Nanomaterial Delivery Systems for mRNA Vaccines

 , , and
, , and
Abstract
:1. Introduction
2. Early Delivery Systems for mRNA Vaccines
3. Polymers for mRNA Delivery
4. Development of Lipid Nanoparticles for the Current SARS-CoV-2 Clinical Trials
5. mRNA Lipid Nanoparticles in the Current SARS-CoV-2 Clinical Trials
5.1. BioNTech/Pfizer
5.2. Moderna
5.3. CureVac
5.4. TranslateBio
5.5. Arcturus
5.6. Imperial College London
5.7. Chulalongkorn University, University of Pennsylvania
5.8. Providence Therapeutics
5.9. Storage and Distribution
6. Lipidoid Nanoparticles
7. Intranasal Delivery of mRNA Lipid Nanoparticles
8. Delivery of mRNA LNPs Encoding Antibodies
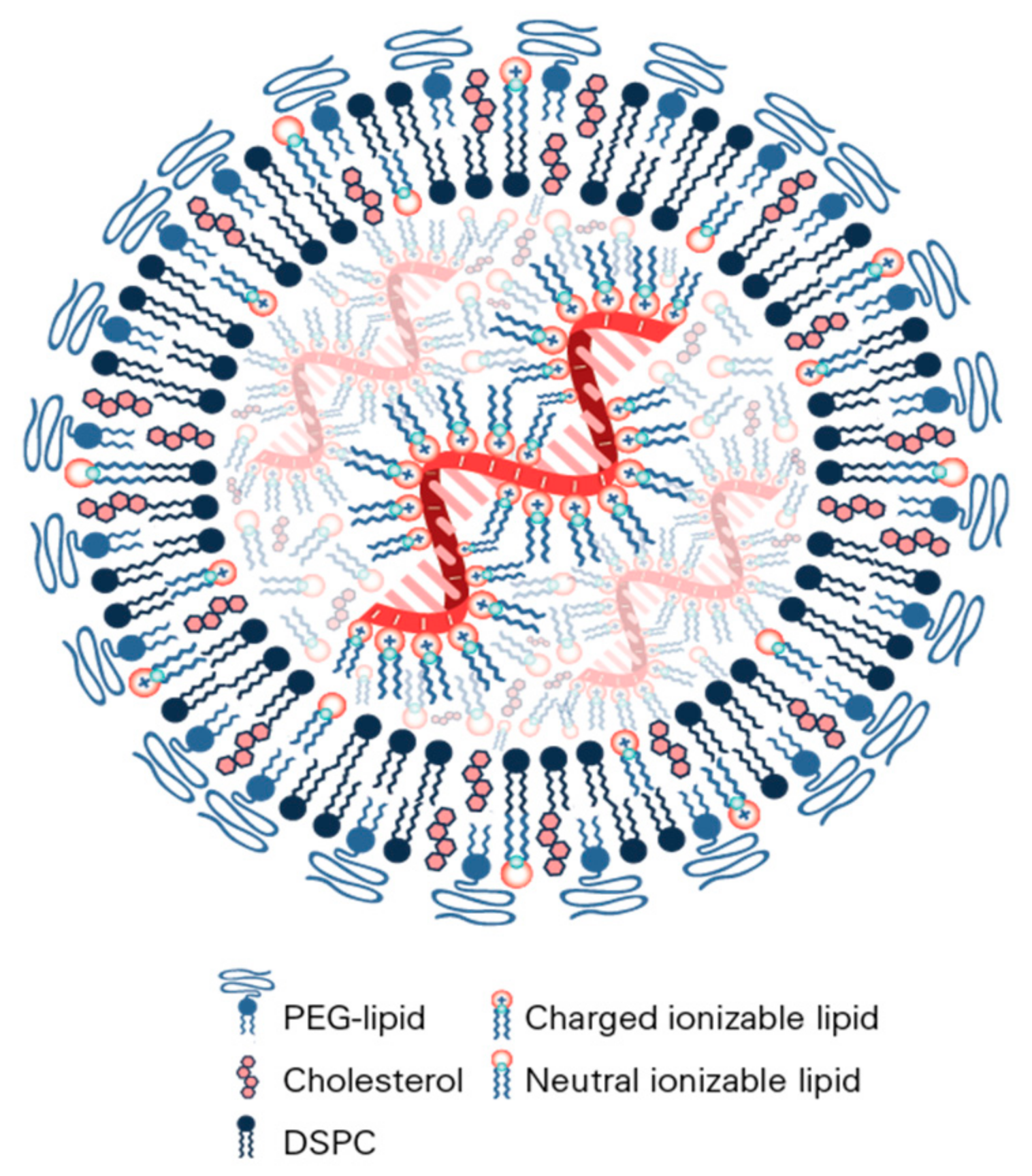
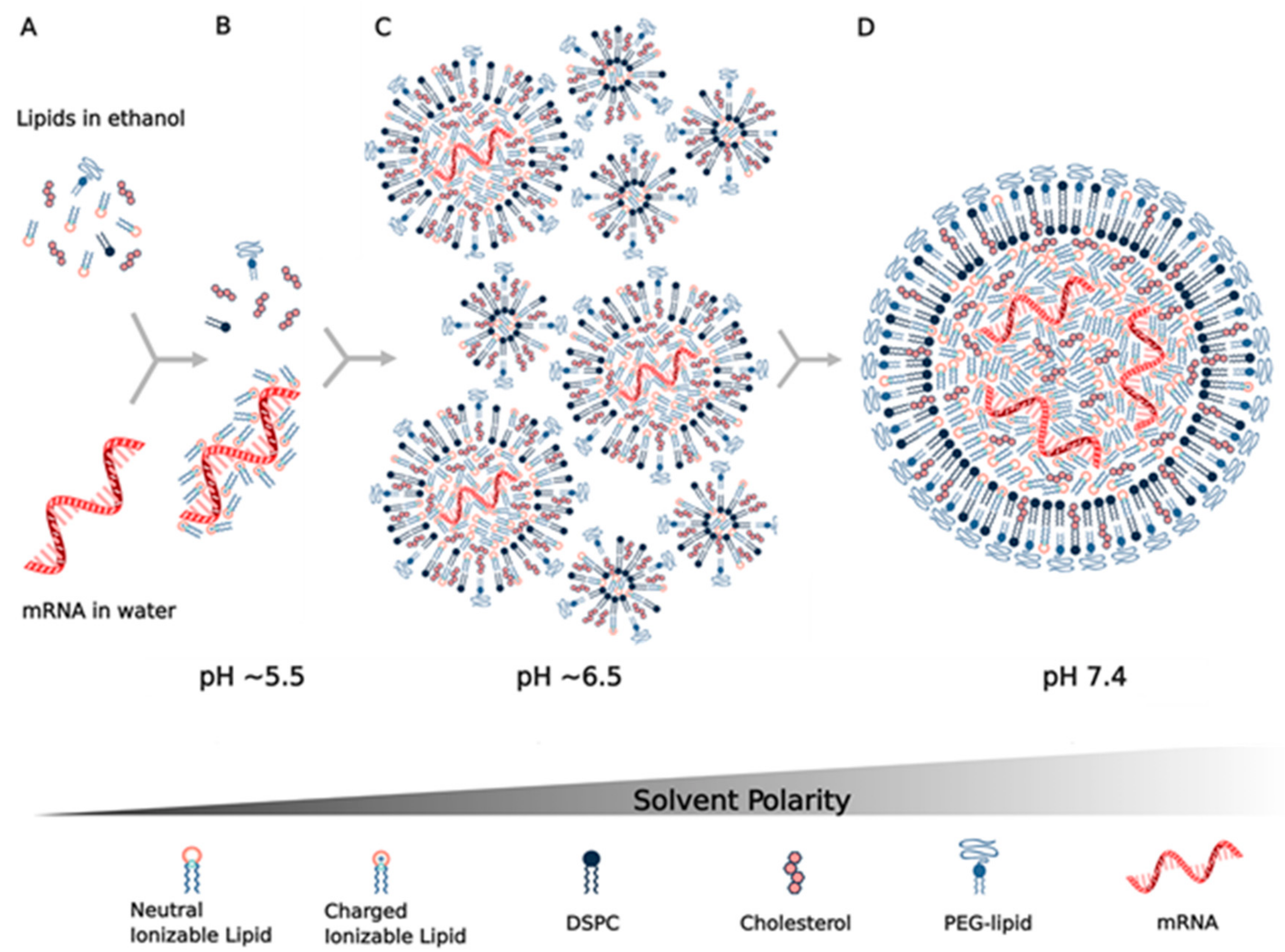
9. Assembly and Structure of Lipid Nanoparticles
10. Determinants of Performance of mRNA Delivery Systems for Vaccines
10.1. Dose
10.2. Potency and Delivery Efficiency
10.3. Endosomal Release
10.4. Charge and Ligand Mediated Targeting
10.5. Adjuvanticity of the Lipid Nanoparticle
10.6. Injection Site Reactions, Safety, Tolerability, Reactogenicity of mRNA LNPs
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zimmer, C.; Corum, J.; Wee, S.-L. Coronavirus Vaccine Tracker. In The New York Times; The New York Times Company: New York, NY, USA, 2020. [Google Scholar]
- Moderna’s COVID-19 Vaccine Candidate Meets its Primary Efficacy Endpoint in the First Interim Analysis of the Phase 3 COVE Study|Moderna, Inc. Available online: https://investors.modernatx.com/news-releases/news-release-details/modernas-covid-19-vaccine-candidate-meets-its-primary-efficacy/ (accessed on 29 November 2020).
- Pfizer and BioNTech Conclude Phase 3 Study of COVID-19 Vaccine Candidate, Meeting All Primary Efficacy Endpoints|BioNTech. Available online: https://investors.biontech.de/news-releases/news-release-details/pfizer-and-biontech-conclude-phase-3-study-covid-19-vaccine/ (accessed on 29 November 2020).
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Solinís, M.Á.; del Pozo-Rodríguez, A. Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef] [Green Version]
- Hajj, K.A.; Whitehead, K.A. Tools for Translation: Non-Viral Materials for Therapeutic mRNA Delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, X.; Dong, Y. Nanoscale platforms for messenger RNA delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, e1530. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Roose, K. How mRNA Therapeutics Are Entering the Monoclonal Antibody Field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [Green Version]
- Wahane, A.; Waghmode, A.; Kapphahn, A.; Dhuri, K.; Gupta, A.; Bahal, R. Role of Lipid-Based and Polymer-Based Non-Viral Vectors in Nucleic Acid Delivery for Next-Generation Gene Therapy. Molecules 2020, 25, 2866. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, C. Emerging Concepts of Nanobiotechnology in mRNA Delivery. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef] [PubMed]
- Witzigmann, D.; Kulkarni, J.A.; Leung, J.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hou, X.; Vick, O.G.; Dong, Y. RNA delivery biomaterials for the treatment of genetic and rare diseases. Biomaterials 2019, 217, 119291. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent Advances in mRNA Vaccine Technology. Curr. Opin. Immunol. 2020, 65, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Alameh, M.-G.; Weissman, D.; Pardi, N. Messenger RNA-Based Vaccines Against Infectious Diseases. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2020; pp. 1–35. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Xia, P.; Li, S.; Zhang, T.; Wang, T.T.; Zhu, J. RNA sensors of the innate immune system and their detection of pathogens. Iubmb Life 2017, 69, 297–304. [Google Scholar] [CrossRef]
