. PEMFC for aeronautic applications: A review on the durability aspects. Open Eng. 2017, 7, 287–302. [Google Scholar] [CrossRef]
- Jha, V.; Hariharan, R.; Krishnamurthy, B. A 3 dimensional numerical model to study the effect of GDL porosity on high temperature PEM fuel cells. Int. J. Heat Mass Transf. 2020, 161, 120311. [Google Scholar] [CrossRef]
- U.S. Drive Partnership. Fuel Cell Technical Team Roadmap. Available online: http://energy.gov/sites/prod/files/2014/02/f8/fctt_roadmap_june2013.pdf (accessed on 8 October 2021).
- Borup, R.; Meyers, J.; Pivova, B.; Kim, Y.S.; Mukundan, R.; Garland, N. Scientific Aspects of Polymer Electrolyte Fuel Cell Durability and Degradation. Chem. Rev. 2007, 107, 3904–3951. [Google Scholar] [CrossRef]
- Huang, X.Y.; Solasi, R.; Zou, Y.; Feshler, M.; Reifsnider, K.; Condit, D.; Burlatsky, S.; Madden, T. Mechanical endurance of polymer electrolyte membrane and PEM fuel cell durability. J. Polym. Sci. Part B. Polym. Phys. 2006, 44, 2346–2357. [Google Scholar] [CrossRef]
- Pan, C.F.; Wu, H.; Wang, C.; Wang, B.; Zhang, L.; Cheng, Z.D.; Hu, P.; Pan, W.; Zhou, Z.Y.; Yang, X.; et al. Nanowire-Based High-Performance “Micro Fuel Cells”: One Nanowire, One Fuel Cell. Adv. Mater. 2008, 20, 1644–1648. [Google Scholar] [CrossRef]
- Wilson, M.S.; Gottesfeld, S. High performance catalyzed membrane of ultra-low Pt loading for PEMFCs. J. Electrochem. Soc. 1992, 139, L28–L30. [Google Scholar] [CrossRef]
- Ticianelli, E.A.; Derouin, C.R.; Redondo, A.; Srinivasan, S. Methods to Advance Technology of Proton Exchange Membrane Fuel Cells. J. Electrochem. Soc. 1988, 135, 2209–2214. [Google Scholar] [CrossRef]
- Schulenburg, H.; Schwanitz, B.; Linse, N.; Scherer, G.G.; Wokaun, A.; Krbanjevic, J.; Grothausmann, R.; Manke, I. 3D Imaging of Catalyst Support Corrosion in Polymer Electrolyte Fuel Cells. J. Phys. Chem. C. 2011, 115, 14236–14243. [Google Scholar] [CrossRef]
- Vielstich, W.; Lamm, A.; Gasteiger, H.A. Handbook of Fuel Cells, Mechanisms of Membrane Degradation; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2003. [Google Scholar]
- Castanheira, L.; Silva, W.O.; Lima, F.H.B.; Crisci, A.; Dubau, L.; Maillard, F. Carbon Corrosion in Proton-Exchange Membrane Fuel Cells: Effect of the Carbon Structure, the Degradation Protocol, and the Gas Atmosphere. ACS Catal. 2015, 5, 2184–2194. [Google Scholar] [CrossRef]
- DOE. DOE Hydrogen and Fuel Cells Program: 2015 Annual Progress Report. Available online: https://www.nrel.gov/docs/fy16osti/64753.pdf (accessed on 8 October 2021).
- Thompson, S.T.; Papageorgopoulos, D. Platinum group metal-free catalysts boost cost competitiveness of fuel cell vehicles. Nat. Catal. 2019, 2, 558–561. [Google Scholar] [CrossRef]
- Shi, J.F.; Zhang, P.C.; Han, Y.T.; Wang, H.Y.; Wang, X.Y.; Yu, Y.S.; Sun, J.C. Investigation on electrochemical behavior and surface conductivity of titanium carbide modified Ti bipolar plate of PEMFC. Int. J. Hydrog. Energy 2020, 45, 10050–10058. [Google Scholar] [CrossRef]
- Sasiwimonrit, K.; Chang, W.C. To improve the high temperature polymer electrolyte membrane fuel cells performance by altering the properties of catalyst layer. Int. J. Hydrog. Energy 2020, 45, 14491–14499. [Google Scholar] [CrossRef]
- Sohn, Y.J.; Yim, S.D.; Park, G.G.; Kim, M.J.; Cha, S.W.; Kim, K. PEMFC modeling based on characterization of effective diffusivity in simulated cathode catalyst layer. Int. J. Hydrog. Energy 2017, 42, 13226–13233. [Google Scholar] [CrossRef]
- Wilson, M.S.; Valerio, J.A.; Gottesfeld, S. Low platinum loading electrodes for polymer electrolyte fuel cells fabricated using thermoplastic ionomers. Electrochim. Acta 1995, 40, 355–363. [Google Scholar] [CrossRef]
- Pu, X.T.; Duan, Y.T.; Li, J.L.; Ru, C.Y.; Zhao, C.J. Understanding of hydrocarbon ionomers in catalyst layers for enhancing the performance and durability of proton exchange membrane fuel cells. J. Power Sources 2021, 493, 229671. [Google Scholar] [CrossRef]
- Cheng, X.L.; Yi, B.L.; Han, M.; Zhang, J.X.; Qiao, Y.G.; Yu, J.R. Investigation of platinum utilization and morphology in catalyst layer of polymer electrolyte fuel cells. J. Power Sources 1999, 79, 75–81. [Google Scholar] [CrossRef]
- Wilson, M.S.; Gottesfeld, S. Thin-film catalyst layers for polymer electrolyte fuel-cell electrodes. J. Appl. Electrochem. 1992, 22, 1–7. [Google Scholar] [CrossRef]
- Passos, R.R.; Paganin, V.A.; Ticianelli, E.A. Studies of the performance of PEM fuel cell cathodes with the catalyst layer directly applied on Nafion membranes. Electrochim. Acta 2006, 51, 5239–5245. [Google Scholar] [CrossRef]
- Hoffmann, E.; Zhang, S.; Thoma, M.; Damm, C.; Peukert, W. Formulation of carbon black-ionomer dispersions for thin film formation in fuel cells. Particuology 2019, 44, 7–21. [Google Scholar] [CrossRef]
- Yang, L.J.; Shui, J.L.; Du, L.; Shao, Y.Y.; Liu, J.; Dai, L.M.; Hu, Z. Carbon-Based Metal-Free ORR Electrocatalysts for Fuel Cells: Past, Present, and Future. Adv. Mater. 2019, 31, e1804799. [Google Scholar] [CrossRef] [PubMed]
- Peera, S.G.; Maiyalagan, T.; Liu, C.; Ashmath, S.; Lee, T.G.; Jiang, Z.Q.; Mao, S. A review on carbon and non-precious metal based cathode catalysts in microbial fuel cells. Int. J. Hydrog. Energy 2021, 46, 3056–3089. [Google Scholar] [CrossRef]
- Litster, S.; McLean, G. PEM fuel cell electrodes. J. Power Sources 2004, 130, 61–76. [Google Scholar] [CrossRef]