- Tatematsu, M.; Funami, K.; Seya, T.; Matsumoto, M. Extracellular RNA Sensing by Pattern Recognition Receptors. J. Innate Immun. 2018, 10, 398–406. [Google Scholar] [CrossRef]
- Devoldere, J.; Dewitte, H.; De Smedt, S.C.; Remaut, K. Evading Innate Immunity in Nonviral mRNA Delivery: Don’t Shoot the Messenger. Drug Discov. Today 2016, 21, 11–25. [Google Scholar] [CrossRef] [Green Version]
- García, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N 1-Methylpseudouridine-Incorporated mRNA Outperforms Pseudouridine-Incorporated mRNA by Providing Enhanced Protein Expression and Reduced Immunogenicity in Mammalian Cell Lines and Mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-Engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heil, F. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.A.; Xu, Y.; Tao, W.; Ubellacker, J.M.; Lim, M.; Aum, D.; Lee, G.Y.; Zhou, K.; Zope, H.; Yu, M.; et al. Restoration of Tumour-Growth Suppression in Vivo via Systemic Nanoparticle-Mediated Delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Behlke, M.A. Chemical Modification of siRNAs for In Vivo Use. Oligonucleotides 2008, 18, 305–320. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Indrisiunaite, G.; DeMirci, H.; Ieong, K.-W.; Wang, J.; Petrov, A.; Prabhakar, A.; Rechavi, G.; Dominissini, D.; He, C.; et al. 2′- O -methylation in mRNA disrupts tRNA decoding during translation elongation. Nat. Struct. Mol. Biol. 2018, 25, 208–216. [Google Scholar] [CrossRef]
- Lorenz, C.; Fotin-Mleczek, M.; Roth, G.; Becker, C.; Dam, T.C.; Verdurmen, W.P.R.; Brock, R.; Probst, J.; Schlake, T. Protein Expression from Exogenous mRNA: Uptake by Receptor-Mediated Endocytosis and Trafficking via the Lysosomal Pathway. RNA Biol. 2011, 8, 627–636. [Google Scholar] [CrossRef]
- Diken, M.; Kreiter, S.; Selmi, A.; Britten, C.M.; Huber, C.; Türeci, Ö.; Sahin, U. Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther. 2011, 18, 702–708. [Google Scholar] [CrossRef] [Green Version]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-Modified mRNA Vaccines Induce Potent T Follicular Helper and Germinal Center B Cell Responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Thran, M.; Jordan, I. mRNA as Novel Technology for Passive Immunotherapy. Cell. Mol. Life Sci. 2019, 76, 301–328. [Google Scholar] [CrossRef] [Green Version]
- CleanCap|TriLink BioTechnologies. Available online: https://www.trilinkbiotech.com/cleancap (accessed on 29 November 2020).
- Yang, Y.; Wang, Z. IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 2019, 11, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the Optimal MRNA for Therapy: HPLC Purification Eliminates Immune Activation and Improves Translation of Nucleoside-Modified, Protein-Encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfizer-BioNTech Covid-19 Vaccine FDA EAU Letter of Authorization. Available online: https://www.fda.gov/media/144412/download (accessed on 14 December 2020).