- Miyazaki, K.; Abe, T.; Nishio, K.; Nakanishi, H.; Ogumi, Z. Use of layered double hydroxides to improve the triple phase boundary in anion-exchange membrane fuel cells. J. Power Sources 2010, 195, 6500–6503. [Google Scholar] [CrossRef]
- Li, J.K.; Brüller, S.; Sabarirajan, D.C.; Ranjbar-Sahraie, N.; Sougrati, M.T.; Cavaliere, S.; Jones, D.; Zenyuk, I.V.; Zitolo, A.; Jaouen, F. Designing the 3D Architecture of PGM-Free Cathodes for H2/Air Proton Exchange Membrane Fuel Cells. ACS Appl. Energy Mater. 2019, 2, 7211–7222. [Google Scholar] [CrossRef]
- Liu, Q.T.; Li, Y.C.; Zheng, L.R.; Shang, J.X.; Liu, X.F.; Yu, R.H.; Shui, J.L. Sequential Synthesis and Active-Site Coordination Principle of Precious Metal Single-Atom Catalysts for Oxygen Reduction Reaction and PEM Fuel Cells. Adv. Energy Mater. 2020, 10, 2000689. [Google Scholar] [CrossRef]
- Chen, M.; Kim, J.; Liu, J.P.; Fan, H.Y.; Sun, S.H. Synthesis of FePt Nanocubes and Their Oriented Self-Assembly. J. Am. Chem. Soc. 2006, 128, 7132–7133. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.H.; Gan, L.; Heggen, M.; Rudi, S.; Strasser, P. Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis. Nat. Mater. 2013, 12, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Xie, S.F.; Shao, M.H.; Odell, J.H.; Lu, N.; Peng, H.C.; Protsailo, L.; Guerrero, S.; Park, J.; Xia, X.H.; et al. Synthesis and characterization of 9 nm Pt-Ni octahedra with a record high activity of 3.3 A/mg(Pt) for the oxygen reduction reaction. Nano Lett. 2013, 13, 3420–3425. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Marín, A.M.; Feliu, J.M. Oxygen reduction on nanostructured platinum surfaces in acidic media: Promoting effect of surface steps and ideal response of Pt(111). Catal. Today 2015, 244, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Hoshi, N.; Nakamura, M.; Hitotsuyanagi, A. Active sites for the oxygen reduction reaction on the high index planes of Pt. Electrochim. Acta 2013, 112, 899–904. [Google Scholar] [CrossRef]
- Wang, Y.J.; Zhao, N.N.; Fang, B.Z.; Li, H.; Bi, X.T.; Wang, H.J. Carbon-supported Pt-based alloy electrocatalysts for the oxygen reduction reaction in polymer electrolyte membrane fuel cells: Particle size, shape, and composition manipulation and their impact to activity. Chem. Rev. 2015, 115, 3433–3467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Mueller, T. Rational Design of Pt3Ni Surface Structures for the Oxygen Reduction Reaction. J. Phys. Chem. C. 2015, 119, 17735–17747. [Google Scholar] [CrossRef]
- Hwang, S.J.; Kim, S.K.; Lee, J.G.; Lee, S.C.; Jang, J.H.; Kim, P.; Lim, T.H.; Sung, Y.E.; Sung, J.Y. Role of electronic perturbation in stability and activity of Pt-based alloy nanocatalysts for oxygen reduction. J. Am. Chem. Soc. 2012, 134, 19508–19511. [Google Scholar] [CrossRef]
- Zhao, D.; Xu, B.Q. Platinum covering of gold nanoparticles for utilization enhancement of Pt in electrocatalysts. Phys. Chem. Chem. Phys. 2006, 8, 5106–5114. [Google Scholar] [CrossRef]
- Wang, G.W.; Huang, B.; Xiao, L.; Ren, Z.D.; Chen, H.; Wang, D.L.; Abruña, H.D.; Lu, J.T.; Zhuang, L. Pt skin on AuCu intermetallic substrate: A strategy to maximize Pt utilization for fuel cells. J. Am. Chem. Soc. 2014, 136, 9643–9649. [Google Scholar] [CrossRef]
- Chen, Y.M.; Liang, Z.X.; Yang, F.; Liu, Y.W.; Chen, S.L. Ni-Pt Core-Shell Nanoparticles as Oxygen Reduction Electrocatalysts: Effect of Pt Shell Coverage. J. Phys. Chem. C. 2011, 115, 24073–24079. [Google Scholar] [CrossRef]
- Shin, D.Y.; Shin, Y.J.; Kim, M.S.; Kwon, J.A.; Lim, D.H. Density functional theory–based design of a Pt-skinned PtNi catalyst for the oxygen reduction reaction in fuel cells. Appl. Surf. Sci. 2021, 565, 150518. [Google Scholar] [CrossRef]
- Toyoda, M.; Yano, T.; Tryba, B.; Mozia, S.; Tsumura, T.; Inagaki, M. Preparation of carbon-coated Magneli phases TinO2n−1 and their photocatalytic activity under visible light. Appl. Catal. B. Environ. 2009, 88, 160–164. [Google Scholar] [CrossRef]
- Shao, Y.Y.; Yin, G.P.; Gao, Y.Z. Understanding and approaches for the durability issues of Pt-based catalysts for PEM fuel cell. J. Power Sources 2007, 171, 558–566. [Google Scholar] [CrossRef]
- Han, D.B.; Tsipoaka, M.; Shanmugam, S. A modified cathode catalyst layer with optimum electrode exposure for high current density and durable proton exchange membrane fuel cell operation. J. Power Sources 2021, 496, 229816. [Google Scholar] [CrossRef]
- Antolini, E.; Gonzalez, E.R. Ceramic materials as supports for low-temperature fuel cell catalysts. Solid State Ion. 2009, 180, 746–763. [Google Scholar] [CrossRef]
- Yao, C.L.; Yu, J.; Yang, S.J.; Shen, Y.H.; Xie, A.J. Porous Co/NPC@TiO2/TiN composite: Facile preparation and excellent catalytic activity for the oxygen reduction reaction. J. Alloy. Compd. 2021, 883, 160838. [Google Scholar] [CrossRef]
- Seger, B.; Kongkanand, A.; Vinodgopal, K.; Kamat, P.V. Platinum dispersed on silica nanoparticle as electrocatalyst for PEM fuel cell. J. Electroanal Chem. 2008, 621, 198–204. [Google Scholar] [CrossRef]
- Bernardis, F.L.; Grant, R.A.; Sherrington, D.C. A review of methods of separation of the platinum-group metals through their chloro-complexes. React. Funct. Polym. 2005, 65, 205–217. [Google Scholar] [CrossRef]
- DOE. Hydrogen and Fuel Cell Technologies Office: Hydrogen and Fuel Cell Technologies Office Multi-Year Research, Development, and Demonstration (2016) Plan.3.4 Fuel Cells. Available online: https://www.energy.gov/sites/default/files/2017/05/f34/fcto_myrdd_fuel_cells.pdf (accessed on 8 October 2021).
- Ram, S.; Dusan, S.; Vojislav, S.; Nenad, M.M. Three Phase Interfaces at Electrified Metal-Solid Electrolyte Systems 1. Study of the Pt(hkl)−Nafion Interface. J. Phys. Chem. C. 2010, 114, 8414–8422. [Google Scholar] [CrossRef]
- Subbaraman, R.; Strmcnik, D.; Paulikas, A.P.; Stamenkovic, V.R.; Markovic, N.M. Oxygen reduction reaction at three-phase interfaces. Chemphyschem J. Chem. Phys. Phys. Chem. 2010, 11, 2825–2833. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Morimoto, Y.; Pivovar, B.S.; Kocha, S.S. Suppression of oxygen reduction reaction activity on Pt-based electrocatalysts from ionomer incorporation. J. Power Sources 2016, 325, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Kodama, K.; Jinnouchi, R.; Suzuki, T.; Murata, H.; Hatanaka, T.; Morimoto, Y. Increase in adsorptivity of sulfonate anions on Pt (111) surface with drying of ionomer. Electrochem. Commun. 2013, 36, 26–28. [Google Scholar] [CrossRef]
- Kodama, K.; Shinohara, A.; Hasegawa, N.; Shinozaki, K.; Jinnouchi, R.; Suzuki, T.; Hatanaka, T.; Morimoto, Y. Catalyst Poisoning Property of Sulfonimide Acid Ionomer on Pt (111) Surface. J. Electrochem. Soc. 2014, 161, F649–F652. [Google Scholar] [CrossRef]
- Kodama, K.; Motobayashi, K.; Shinohara, A.; Hasegawa, N.; Kudo, K.; Jinnouchi, R.; Osawa, M.; Morimoto, Y. Effect of the Side-Chain Structure of Perfluoro-Sulfonic Acid Ionomers on the Oxygen Reduction Reaction on the Surface of Pt. ACS Catal. 2018, 8, 694–700. [Google Scholar] [CrossRef]
- Huang, Z.; Tu, H.L.; Zhang, J.Q.; Zhan, F. The Research and Applications of Proton Exchange Membrane Fuel Cells; Publishing Company of Metallurgical Industry: Beijing, China, 2000. (In Chinese) [Google Scholar]
- Vinothkannan, M.; Kim, A.R.; Yoo, D.J. Potential carbon nanomaterials as additives for state-of-the-art Nafion electrolyte in proton-exchange membrane fuel cells: A concise review. RSC Adv. 2021, 11, 18351–18370. [Google Scholar] [CrossRef]
- Savadogo, O. Emerging membranes for electrochemical systems. J. Power Sources 2004, 127, 135–161. [Google Scholar] [CrossRef]
- Halim, J.; Büchi, F.N.; Haas, O.; Stamma, M.; Scherer, G.G. Characterization of perfluorosulfonic acid membranes by conductivity measurements and small-angle X-ray scattering. Electrochim. Acta 1994, 39, 1303–1307. [Google Scholar] [CrossRef]
- Hsu, W.Y.; Gierke, T.D. Ion transport and clustering in Nafion perfluorinated membranes. J. Membr. Sci. 1983, 1, 307–326. [Google Scholar] [CrossRef]
- Yi, B.L. Fuel Cell: Principle, Technology and Application; Chemical Industry Press: Beijing, China, 2003. [Google Scholar]
- Yu, X.; Yuan, J.; Sundén, B. Review on the Properties of Nano-/Microstructures in the Catalyst Layer of PEMFC. J. Fuel Cell Sci. Technol. 2011, 8, 034001. [Google Scholar] [CrossRef]
- Liu, H.; Epting, W.K.; Litster, S. Gas Transport Resistance in Polymer Electrolyte Thin Films on Oxygen Reduction Reaction Catalysts. Langmuir ACS J. Surf. Colloids 2015, 31, 9853–9858. [Google Scholar] [CrossRef] [PubMed]
- More, K.; Borup, R.; Reeves, K. Identifying Contributing Degradation Phenomena in PEM Fuel Cell Membrane Electrode Assemblies Via Electron Microscopy. ECS Trans. 2006, 3, 717–733. [Google Scholar] [CrossRef]
- Zhang, S.S.; Yuan, X.Z.; Hin, J.N.C.; Wang, H.J.; Friedrich, K.A.; Schulze, M. A review of platinum-based catalyst layer degradation in proton exchange membrane fuel cells. J. Power Sources 2009, 194, 588–600. [Google Scholar] [CrossRef]
- Pharoah, J.G.; Peppley, B.; Atiyeh, H.; Halliop, E.; Karan, K.; Phoenix, A. Investigating the Role of a Microporous Layer on the Water Transport and Performance of a PEMFC. ECS Trans. 2006, 3, 1227–1237. [Google Scholar] [CrossRef]
- Uchida, M.; Aoyama, Y.; Eda, N.; Ohta, A. Investigation of the Microstructure in the Catalyst Layer and Effects of Both Perfluorosulfonate Ionomer and PTFE-Loaded Carbon on the Catalyst Layer of Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 1995, 142, 4143–4149. [Google Scholar] [CrossRef]
- Zeng, R.; Zhang, H.Y.; Jiang, L.J.; Liua, X.P. Investigation of Microstructure and Pt Distribution in Catalyst Layer after Accelerated Degradation Test in PEMFC. ECS Trans. 2017, 80, 215–223. [Google Scholar] [CrossRef]
- Lee, S.J.; Mukerjee, S.; McBreen, J.; Rho, Y.W.; Kho, Y.T.; Lee, T.H. Effects of Nafion impregnation on performances of PEMFC electrodes. Electrochi. Acta 1998, 43, 3693–3701. [Google Scholar] [CrossRef]
- Fultz, D.W.; Chuang, P.Y.A. The Property and Performance Differences Between Catalyst Coated Membrane and Catalyst Coated Diffusion Media. J. Fuel Cell Sci. Technol. 2011, 8, 41010. [Google Scholar] [CrossRef]
- Paul, M.T.Y.; Saha, M.S.; Qi, W.L.; Stumper, J.; Gates, B.D. Microstructured membranes for improving transport resistances in proton exchange membrane fuel cells. Int. J. Hydrog. Energy 2020, 45, 1304–1312. [Google Scholar] [CrossRef]
- Omosebi, A.; Besser, R.S. Ultra-low Mass Sputtered and Conventional Catalyst Layers on Plasma-etched Nafion for PEMFC Applications. Fuel Cells 2017, 17, 762–769. [Google Scholar] [CrossRef]