- mRNA-1273-P301-Protocol. Available online: https://www.modernatx.com/sites/default/files/mRNA-1273-P301-Protocol.pdf (accessed on 1 December 2020).
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schafer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA Vaccine Development Enabled by Prototype Pathogen Preparedness. bioRxiv 2020. [Google Scholar] [CrossRef]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Novel Lipids and Lipid Nanoparticle Formulations for Delivery of Nucleic Acids. Available online: https://patents.google.com/patent/WO2017075531A1/en (accessed on 18 December 2020).
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.P.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase 1/2 Study to Describe the Safety and Immunogenicity of a COVID-19 RNA Vaccine Candidate (BNT162b1) in Adults 18 to 55 Years of Age: Interim Report. medRxiv 2020. [Google Scholar] [CrossRef]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. A prefusion SARS-CoV-2 spike RNA vaccine is highly immunogenic and prevents lung infection in non-human primates. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and T H 1 T cell responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.W.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Kremsner, P.; Mann, P.; Bosch, J.; Fendel, R.; Gabor, J.J.; Kreidenweiss, A.; Kroidl, A.; Leroux-Roels, I.; Leroux-Roels, G.; Schindler, C.; et al. Phase 1 Assessment of the Safety and Immunogenicity of an mRNA- Lipid Nanoparticle Vaccine Candidate Against SARS-CoV-2 in Human Volunteers. medRxiv 2020. [Google Scholar] [CrossRef]
- Rauch, S.; Roth, N.; Schwendt, K.; Fotin-Mleczek, M.; Mueller, S.O.; Petsch, B. mRNA Based SARS-CoV-2 Vaccine Candidate CVnCoV Induces High Levels of Virus Neutralizing Antibodies and Mediates Protection in Rodents. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ice-Based Lipid Nanoparticle Formulation for Delivery of mRNA. Available online: https://patents.google.com/patent/US20200155691A1/ (accessed on 18 January 2021).
- Derosa, F. Cystine Cationic Lipids. Available online: https://patents.google.com/patent/WO2020214946A1/ (accessed on 18 January 2021).
- Kalnin, K.V.; Plitnik, T.; Kishko, M.; Zhang, J.; Zhang, D.; Beauvais, A.; Anosova, N.G.; Tibbitts, T.; DiNapoli, J.M.; Huang, P.-W.D.; et al. Immunogenicity of Novel mRNA COVID-19 Vaccine MRT5500 in Mice and Non-Human Primates. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ionizable Cationic Lipid for Rna Delivery. Available online: https://patents.google.com/patent/US9670152B2/en (accessed on 18 January 2021).
- De Alwis, R.; Gan, E.S.; Chen, S.; Leong, Y.S.; Tan, H.C.; Zhang, S.L.; Yau, C.; Matsuda, D.; Allen, E.; Hartman, P.; et al. A Single Dose of Self-Transcribing and Replicating RNA Based SARS-CoV-2 Vaccine Produces Protective Adaptive Immunity In Mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lipids and Lipid Nanoparticle Formulations for Delivery of Nucleic Acids. Available online: https://patents.google.com/patent/US10221127B2/en (accessed on 18 January 2021).
- McKay, P.F.; Hu, K.; Blakney, A.K.; Samnuan, K.; Brown, J.C.; Penn, R.; Zhou, J.; Bouton, C.R.; Rogers, P.; Polra, K.; et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 2020, 11, 3523. [Google Scholar] [CrossRef]
- Lipid Nanoparticle Formulations. Available online: https://patents.google.com/patent/WO2020219941A1/ (accessed on 18 January 2021).