- Bae, J.W.; Cho, Y.H.; Sung, Y.E.; Shin, K.; Jho, J.Y. Performance enhancement of polymer electrolyte membrane fuel cell by employing line-patterned Nafion membrane. J. Ind. Eng. Chem. 2012, 18, 876–879. [Google Scholar] [CrossRef]
- Kim, S.M.; Kang, Y.S.; Ahn, C.; Jang, S.; Kim, M.; Sung, Y.E.; Yoo, S.J.; Choi, M. Prism-patterned Nafion membrane for enhanced water transport in polymer electrolyte membrane fuel cell. J. Power Sources 2016, 317, 19–24. [Google Scholar] [CrossRef]
- Cho, H.; Kim, S.M.; Kang, Y.S.; Kim, J.; Jang, S.; Kim, M.; Park, H.; Bang, J.W.; Seo, S.; Suh, K.Y.; et al. Multiplex lithography for multilevel multiscale architectures and its application to polymer electrolyte membrane fuel cell. Nat. Commun. 2015, 6, 8484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.J.; Huang, X.M.; Chang, H.W.; Hwa Chan, S.; Tu, Z.K. Effects of operating temperature on the carbon corrosion in a proton exchange membrane fuel cell under high current density. Energ. Convers. Mange. 2021, 10, 100087. [Google Scholar] [CrossRef]
- Zhang, W.Q.; Yao, D.M.; Tian, L.L.; Xie, Z.; Ma, Q.; Xu, Q.; Pasupathi, S.; Xing, L.; Su, H.N. Enhanced performance of high temperature polymer electrolyte membrane fuel cell using a novel dual catalyst layer structured cathode. J. Taiwan Inst. Chem. Eng. 2021, 125, 285–290. [Google Scholar] [CrossRef]
- Lin, R.; Yu, X.T.; Chen, L.; Tang, S.H.; Yin, X.L.; Hao, Z.X. Structure majorization on the surface of microporous layer in polymer electrolyte membrane fuel cells to optimize performance and durability. Energy Convers. Manag. 2021, 243, 114319. [Google Scholar] [CrossRef]
- Schneider, I.A.; Kuhn, H.; Wokaun, A.; Scherer, G.G. Study of Water Balance in a Polymer Electrolyte Fuel Cell by Locally Resolved Impedance Spectroscopy. J. Electrochem. 2005, 152, A2383–A2389. [Google Scholar] [CrossRef]
- Kannan, A.M.; Cindrella, L.; Munukutla, L. Functionally graded nano-porous gas diffusion layer for proton exchange membrane fuel cells under low relative humidity conditions. Electrochim. Acta 2008, 53, 2416–2422. [Google Scholar] [CrossRef]
- Chen, J.H.; Matsuura, T.; Hori, M. Novel gas diffusion layer with water management function for PEMFC. J. Power Sources 2004, 131, 155–161. [Google Scholar] [CrossRef]
- Li, B.; Xie, M.; Ji, H.; Chu, T.K.; Yang, D.J.; Ming, P.W.; Zhang, C.M. Optimization of cathode microporous layer materials for proton exchange membrane fuel cell. Int. J. Hydrog. Energy 2021, 46, 14674–14686. [Google Scholar] [CrossRef]
- Mao, Z.Q. Fuel Cell; Chemical Industry Press: Beijing, China, 2005. [Google Scholar]
- Damjanovic, A.; Brusic, V. Electrode kinetics of oxygen reduction on oxide free platinum electrodes. Electrochim. Acta 1967, 12, 615–628. [Google Scholar] [CrossRef]
- Vielstich, W.; Lamm, A.; Gasteiger, H.A. Handbook of Fuel Cells; John and Wiley and Sons: Hoboken, NY, USA, 2003; Volume 2. [Google Scholar]
- Berger, C. Handbook of Fuel Cell Technology; Prentice-Hall: Englewood Cliffs, NJ, USA, 1968. [Google Scholar]
- Eslamibidgoli, M.J.; Huang, J.; Kadyk, T.; Malek, A.; Eikerling, M. How theory and simulation can drive fuel cell electrocatalysis. Nano Energy 2016, 29, 334–361. [Google Scholar] [CrossRef]
- Yuan, Y.; Qu, Z.G.; Wang, W.K.; Ren, G.F.; Hu, B.B. Illustrative Case Study on the Performance and Optimization of Proton Exchange Membrane Fuel Cell. Chem. Eng. 2019, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Middelman, E. Improved PEM fuel cell electrodes by controlled self-assembly. Fuel Cells Bull. 2002, 1, 9–12. [Google Scholar] [CrossRef]
- Rajalakshmi, N.; Dhathathreyan, K.S. Catalyst layer in PEMFC electrodes—Fabrication, characterisation and analysis. Chem. Eng. J. 2007, 129, 31–40. [Google Scholar] [CrossRef]
- Kulkarni, A.; Siahrostami, S.; Patel, A. Understanding catalytic activity trends in the oxygen reduction reaction. Chem. Rev. 2018, 118, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; TWagner, F. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl Catal. B. Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Wang, W.T.; Chen, S.Q.; Li, J.J.; Wang, W. Fabrication of catalyst coated membrane with screen printing method in a proton exchange membrane fuel cell. Int. J. Hydrog. Energy 2015, 40, 4649–4658. [Google Scholar] [CrossRef]
- Kundu, S.; Fowler, M.W.; Simon, L.C.; Grot, S. Morphological features (defects) in fuel cell membrane electrode assemblies. J. Power Sources 2006, 157, 650–656. [Google Scholar] [CrossRef]
- Huang, D.C.; Yu, P.J.; Liu, F.J.; Huang, S.L.; Hsueh, K.L.; Chen, Y.C.; Wu, C.H.; Chang, W.C.; Tsau, F.H. Effect of dispersion solvent in catalyst ink on proton exchange membrane fuel cell performance. Int. J. Electrochem. Sci. 2011, 6, 2551–2565. [Google Scholar]
- Therdthianwong, A.; Ekdharmasuit, P.; Therdthianwong, S. Fabrication and Performance of Membrane Electrode Assembly Prepared by a Catalyst-Coated Membrane Method: Effect of Solvents Used in a Catalyst Ink Mixture. Energy Fuels 2010, 24, 1191–1196. [Google Scholar] [CrossRef]
- Komoda, Y.; Okabayashi, K.; Nishimura, H.; Hiromitsu, M.; Oboshi, T.; Usui, H. Dependence of polymer electrolyte fuel cell performance on preparation conditions of slurry for catalyst layers. J. Power Sources 2009, 193, 488–494. [Google Scholar] [CrossRef]
- Dixit, M.B.; Harkey, B.A.; Shen, F.Y.; Hatzell, K.B. Catalyst Layer Ink Interactions That Affect Coatability. J. Electrochem. Soc. 2018, 165, F264–F271. [Google Scholar] [CrossRef]
- Kusano, T.; Hiroi, T.; Amemiya, K.; Ando, M.; Takahashi, T.; Shibayama, M. Structural evolution of a catalyst ink for fuel cells during the drying process investigated by CV-SANS. Polym. J. 2015, 47, 546–555. [Google Scholar] [CrossRef]
- Shi, J.R.; Zhan, Z.G.; Zhang, D.; Yu, Y.; Yang, X.X.; He, L.Y.; Pan, M. Effects of Cracks on the Mass Transfer of Polymer Electrolyte Membrane Fuel Cell with High Performance Membrane Electrode Assembly. J. Wuhan Univ. Technol. Mat. Sci. Edit. 2021, 36, 318–330. [Google Scholar] [CrossRef]
- Ahn, C.Y.; Jang, S.; Cho, Y.H.; Choi, J.; Kim, S.; Kim, S.M.; Sung, Y.E.; Choi, M. Guided cracking of electrodes by stretching prism-patterned membrane electrode assemblies for high-performance fuel cells. Sci. Rep. 2018, 8, 1257. [Google Scholar] [CrossRef]
- Kim, S.M.; Ahn, C.Y.; Cho, Y.H.; Kim, S.; Hwang, W.; Jang, S.; Shin, S.; Lee, G.; Sung, Y.E.; Choi, M. High-performance Fuel Cell with Stretched Catalyst-Coated Membrane: One-step Formation of Cracked Electrode. Sci. Rep. 2016, 6, 26503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsushima, S.; Hirai, S. An overview of cracks and interfacial voids in membrane electrode assemblies in polymer electrolyte fuel cells. J. Therm. Sci. Technol. 2015, 10, JTST0002. [Google Scholar] [CrossRef] [Green Version]
- Ramani, D.; Singh, Y.; Orfino, F.P.; Dutta, M.; Kjeang, E. Characterization of Membrane Degradation Growth in Fuel Cells Using X-ray Computed Tomography. J. Electrochem. Soc. 2018, 165, F3200–F3208. [Google Scholar] [CrossRef]
- Singh, Y.; Orfino, F.P.; Dutta, M.; Kjeang, E. 3D Failure Analysis of Pure Mechanical and Pure Chemical Degradation in Fuel Cell Membranes. J. Electrochem. Soc. 2017, 164, F1331–F1341. [Google Scholar] [CrossRef]
- White, R.T.; Wu, A.; Najm, M.; Orfino, F.P.; Dutta, M.; Kjeang, E. 4D in situ visualization of electrode morphology changes during accelerated degradation in fuel cells by X-ray computed tomography. J. Power Sources 2017, 350, 94–102. [Google Scholar] [CrossRef]
- Singh, Y.; Orfino, F.P.; Dutta, M.; Kjeang, E. 3D visualization of membrane failures in fuel cells. J. Power Sources 2017, 345, 1–11. [Google Scholar] [CrossRef]
- Pestrak, M.; Li, Y.Q.; Case, S.W.; Dillard, D.A.; Ellis, M.W.; Lai, Y.H.; Gittleman, C.S. The Effect of Mechanical Fatigue on the Lifetimes of Membrane Electrode Assemblies. J. Fuel Cell Sci. Technol. 2010, 7, 041009. [Google Scholar] [CrossRef]
- Banan, R.; Zu, J.; Bazylak, A. Humidity and Temperature Cycling Effects on Cracks and Delaminations in PEMFCs. Fuel Cells 2015, 15, 327–336. [Google Scholar] [CrossRef]
- You, X.G.; Feng, Z.L.; Ting, L. Study on the Effect of Cracks in Catalyst Layer on the Durability of PEMFC Membrane Electrode Assembly. Dongfang Electr. Rev. 2021, 35, 9–12. [Google Scholar] [CrossRef]
- Zhang, X.X.; Jiang, Y.; Sun, T.F.; Chen, W.R. Review on Fault Diagnosis for Flooding and Drying in Proton Exchange Membrane Fuel Cells. J. Southwest Jiaotong Univ. 2020, 55, 828–838. [Google Scholar] [CrossRef]
- Gopalan, P.; Kandlikar, S.G. Effect of channel materials and trapezoidal corner angles on emerging droplet behavior in Proton Exchange Membrane Fuel Cell gas channels. J. Power Sources 2014, 248, 230–238. [Google Scholar] [CrossRef]
- Yang, X.G.; Ye, Q.; Cheng, P. In-plane transport effects on hydrogen depletion and carbon corrosion induced by anode flooding in proton exchange membrane fuel cells. Int. J. Heat Mass Transf. 2012, 55, 4754–4765. [Google Scholar] [CrossRef]
- Patterson, T.W.; Darling, R.M. Damage to the Cathode Catalyst of a PEM Fuel Cell Caused by Localized Fuel Starvation. Electrochem. Solid State Lett. 2006, 9, A183–A185. [Google Scholar] [CrossRef]
- Paul, C.O.; Ikram, B.B.; Wilfred, E.; Paul, C.U. Nafion degradation mechanisms in proton exchange membrane fuel cell (PEMFC) system: A review. Int. J. Hydrog. Energy 2021, 46, 27956–27973. [Google Scholar] [CrossRef]
- Inaba, M.; Kinumoto, T.; Kiriake, M.; Umebayashi, R.; Tasaka, A.; Ogumi, Z. Gas crossover and membrane degradation in polymer electrolyte fuel cells. Electrochim. Acta 2006, 51, 5746–5753. [Google Scholar] [CrossRef]
- Liu, W.; David, Z. In Situ Detection of Hydrogen Peroxide in PEM Fuel Cells. J. Electrochem. Soc. 2005, 152, A1165–A1170. [Google Scholar] [CrossRef]
- Curtin, D.E.; Lousenberg, R.D.; Henry, T.J.; Tangeman, P.C.; Tisack, M.E. Advanced materials for improved PEMFC performance and life. J. Power Sources 2004, 131, 41–48. [Google Scholar] [CrossRef]