- Hoerr, I.; Obst, R.; Rammensee, H.-G.; Jung, G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000, 30, 1–7. [Google Scholar] [CrossRef]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkić-Zrna, S.; Probst, J.; Kallen, K.-J. Messenger RNA-based Vaccines With Dual Activity Induce Balanced TLR-7 Dependent Adaptive Immune Responses and Provide Antitumor Activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef]
- Scheel, B.; Braedel, S.; Probst, J.; Carralot, J.-P.; Wagner, H.; Schild, H.; Jung, G.; Rammensee, H.-G.; Pascolo, S. Immunostimulating capacities of stabilized RNA molecules. Eur. J. Immunol. 2004, 34, 537–547. [Google Scholar] [CrossRef]
- Armbruster, N.; Jasny, E.; Petsch, B. Advances in RNA Vaccines for Preventive Indications: A Case Study of a Vaccine against Rabies. Vaccines 2019, 7, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnee, M.; Vogel, A.B.; Voss, D.; Petsch, B.; Baumhof, P.; Kramps, T.; Stitz, L. An mRNA Vaccine Encoding Rabies Virus Glycoprotein Induces Protection against Lethal Infection in Mice and Correlates of Protection in Adult and Newborn Pigs. Plos Negl. Trop. Dis. 2016, 10, e0004746. [Google Scholar] [CrossRef]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and Immunogenicity of a mRNA Rabies Vaccine in Healthy Adults: An Open-Label, Non-Randomised, Prospective, First-in-Human Phase 1 Clinical Trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, J.; Lazzaro, S.; Habbeddine, M.; Schmidt, K.E.; Baumhof, P.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Heidenreich, R.; et al. Unmodified mRNA in LNPs Constitutes a Competitive Technology for Prophylactic Vaccines. NPJ Vaccines 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erasmus, J.H.; Khandhar, A.P.; Guderian, J.; Granger, B.; Archer, J.; Archer, M.; Gage, E.; Fuerte-Stone, J.; Larson, E.; Lin, S.; et al. A Nanostructured Lipid Carrier for Delivery of a Replicating Viral RNA Provides Single, Low-Dose Protection against Zika. Mol. Ther. 2018, 26, 2507–2522. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, J.C.; Patel, A.K.; Kauffman, K.J.; Fenton, O.S.; Webber, M.J.; Heartlein, M.W.; DeRosa, F.; Anderson, D.G. Polymer–Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew. Chem. Int. Ed. 2016, 55, 13808–13812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Gaudin, A.; Zhang, J.; Agarwal, T.; Song, E.; Kauffman, A.C.; Tietjen, G.T.; Wang, Y.; Jiang, Z.; Cheng, C.J.; et al. A “Top-down” Approach to Actuate Poly(Amine-Co-Ester) Terpolymers for Potent and Safe mRNA Delivery. Biomaterials 2018, 176, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.K.; Kaczmarek, J.C.; Bose, S.; Kauffman, K.J.; Mir, F.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv. Mater. 2019, 31, 1805116. [Google Scholar] [CrossRef] [Green Version]
- Blakney, A.K.; Zhu, Y.; McKay, P.F.; Bouton, C.R.; Yeow, J.; Tang, J.; Hu, K.; Samnuan, K.; Grigsby, C.L.; Shattock, R.J.; et al. Big Is Beautiful: Enhanced saRNA Delivery and Immunogenicity by a Higher Molecular Weight, Bioreducible, Cationic Polymer. ACS Nano 2020, 14, 5711–5727. [Google Scholar] [CrossRef] [Green Version]
- Lallana, E.; Rios de la Rosa, J.M.; Tirella, A.; Pelliccia, M.; Gennari, A.; Stratford, I.J.; Puri, S.; Ashford, M.; Tirelli, N. Chitosan/Hyaluronic Acid Nanoparticles: Rational Design Revisited for RNA Delivery. Mol. Pharm. 2017, 14, 2422–2436. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Wang, W.; Wu, C.; Hong, C.; Hammond, P.T. Polyamine-Mediated Stoichiometric Assembly of Ribonucleoproteins for Enhanced mRNA Delivery. Angew. Chem. Int. Ed. 2017, 56, 13709–13712. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, W.; He, Y.; Li, Y.; Yan, E.Z.; Zhang, K.; Irvine, D.J.; Hammond, P.T. Structurally Programmed Assembly of Translation Initiation Nanoplex for Superior mRNA Delivery. ACS Nano 2017, 11, 2531–2544. [Google Scholar] [CrossRef]
- McKinlay, C.J.; Benner, N.L.; Haabeth, O.A.; Waymouth, R.M.; Wender, P.A. Enhanced mRNA Delivery into Lymphocytes Enabled by Lipid-Varied Libraries of Charge-Altering Releasable Transporters. Proc. Natl. Acad. Sci. USA 2018, 115, E5859–E5866. [Google Scholar] [CrossRef] [Green Version]
- Soliman, O.Y.; Alameh, M.G.; De Cresenzo, G.; Buschmann, M.D.; Lavertu, M. Efficiency of Chitosan/Hyaluronan-Based mRNA Delivery Systems In Vitro: Influence of Composition and Structure. J. Pharm. Sci. 2020, 109, 1581–1593. [Google Scholar] [CrossRef]
- Uchida, H.; Itaka, K.; Uchida, S.; Ishii, T.; Suma, T.; Miyata, K.; Oba, M.; Nishiyama, N.; Kataoka, K. Synthetic Polyamines to Regulate mRNA Translation through the Preservative Binding of Eukaryotic Initiation Factor 4E to the Cap Structure. J. Am. Chem. Soc. 2016, 138, 1478–1481. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, J.; Yang, Y.; Song, H.; Fu, J.; Gu, Z.; Yu, C. Functional Nanoparticles with a Reducible Tetrasulfide Motif to Upregulate mRNA Translation and Enhance Transfection in Hard-to-Transfect Cells. Angew. Chem. Int. Ed. 2020, 59, 2695–2699. [Google Scholar] [CrossRef]
- Yoshinaga, N.; Cho, E.; Koji, K.; Mochida, Y.; Naito, M.; Osada, K.; Kataoka, K.; Cabral, H.; Uchida, S. Bundling mRNA Strands to Prepare Nano-Assemblies with Enhanced Stability Towards RNase for In Vivo Delivery. Angew. Chem. Int. Ed. 2019, 58, 11360–11363. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, N.; Uchida, S.; Naito, M.; Osada, K.; Cabral, H.; Kataoka, K. Induced Packaging of mRNA into Polyplex Micelles by Regulated Hybridization with a Small Number of Cholesteryl RNA Oligonucleotides Directed Enhanced in Vivo Transfection. Biomaterials 2019, 197, 255–267. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [Green Version]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, J.J.; Palmer, L.; Ossanlou, M.; MacLachlan, I.; Graham, R.W.; Zhang, Y.P.; Hope, M.J.; Scherrer, P.; Cullis, P.R. Stabilized plasmid-lipid particles: Construction and characterization. Gene Ther. 1999, 6, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Heyes, J.; Palmer, L.; Bremner, K.; MacLachlan, I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release 2005, 107, 276–287. [Google Scholar] [CrossRef]
- Arteta, M.Y.; Kjellman, T.; Bartesaghi, S.; Wallin, S.; Wu, X.; Kvist, A.J.; Dabkowska, A.; Székely, N.; Radulescu, A.; Bergenholtz, J.; et al. Successful Reprogramming of Cellular Protein Production through mRNA Delivered by Functionalized Lipid Nanoparticles. Proc. Natl. Acad. Sci. USA 2018, 115, E3351–E3360. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Lee, R.J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef]
- Gruner, S.M.; Cullis, P.R.; Hope, M.J.; Tilcock, C.P.S. Lipid Polymorphism:The Molecular Basis of Nonbilayer Phases. Annu. Rev. Biophys. Biophys. Chem. 1985, 14, 211–238. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the Potency of siRNA Lipid Nanoparticles for Hepatic Gene Silencing In Vivo. Angew. Chem. Int. Ed. 2012, 51, 8529–8533. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational Design of Cationic Lipids for siRNA Delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Evers, M.J.W.; Kulkarni, J.A.; van der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [Green Version]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, Ö.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Lindgren, G.; Lin, A.; Thompson, E.A.; Ols, S.; Röhss, J.; John, S.; Hassett, K.; Yuzhakov, O.; Bahl, K.; et al. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified mRNA Vaccine Administration in Rhesus Macaques. Mol. Ther. 2017, 25, 2635–2647. [Google Scholar] [CrossRef] [Green Version]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125.e10. [Google Scholar] [CrossRef] [Green Version]
- Feldman, R.A.; Fuhr, R.; Smolenov, I.; Ribeiro, A.M.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, Ö; Pujar, H.S.; et al. mRNA Vaccines against H10N8 and H7N9 Influenza Viruses of Pandemic Potential Are Immunogenic and Well Tolerated in Healthy Adults in Phase 1 Randomized Clinical Trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-Human Primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, M.S.; Fromherz, P. Lipoid pH indicators as probes of electrical potential and polarity in micelles. J. Phys. Chem. 1977, 81, 1755–1761. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Jia, Y.; Farbiak, L.; Zhou, K.; Zhang, S.; Wei, Y.; Zhu, H.; Siegwart, D.J. Dendrimer-Based Lipid Nanoparticles Deliver Therapeutic FAH mRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I. Adv. Mater. 2018, 30, 1805308. [Google Scholar] [CrossRef]
- Hajj, K.A.; Ball, R.L.; Deluty, S.B.; Singh, S.R.; Strelkova, D.; Knapp, C.M.; Whitehead, K.A. Branched-Tail Lipid Nanoparticles Potently Deliver mRNA In Vivo Due to Enhanced Ionization at Endosomal PH. Small 2019, 15, 1805097. [Google Scholar] [CrossRef]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-Based Analysis of Lipid Nanoparticle–Mediated siRNA Delivery, Intracellular Trafficking and Endosomal Escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Development and Licensure of Vaccines to Prevent COVID-19; Guidance for Industry. U.S. Dep. Health Hum. Serv. Food Drug Adm. Cent. Biol. Eval. Res. 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-and-licensure-vaccines-prevent-covid-19 (accessed on 1 December 2020).
- Wadman, M. Public needs to prep for vaccine side effects. Science 2020, 370, 1022. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA Vaccine Delivery Using Lipid Nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Cleavable Lipids. Available online: https://patents.google.com/patent/US20200237671A1/ (accessed on 18 January 2021).
- Rajappan, K.; Tanis, S.P.; Mukthavaram, R.; Roberts, S.; Nguyen, M.; Tachikawa, K.; Sagi, A.; Sablad, M.; Limphong, P.; Leu, A.; et al. Property-Driven Design and Development of Lipids for Efficient Delivery of siRNA. J. Med. Chem. 2020, 63, 12992–13012. [Google Scholar] [CrossRef]
- Arcturus Therapeutics Announces Positive Interim ARCT-021 (LUNAR-COV19) Phase 1/2 Study Results for Both Single Shot and Prime-Boost Regimens, and Up to $220 Million in Additional Financial Commitments from Singapore|Arcturus Therapeutics, Inc. Available online: https://ir.arcturusrx.com/news-releases/news-release-details/arcturus-therapeutics-announces-positive-interim-arct-021-lunar (accessed on 28 November 2020).
- Providence Therapeutics COVID-19 Vaccine Receives Health Canada Authorization to Begin Clinical Trials. Available online: http://www.providencetherapeutics.com/providence-therapeutics-covid-19-vaccine-receives-health-canada-authorization-to-begin-clinical-trials (accessed on 10 January 2021).
- Providence Therapeutics Reports Supportive Preclinical Data for its COVID-19 Vaccine Candidate (PTX-COVID19-B). Available online: http://www.providencetherapeutics.com/providence-therapeutics-reports-supportive-preclinical-data-for-its-covid-19-vaccine-candidate-ptx-covid19-b (accessed on 10 January 2021).