- Sethuraman, V.A.; Weidner, J.W.; Haug, A.T.; Motupally, S.; Protsailo, L.V. Hydrogen Peroxide Formation Rates in a PEMFC Anode and Cathode. J. Electrochem. Soc. 2008, 155, B50–B57. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, S.B.; Zhang, J.B.; Li, J.Q.; Wang, J.L.; Yang, M. The Durability Research on the Proton Exchange Membrane Fuel Cell for Automobile Application. Prog. Chem. 2015, 27, 424–435. [Google Scholar] [CrossRef]
- Zhang, F.Y.; Advani, S.G.; Prasad, A.K.; Boggs, M.E.; Sullivan, S.P.; Beebe, T.P. Quantitative characterization of catalyst layer degradation in PEM fuel cells by X-ray photoelectron spectroscopy. Electrochim. Acta 2009, 54, 4025–4030. [Google Scholar] [CrossRef]
- Maass, S.; Finsterwalder, F.; Frank, G.; Hartmann, R.; Merten, C. Carbon support oxidation in PEM fuel cell cathodes. J. Power Sources 2008, 176, 444–451. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, P.Q.; Chen, H.C.; Pei, P.C. A review of automotive proton exchange membrane fuel cell degradation under start-stop operating condition. Appl. Energy 2018, 223, 249–262. [Google Scholar] [CrossRef]
- Reiser, C.A.; Bregoli, L.; Patterson, T.W.; Yi, J.S.; Yang, J.D.; Perry, M.L.; Jarvi, T.D. A Reverse-Current Decay Mechanism for Fuel Cells. Electrochem. Solid-State Letters 2005, 8, A273–A276. [Google Scholar] [CrossRef]
- Meie, J.C.; Galeano, C.; Katsounaros, I.; Witte, J.; Bongard, H.J.; Topalov, A.A.; Baldizzone, C.; Mezzavilla, S.; Schüth, F.; Mayrhofer, K.J.J. Design criteria for stable Pt/C fuel cell catalysts. Beilstein J. Nanotechnol. 2014, 5, 44–67. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Qi, Z.G.; Ramani, M.; Elter, J.F. PEM fuel cell cathode carbon corrosion due to the formation of air/fuel boundary at the anode. J. Power Sources 2006, 158, 1306–1312. [Google Scholar] [CrossRef]
- Baschuk, J.J.; Li, X.G. Carbon monoxide poisoning of proton exchange membrane fuel cells. Int. J. Energy Res. 2001, 25, 695–713. [Google Scholar] [CrossRef]
- Fischer, T.E.; Kelemen, S.R. Model experiments on the poisoning of Pt catalysts by sulfur. J. Catal. 1978, 53, 24–34. [Google Scholar] [CrossRef]
- Chen, M.; Du, C.Y.; Zhang, J.; Wang, P.P.; Zhu, T. Effect, mechanism and recovery of nitrogen oxides poisoning on oxygen reduction reaction at Pt/C catalysts. J. Power Sources 2011, 196, 620–626. [Google Scholar] [CrossRef]
- Paul, C.O.; Oladeji, O.I.; El, M.B.; Paul, C.U.; Wilfred, E.; Abdelbaki, B.; Abdullah, A.M. Platinum degradation mechanisms in proton exchange membrane fuel cell (PEMFC) system: A review. Int. J. Hydrog. Energy 2021, 46, 15850–15865. [Google Scholar] [CrossRef]
- Voorhees, P.W. The theory of Ostwald ripening. J. Stat. Phys. 1985, 38, 231–252. [Google Scholar] [CrossRef] [Green Version]
- Darling, R.M.; Meyers, J.P. Kinetic Model of Platinum Dissolution in PEMFCs. J. Electrochem. Soc. 2003, 150, A1523. [Google Scholar] [CrossRef]
- Cheng, X.; Shi, Z.; Glass, N. A review of PEM hydrogen fuel cell contamination: Impacts, mechanisms and mitigation. J. Power Sources 2007, 165, 739–756. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, Z.; Zhao, X.; Xin, Q.; Sun, G.; Yi, B. Studies on performance degradation of a direct methanol fuel cell (DMFC) in life test. Phys. Chem. Chem. Phys. 2004, 6, 134–137. [Google Scholar] [CrossRef]
- Jian, X.; David, L.W.; Karren, L.M.; Plamen, A.; Rodney, L.B. Microstructural Changes of Membrane Electrode Assemblies during PEFC Durability Testing at High Humidity Conditions. J. Electrochem. Soc. 2005, 152, A1011–A1020. [Google Scholar] [CrossRef]
- Knights, S.D.; Colbow, K.M.; St-Pierre, J.; Wilkinson, D.P. Aging mechanisms and lifetime of PEFC and DMFC. J. Power Sources 2004, 127, 127–134. [Google Scholar] [CrossRef]
- Meier, J.C.; Galeano, C.; Katsounaros, I.; Topalov, A.A.; Kostka, A.; Schüth, F.; Mayrhofer, K.J.J. Degradation Mechanisms of Pt/C Fuel Cell Catalysts under Simulated Start–Stop Conditions. ACS Catal. 2012, 2, 832–843. [Google Scholar] [CrossRef]
- Cao, M.; Wu, D.S.; Cao, R. Recent Advances in the Stabilization of Platinum Electrocatalysts for Fuel-Cell Reactions. Chem. Cat. Chem. 2014, 6, 26–45. [Google Scholar] [CrossRef]
- Okaya, K.; Yano, H.; Uchida, H.; Watanabe, M. Control of particle size of pt and pt alloy electrocatalysts supported on carbon black by the nanocapsule method. ACS Appl. Mater. Interfaces 2010, 2, 888–895. [Google Scholar] [CrossRef]
- Tritsaris, G.A.; Greeley, J.; Rossmeisl, J.; Nørskov, J.K. Atomic-Scale Modeling of Particle Size Effects for the Oxygen Reduction Reaction on Pt. Catal. Lett. 2011, 141, 909–913. [Google Scholar] [CrossRef]
- Qi, Z.G.; Lefebvre, M.C.; Pickup, P.G. Electron and proton transport in gas diffusion electrodes containing electronically conductive proton exchange polymers. J. Electroanal. Chem. 1998, 495, 9–14. [Google Scholar] [CrossRef]
- Ye, S.Y.; Vijh, A.K.; Dao, L.H. A new fuel cell electrolyte based on highly porous carbonized polyacrylonitrite foam with very low platinum loading. J. Electrochem. Soc. 1996, 143, L7–L9. [Google Scholar] [CrossRef]