- Laczkó, D.; Hogan, M.J.; Toulmin, S.A.; Hicks, P.; Lederer, K.; Gaudette, B.T.; Castaño, D.; Amanat, F.; Muramatsu, H.; Oguin, T.H.; et al. A Single Immunization with Nucleoside-Modified mRNA Vaccines Elicits Strong Cellular and Humoral Immune Responses against SARS-CoV-2 in Mice. Immunity 2020, 53, 724–732.e7. [Google Scholar] [CrossRef]
- Zhao, P.; Hou, X.; Yan, J.; Du, S.; Xue, Y.; Li, W.; Xiang, G.; Dong, Y. Long-term storage of lipid-like nanoparticles for mRNA delivery. Bioact. Mater. 2020, 5, 358–363. [Google Scholar] [CrossRef]
- Gindy, M.E.; Feuston, B.; Glass, A.; Arrington, L.; Haas, R.M.; Schariter, J.; Stirdivant, S.M. Stabilization of Ostwald Ripening in Low Molecular Weight Amino Lipid Nanoparticles for Systemic Delivery of siRNA Therapeutics. Mol. Pharm. 2014, 11, 4143–4153. [Google Scholar] [CrossRef]
- Fact Sheet for Healthcare Providers Administering Vaccine (Vaccination Providers) Emergency Use Authorization (EUA) of the Moderna COVID-19 Vaccine to Prevent Coronavirus Disease 2019 (COVID-19). Available online: https://www.fda.gov/media/144637/download (accessed on 1 January 2021).
- Fact Sheet for Healthcare Providers Administering Vaccine (Vaccination Providers) Emergency Use Authorization (EUA) of the Pfizer-Biontech COVID-19 Vaccine to Prevent Coronavirus Disease 2019 (COVID-19). Available online: https://www.fda.gov/media/144413/download (accessed on 1 January 2021).
- Gómez, G.; Pikal, M.J.; Rodríguez-Hornedo, N. Effect of Initial Buffer Composition on pH Changes During Far-From-Equilibrium Freezing of Sodium Phosphate Buffer Solutions. Pharm. Res. 2001, 18, 90–97. [Google Scholar] [CrossRef]
- Tang, X.C.; Pikal, M.J. Design of Freeze-Drying Processes for Pharmaceuticals: Practical Advice. Pharm. Res. 2004, 21, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Tides Presentation—Arcturus Therapeutics, Inc. Available online: https://ir.arcturusrx.com/static-files/5f0c040f-872c-4e16-82ce-523ff083b708 (accessed on 1 January 2021).
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, In Vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic mRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Nguyen, L.H.; Miller, J.B.; Yan, Y.; Kos, P.; Xiong, H.; Li, L.; Hao, J.; Minnig, J.T.; Zhu, H.; et al. Modular degradable dendrimers enable small RNAs to extend survival in an aggressive liver cancer model. Proc. Natl. Acad. Sci. USA 2016, 113, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef] [Green Version]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jozic, A.; Sahay, G. Naturally Derived Membrane Lipids Impact Nanoparticle-Based Messenger RNA Delivery. Cell. Mol. Bioeng. 2020. [Google Scholar] [CrossRef]
- Mai, Y.; Guo, J.; Zhao, Y.; Ma, S.; Hou, Y.; Yang, J. Intranasal Delivery of Cationic Liposome-Protamine Complex mRNA Vaccine Elicits Effective Anti-Tumor Immunity. Cell. Immunol. 2020, 354, 104143. [Google Scholar] [CrossRef]
- Zhuang, X.; Qi, Y.; Wang, M.; Yu, N.; Nan, F.; Zhang, H.; Tian, M.; Li, C.; Lu, H.; Jin, N. mRNA Vaccines Encoding the HA Protein of Influenza A H1N1 Virus Delivered by Cationic Lipid Nanoparticles Induce Protective Immune Responses in Mice. Vaccines 2020, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of Nucleoside-Modified mRNA Encoding Broadly Neutralizing Antibody Protects Humanized Mice from HIV-1 Challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef]
- Thran, M.; Mukherjee, J.; Pönisch, M.; Fiedler, K.; Thess, A.; Mui, B.L.; Hope, M.J.; Tam, Y.K.; Horscroft, N.; Heidenreich, R.; et al. mRNA Mediates Passive Vaccination against Infectious Agents, Toxins, and Tumors. EMBO Mol. Med. 2017, 9, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Stadler, C.R.; Bähr-Mahmud, H.; Celik, L.; Hebich, B.; Roth, A.S.; Roth, R.P.; Karikó, K.; Türeci, Ö.; Sahin, U. Elimination of Large Tumors in Mice by mRNA-Encoded Bispecific Antibodies. Nat. Med. 2017, 23, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, L.; Verbeke, R.; De Vlieger, D.; Dewitte, H.; Roose, K.; Van Nevel, S.; Krysko, O.; Bachert, C.; Schepens, B.; Lentacker, I.; et al. mRNA Encoding a Bispecific Single Domain Antibody Construct Protects against Influenza A Virus Infection in Mice. Mol. Ther. Nucleic Acids 2020, 20, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Kose, N.; Fox, J.M.; Sapparapu, G.; Bombardi, R.; Tennekoon, R.N.; de Silva, A.D.; Elbashir, S.M.; Theisen, M.A.; Humphris-Narayanan, E.; Ciaramella, G.; et al. A Lipid-Encapsulated mRNA Encoding a Potently Neutralizing Human Monoclonal Antibody Protects against Chikungunya Infection. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Moderna Announces Positive Phase 1 Results for the First Systemic Messenger RNA Therapeutic Encoding a Secreted Protein (mRNA-1944)|Moderna, Inc. Available online: https://investors.modernatx.com/news-releases/news-release-details/moderna-announces-positive-phase-1-results-first-systemic/ (accessed on 5 December 2020).
- Belliveau, N.M.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-Size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; van der Meel, R.; Zaifman, J.; Darjuan, M.M.; Grisch-Chan, H.M.; Thöny, B.; Tam, Y.Y.; Cullis, P.R. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale 2019, 11, 9023–9031. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Freyn, A.W.; da Silva, J.R.; Rosado, V.C.; Bliss, C.M.; Pine, M.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; de Souza Ferreira, L.C.; Weissman, D.; et al. A Multi-Targeting, Nucleoside-Modified mRNA Influenza Virus Vaccine Provides Broad Protection in Mice. Mol. Ther. 2020, 28, 1569–1584. [Google Scholar] [CrossRef]
- Chen, S.; Tam, Y.Y.C.; Lin, P.J.C.; Sung, M.M.H.; Tam, Y.K.; Cullis, P.R. Influence of Particle Size on the in Vivo Potency of Lipid Nanoparticle Formulations of siRNA. J. Control. Release 2016, 235, 236–244. [Google Scholar] [CrossRef]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; DuRoss, A.; Sun, C.; Murphy-Benenato, K.E.; Mihai, C.; Almarsson, Ö.; Sahay, G. Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated mRNA. Nano Lett. 2017, 17, 5711–5718. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA Delivery by Lipid Nanoparticles Is Limited by Endocytic Recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.C.; Hocking, K.M.; Kilchrist, K.V.; Wise, E.S.; Brophy, C.M.; Duvall, C.L. Endosomolytic Nano-Polyplex Platform Technology for Cytosolic Peptide Delivery To Inhibit Pathological Vasoconstriction. ACS Nano 2015, 9, 5893–5907. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Lu, Q.; Wang, Y.; Xu, E.; Ho, A.; Singh, P.; Wang, Y.; Jiang, Z.; Yang, F.; Tietjen, G.T.; et al. Quantitating Endosomal Escape of a Library of Polymers for mRNA Delivery. Nano Lett. 2020, 20, 1117–1123. [Google Scholar] [CrossRef]
- Kilchrist, K.V.; Dimobi, S.C.; Jackson, M.A.; Evans, B.C.; Werfel, T.A.; Dailing, E.A.; Bedingfield, S.K.; Kelly, I.B.; Duvall, C.L. Gal8 Visualization of Endosome Disruption Predicts Carrier-Mediated Biologic Drug Intracellular Bioavailability. ACS Nano 2019. [Google Scholar] [CrossRef]
- Miao, L.; Lin, J.; Huang, Y.; Li, L.; Delcassian, D.; Ge, Y.; Shi, Y.; Anderson, D.G. Synergistic Lipid Compositions for Albumin Receptor Mediated Delivery of mRNA to the Liver. Nat. Commun. 2020, 11, 2424. [Google Scholar] [CrossRef]
- Patel, S.; Kim, J.; Herrera, M.; Mukherjee, A.; Kabanov, A.V.; Sahay, G. Brief update on endocytosis of nanomedicines. Adv. Drug Deliv. Rev. 2019, 144, 90–111. [Google Scholar] [CrossRef]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Hölttä, M.; Skantze, P.; Johansson, S.; Sundqvist, M.; et al. Linkage between Endosomal Escape of LNP-mRNA and Loading into EVs for Transport to Other Cells. Nat. Commun. 2019, 10, 4333. [Google Scholar] [CrossRef] [Green Version]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Fenton, O.S.; Kauffman, K.J.; Kaczmarek, J.C.; McClellan, R.L.; Jhunjhunwala, S.; Tibbitt, M.W.; Zeng, M.D.; Appel, E.A.; Dorkin, J.R.; Mir, F.F.; et al. Synthesis and Biological Evaluation of Ionizable Lipid Materials for the In Vivo Delivery of Messenger RNA to B Lymphocytes. Adv. Mater. 2017, 29, 1606944. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective Organ Targeting (SORT) Nanoparticles for Tissue-Specific mRNA Delivery and CRISPR–Cas Gene Editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Kuipers, F.; Havekes, L.M.; Havinga, R.; Dontje, B.; Poelstra, K.; Scherphof, G.L.; Kamps, J.A.A.M. The role of apolipoprotein E in the elimination of liposomes from blood by hepatocytes in the mouse. Biochem. Biophys. Res. Commun. 2005, 328, 57–62. [Google Scholar] [CrossRef]
- Hajj, K.A.; Melamed, J.R.; Chaudhary, N.; Lamson, N.G.; Ball, R.L.; Yerneni, S.S.; Whitehead, K.A. A Potent Branched-Tail Lipid Nanoparticle Enables Multiplexed mRNA Delivery and Gene Editing In Vivo. Nano Lett. 2020, 20, 5167–5175. [Google Scholar] [CrossRef]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression Kinetics of Nucleoside-Modified mRNA Delivered in Lipid Nanoparticles to Mice by Various Routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Parhiz, H.; Shuvaev, V.V.; Pardi, N.; Khoshnejad, M.; Kiseleva, R.Y.; Brenner, J.S.; Uhler, T.; Tuyishime, S.; Mui, B.L.; Tam, Y.K.; et al. PECAM-1 Directed Re-Targeting of Exogenous mRNA Providing Two Orders of Magnitude Enhancement of Vascular Delivery and Expression in Lungs Independent of Apolipoprotein E-Mediated Uptake. J. Control. Release 2018, 291, 106–115. [Google Scholar] [CrossRef]
- Marcos-Contreras, O.A.; Greineder, C.F.; Kiseleva, R.Y.; Parhiz, H.; Walsh, L.R.; Zuluaga-Ramirez, V.; Myerson, J.W.; Hood, E.D.; Villa, C.H.; Tombacz, I.; et al. Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood–brain barrier. Proc. Natl. Acad. Sci. USA 2020, 117, 3405–3414. [Google Scholar] [CrossRef]
- Perche, F.; Gosset, D.; Mével, M.; Miramon, M.-L.; Yaouanc, J.-J.; Pichon, C.; Benvegnu, T.; Jaffrès, P.-A.; Midoux, P. Selective gene delivery in dendritic cells with mannosylated and histidylated lipopolyplexes. J. Drug Target. 2011, 19, 315–325. [Google Scholar] [CrossRef]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell Specific Delivery of Modified mRNA Expressing Therapeutic Proteins to Leukocytes. Nat. Commun. 2018, 9, 4493. [Google Scholar] [CrossRef]