- Zhang, W.M.; Chen, J.; Minett, A.I.; Swiegers, G.F.; Too, C.O.; Wallace, G.G. Novel ACNT arrays based MEA structure-nano-Pt loaded ACNT/Nafion/ACNT for fuel cell applications. Chem. Commun. 2010, 46, 4824–4826. [Google Scholar] [CrossRef]
- Zeng, Y.C.; Shao, Z.G.; Zhang, H.J.; Wang, Z.Q.; Hong, S.J.; Yu, H.M.; Yi, B.L. Nanostructured ultrathin catalyst layer based on open-walled PtCo bimetallic nanotube arrays for proton exchange membrane fuel cells. Nano Energy 2017, 34, 344–355. [Google Scholar] [CrossRef]
- Shao, Z.G.; Yi, B.L.; Han, M. Bifunctional electrodes with a thin catalyst layer for ‘unitized’ proton exchange membrane regenerative fuel cell. J. Power Sources 1999, 79, 82–85. [Google Scholar] [CrossRef]
- Shao, Z.G.; Yi, B.L.; Han, M. The preparation method of thin-layer hydrophobic catalytic layer electrode and membrane electrode three-into-one component. Chinese Patent CN1269429A, 11 October 2000. [Google Scholar]
- Zhang, H.; Chung, H.T.; Cullen, D.A.; Wagner, S.; Kramm, U.I.; More, K.L.; Zelenay, P.; Wu, G. High-performance fuel cell cathodes exclusively containing atomically dispersed iron active sites. Energy Environ. Sci. 2019, 12, 2548–2558. [Google Scholar] [CrossRef]
- Su, H.N.; Liao, S.J.; Wu, Y.N. Significant improvement in cathode performance for proton exchange membrane fuel cell by a novel double catalyst layer design. J. Power Sources 2010, 195, 3477–3480. [Google Scholar] [CrossRef]
- Taylor, A.D.; Kim, E.Y.; Humes, V.P.; Kizuka, J.; Thompson, L.T. Inkjet printing of carbon supported platinum 3-D catalyst layers for use in fuel cells. J. Power Sources 2007, 171, 101–106. [Google Scholar] [CrossRef]
- Jain, P.; Biegler, L.T.; Jhon, M.S. Optimization of Polymer Electrolyte Fuel Cell Cathodes. Electrochem. Solid State Lett. 2008, 11, B193–B196. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, H.J.; Lee, K.Y.; Jang, J.H.; Lee, S.Y.; Cho, E.; Oh, I.H.; Lim, T.H. Effect of Nafion® gradient in dual catalyst layer on proton exchange membrane fuel cell performance. Int. J. Hydrog. Energy 2008, 33, 2783–2789. [Google Scholar] [CrossRef]
- Xie, Z.; Navessin, T.; Shi, K.; Chow, R.; Wang, Q.P.; Song, D.T.; Andreaus, B.; Eikerling, M.; Liu, Z.; Holdcroft, S.; et al. Functionally Graded Cathode Catalyst Layers for Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 2005, 152, A1171–A1179. [Google Scholar] [CrossRef]
- Qiu, Y.L.; Zhang, H.M.; Zhong, H.X.; Zhang, F.X. A novel cathode structure with double catalyst layers and low Pt loading for proton exchange membrane fuel cells. Int. J. Hydrog. Energy 2013, 38, 5836–5844. [Google Scholar] [CrossRef]
- Ye, L.C.; Gao, Y.; Zhu, S.Y.; Zheng, J.S.; Li, P.; Zheng, J.P. A Pt content and pore structure gradient distributed catalyst layer to improve the PEMFC performance. Int. J. Hydrog. Energy 2017, 42, 7241–7245. [Google Scholar] [CrossRef]
- Debe, M.K. Nanostructured thin film electrocatalysts for PEM fuel cells-a tutorial on the fundamental characteristics and practical properties of NSTF catalysts. Ecs. Trans. 2012, 45, 47–68. [Google Scholar] [CrossRef]
- Jung, U.H.; Park, K.T.; Park, E.H.; Kim, S.H. Improvement of low-humidity performance of PEMFC by addition of hydrophilic SiO2 particles to catalyst layer. J. Power Sources 2006, 159, 529–532. [Google Scholar] [CrossRef]
- Lin, C.L.; Hsu, S.C.; Ho, W.Y. Using SiO2 nanopowders in anode catalyst layer to improve the performance of a proton exchange membrane fuel cell at low humidity. J. Mater. Sci. Chem. Eng. 2015, 3, 72–79. [Google Scholar]
- Huang, R.H.; Chiu, T.W.; Lin, T.J.; Sun, C.H.; Chao, W.K.; Tsai, D.C.; Hsueh, K.L.; Shieu, F.S. Improvement of proton exchange membrane fuel cells performance by coating hygroscopic zinc oxide on the anodic catalyst layer. J. Power Sources 2013, 227, 229–236. [Google Scholar] [CrossRef]
- Chao, W.K.; Lee, C.M.; Tsai, D.C.; Chou, C.C.; Hsueh, K.L.; Shieu, F.S. Improvement of the proton exchange membrane fuel cell (PEMFC) performance at low-humidity conditions by adding hygroscopic γ-Al2O3 particles into the catalyst layer. J. Power Sources 2008, 185, 136–142. [Google Scholar] [CrossRef]
- Lindstrom, R.W. Electrocatalytic gas diffusion electrode employing thin carbon cloth layer. US Patent 46473591987, 25 June 1992. [Google Scholar]
- Ticianelli, E.A.; Derouin, C.R.; Srinivasan, S. Localization of platinum in low catalyst loading electrodes to attain high power densities in SPE fuel cells. J. Electroanal. Chem. 1988, 251, 275–295. [Google Scholar] [CrossRef]
- Wakizoe, M.; Velev, O.A.; Srinivasan, S. Analysis of proton exchange membrane fuel cell performance with alternate membranes. Electrochim. Acta 1995, 40, 335–344. [Google Scholar] [CrossRef]
- SasiKumar, G.; Raja, M.; Parthasarathy, S. High performance electrodes with very low platinum loading for polymer electrolyte fuel cells. Electrochim. Acta 1995, 40, 285–290. [Google Scholar] [CrossRef]
- Fuel Cell Technologies Office: DOE Technical Targets for Polymer Electrolyte Membrane Fuel Cell Components. Available online: https://www.energy.gov/eere/fuelcells/doe-technical-targets-polymer-electrolyte-membrane-fuel-cell-components#mea (accessed on 8 October 2021).
- Shahgaldi, S.; Alaefour, I.; Li, X.G. Impact of manufacturing processes on proton exchange membrane fuel cell performance. Appl. Energy 2018, 225, 1022–1032. [Google Scholar] [CrossRef]
- Lim, B.H.; Majlan, E.H.; Tajuddin, A.; Husaini, T.; Wan Daud, W.R.; Mohd Radzuan, N.A.; Haque, M.A. Comparison of catalyst-coated membranes and catalyst-coated substrate for PEMFC membrane electrode assembly: A review. Chin. J. Chem. Eng. 2021, 33, 1–16. [Google Scholar] [CrossRef]
- Shahgaldi, S.; Alaefour, I.; Unsworth, G.; Li, X.G. Development of a low temperature decal transfer method for the fabrication of proton exchange membrane fuel cells. Int. J. Hydrog. Energy 2017, 42, 11813–11822. [Google Scholar] [CrossRef]
- Zhou, C.; Liao, S.J. Membrane electrode assembly (MEA) of PEMFC prepared by catalyst coated membrane (CCM) technique. Chin. J. Power Sources 2007, 31, 469–471. [Google Scholar]
- Millington, B.; Whipple, V.; Pollet, B.G. A novel method for preparing proton exchange membrane fuel cell electrodes by the ultrasonic-spray technique. J. Power Sources 2011, 196, 8500–8508. [Google Scholar] [CrossRef]
- Zhang, Z.; Pan, M. A Comparison of Hydrogen-oxygen PEMFC MEAs Prepared by GDE and CCM Technology. J. Wuhan Univ. Technol. 2018, 40, 8–12. [Google Scholar] [CrossRef]
- Su, K.H.; Sui, S.; Yao, X.Y.; Wei, Z.X.; Zhang, J.L.; Du, S.F. Controlling Pt loading and carbon matrix thickness for a high performance Pt-nanowire catalyst layer in PEMFCs. Int. J. Hydrog. Energy 2014, 39, 3397–3403. [Google Scholar] [CrossRef]
- Su, H.N.; Jao, T.C.; Barron, O.; Pollet, B.G.; Pasupathi, S. Low platinum loading for high temperature proton exchange membrane fuel cell developed by ultrasonic spray coating technique. J. Power Sources 2014, 267, 155–159. [Google Scholar] [CrossRef]
- Pollet, B.G. The use of ultrasound for the fabrication of fuel cell materials. Int. J. Hydrog. Energy 2010, 35, 11986–12004. [Google Scholar] [CrossRef]
- Campana, D.M.; Valdez Silva, L.D.; Carvalho, M.S. Slot coating flows of non-colloidal particle suspensions. AIChE J. 2017, 63, 1122–1131. [Google Scholar] [CrossRef]
- Ding, X.; Liu, J.; Harris, T.A.L. A review of the operating limits in slot die coating processes. AIChE J. 2016, 62, 2508–2524. [Google Scholar] [CrossRef]
- Ma, J.X.; Yi, B.L.; Yu, H.M.; Hong, Z.J.; Zhang, H.M. Review on preparation method of Membrane Electrode Assembly(MEA) for PEMFC. Prog. Chem. 2004, 16, 804–812. [Google Scholar] [CrossRef]
- Xie, J.; Xu, F.; Wood, D.L.; More, K.L.; Zawodzinski, T.A.; Smith, W.H. Influence of ionomer content on the structure and performance of PEFC membrane electrode assemblies. Electrochim. Acta 2010, 55, 7404–7412. [Google Scholar] [CrossRef]
- Bevers, D.; Wagner, N.; Bradke, M.V. Innovative production procedure for low cost PEFC electrodes and electrode/membrane structures. Int. J. Hydrog. Energy 1998, 23, 57–63. [Google Scholar] [CrossRef]
- Gulzow, E.; Schulze, M.; Wagner, N.; Kaz, T.; Reissner, R.; Steinhilber, G. Dry layer preparation and characterisation of polymer electrolyte fuel cell components. J. Power Sources 2000, 86, 352–362. [Google Scholar] [CrossRef]
- Yen, Y.T.; Fang, T.H.; Lin, Y.C. Optimization of screen-printing parameters of SN9000 ink for pinholes using Taguchi method in chip on film packaging. Robot. Comput. Intergr. Manuf. 2011, 27, 531–537. [Google Scholar] [CrossRef]
- Wee, J.H.; Lee, K.Y.; Kim, S.H. Fabrication methods for low-Pt-loading electrocatalysts in proton exchange membrane fuel cell systems. J. Power Sources 2007, 165, 667–677. [Google Scholar] [CrossRef]
- Taylor, E.J.; Anderson, E.B.; Vilambi, N.R.K. Preparation of high-Pt-utilization gas diffusion electrodes for proton-exchange-membrane fuel cells. J. Electrochem. Soc. 1992, 139, L45–L46. [Google Scholar] [CrossRef]
- Cooper, C.D.; Burk, J.J.; Taylor, C.P.; Buratto, S.K. Ultra-low Pt loading catalyst layers prepared by pulse electrochemical deposition for PEM fuel cells. J. Appl. Electrochem. 2017, 47, 699–709. [Google Scholar] [CrossRef]
- Ahn, S.H.; Lee, B.S.; Choi, I.; Yoo, S.J.; Kim, H.J.; Cho, E.; Henkensmeiera, D.; Nama, S.W.; Kimb, S.-K.; Janga, J.H. Development of a membrane electrode assembly for alkaline water electrolysis by direct electrodeposition of nickel on carbon papers. Appl. Catal. B. Environ. 2014, 154, 197–205. [Google Scholar] [CrossRef]
- Watanabe, J.; Lepoutre, P. A mechanism for the consolidation of the structure of clay-latex coating. Appl. Polym. Sci. 1982, 27, 4207–4219. [Google Scholar] [CrossRef]
- Stanislawsica, A.; Lepoutre, P. Consolidation of pigmented coatings: Development of porous structure. TAPPI J. 1996, 79, 117–125. [Google Scholar]
- Zang, Y.H.; Liu, Z.; Cao, Z.L. A New Mechanism for the Consolidation of the Structure of Latex-pigment Coatings. China Pulp Pap. 2008, 2, 34–40. [Google Scholar] [CrossRef]
- Talukdar, K.; Delgado, S.; Lagarteira, T.; Gazdzicki, P.; Friedrich, K.A. Minimizing mass-transport loss in proton exchange membrane fuel cell by freeze-drying of cathode catalyst layers. J. Power Sources 2019, 427, 309–317. [Google Scholar] [CrossRef]
- Zang, Y.H.; Sun, J.T.; Du, J.; Du, Y.F.; Xu, Y. Influence of coating process variables on the structure and ink absorbency of model clay coatings. China Pulp Pap. 2010, 29, 15–18. [Google Scholar]
- Croll, S.G. Drying of latex paint. Coat. Technol. 1986, 58, 41–49. [Google Scholar]
- Sun, J.T. Study on coating consolidation mechanism of coated paper. Master’s Thesis, Tianjin University of Science and Technology, Tianjin, China, 2011. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, M.; Chu, T.; Wang, T.; Wan, K.; Yang, D.; Li, B.; Ming, P.; Zhang, C. Preparation, Performance and Challenges of Catalyst Layer for Proton Exchange Membrane Fuel Cell. Membranes 2021, 11, 879. https://doi.org/10.3390/membranes11110879
Xie M, Chu T, Wang T, Wan K, Yang D, Li B, Ming P, Zhang C. Preparation, Performance and Challenges of Catalyst Layer for Proton Exchange Membrane Fuel Cell. Membranes. 2021; 11(11):879. https://doi.org/10.3390/membranes11110879
Chicago/Turabian StyleXie, Meng, Tiankuo Chu, Tiantian Wang, Kechuang Wan, Daijun Yang, Bing Li, Pingwen Ming, and Cunman Zhang. 2021. "Preparation, Performance and Challenges of Catalyst Layer for Proton Exchange Membrane Fuel Cell" Membranes 11, no. 11: 879. https://doi.org/10.3390/membranes11110879