- Kedmi, R.; Veiga, N.; Ramishetti, S.; Goldsmith, M.; Rosenblum, D.; Dammes, N.; Hazan-Halevy, I.; Nahary, L.; Leviatan-Ben-Arye, S.; Harlev, M.; et al. A modular platform for targeted RNAi therapeutics. Nat. Nanotechnol. 2018, 13, 214–219. [Google Scholar] [CrossRef]
- Swaminathan, G.; Thoryk, E.A.; Cox, K.S.; Meschino, S.; Dubey, S.A.; Vora, K.A.; Celano, R.; Gindy, M.; Casimiro, D.R.; Bett, A.J. A novel lipid nanoparticle adjuvant significantly enhances B cell and T cell responses to sub-unit vaccine antigens. Vaccine 2016, 34, 110–119. [Google Scholar] [CrossRef]
- Swaminathan, G.; Thoryk, E.A.; Cox, K.S.; Smith, J.S.; Wolf, J.J.; Gindy, M.E.; Casimiro, D.R.; Bett, A.J. A Tetravalent Sub-unit Dengue Vaccine Formulated with Ionizable Cationic Lipid Nanoparticle induces Significant Immune Responses in Rodents and Non-Human Primates. Sci. Rep. 2016, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.J.; Souto, E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv. Drug Deliv. Rev. 2007, 59, 478–490. [Google Scholar] [CrossRef]
- Yuki, Y.; Kiyono, H. New generation of mucosal adjuvants for the induction of protective immunity. Rev. Med. Virol. 2003, 13, 293–310. [Google Scholar] [CrossRef]
- Sedic, M.; Senn, J.J.; Lynn, A.; Laska, M.; Smith, M.; Platz, S.J.; Bolen, J.; Hoge, S.; Bulychev, A.; Jacquinet, E.; et al. Safety Evaluation of Lipid Nanoparticle–Formulated Modified mRNA in the Sprague-Dawley Rat and Cynomolgus Monkey. Vet. Pathol. 2018, 55, 341–354. [Google Scholar] [CrossRef]
- Barros, S.A.; Gollob, J.A. Safety profile of RNAi nanomedicines. Adv. Drug Deliv. Rev. 2012, 64, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A. The physiology of the lymphatic system. Adv. Drug Deliv. Rev. 2001, 50, 3–20. [Google Scholar] [CrossRef]
- Castells, M.C.; Phillips, E.J. Maintaining Safety with SARS-CoV-2 Vaccines. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Muskula, P.R.; Main, M.L. Safety With Echocardiographic Contrast Agents. Circ. Cardiovasc. Imaging 2017, 10, e005459. [Google Scholar] [CrossRef] [Green Version]
- Parhiz, H.; Khoshnejad, M.; Myerson, J.W.; Hood, E.; Patel, P.N.; Brenner, J.S.; Muzykantov, V.R. Unintended effects of drug carriers: Big issues of small particles. Adv. Drug Deliv. Rev. 2018, 130, 90–112. [Google Scholar] [CrossRef]
- NIH Devising Study on Rare Allergic Reactions to Coronavirus Vaccine—The Washington Post. Available online: https://www.washingtonpost.com/health/covid-vaccine-allergic-reactions-study/2020/12/21/e01001d2-431a-11eb-b0e4-0f182923a025_story.html (accessed on 10 January 2021).



{kind=link}
{kind=link}
| Company | mRNA Type | Immunogen | mRNA Dose (µg) | Confirmed or Probable LNP Class | Publications |
|---|---|---|---|---|---|
| Moderna | nucleoside modified mRNA | membrane bound prefusion stabilized spike | 100 | Lipid H [42] confirmed in [41] | [43,44,45,46] |
| BioNTech Pfizer | nucleoside modified mRNA | membrane bound prefusion stabilized spike | 30 | Acuitas ALC-0315 [47] confirmed in [40] | [48,49,50,51] |
| CureVac | unmodified mRNA | membrane bound prefusion stabilized spike | 12 | Acuitas ALC-0315 [47] | [52,53] |
| TranslateBio Sanofi | unmodified mRNA | prefusion stabilized double mutant spike | 7.5 | ICE [54] or Cysteine [55] | [56] |
| Arcturus | self-amplifying mRNA | full length spike | 1–10 | Lipid 2,2 (8,8) 4C CH3 [57] | [58] |
| Imperial College | self-amplifying mRNA | membrane bound prefusion stabilized spike | 1–10 | Acuitas A9 [59] | [60] |
| Chulalongkorn | nucleoside modified mRNA | secreted wild type spike | Not available | Genevant CL1 [61] | NA |
| Name | Ionizable Lipid Structure and Theoretical pKas | TNS pKa |
|---|---|---|
| MC3 [92] |  | 6.4 |
| Lipid 319 [68] |  | 6.38 |
| C12-200 [103] |  | 6.96 |
| 5A2-SC8 [104] |  | 6.67 |
| 306Oi10 [105] |  | 6.4 |
| Moderna Lipid 5 [101] |  | 6.56 |
| Moderna Lipid H, SM-102 [42] |  | 6.75 |
| Acuitas A9 [59] |  | 6.27 |
| Acuitas ALC-0315 [47] |  | 6.09 |
| Arcturus Lipid 2,2 (8,8) 4C CH3 [57] |  | 6.69 |
| Genevant CL1[61] |  | NA |
| Delivery System | mRNA Type | Species | Dose | Readout | Reference |
|---|---|---|---|---|---|
| Naked mRNA | non-modified | mouse | 80 µg twice | protection | [140] |
| Naked mRNA | self-amplifying | mouse | 1.25 µg twice | protection | [140] |
| Protamine | non-modified | mouse | 10 µg twice | neutralizing titers | [66] |
| Protamine | non-modified | mouse | 80 µg twice | protection | [66] |
| Modified Dendrimer | self-amplifying | mouse | 40 µg once or 4 µg twice | neutralizing titers | [126] |
| DOTAP/DOPE/PEG | nucleoside modified | mouse | 12 µg twice intranasal | neutralizing titers and protection | [130] |
| Cationic Nanoemulsion | self-amplifying | mouse | 15 µg twice | neutralizing titers | [70] |
| Nanostructured Lipid Carrier | self-amplifying | mouse | 0.1 µg once | neutralizing titers | [71] |
| LNP (Acuitas) | non-modified | mouse | 0.5 µg twice | neutralizing titer | [69] |
| LNP (MC3) | nucleoside-modified | mouse | 10 µg once or 2 µg twice | protection | [99] |
| LNP (MC3) | nucleoside-modified | mouse | 0.4 µg once | protection | [97] |
| LNP (Acuitas) | nucleoside-modified | mouse | 0.5 µg once | protection | [141] |
| LNP (Acuitas) | nucleoside-modified | mouse | 1 µg twice | neutralizing titers | [49] |
| LNP (Moderna) | nucleoside-modified | mouse | 1 µg twice | neutralizing titers and protection | [45] |
| LNP (Acuitas) | non-modified | mouse | 0.25 µg twice | neutralizing titers | [53] |
| LNP (Translate Bio) | non-modified | mouse | 0.2 µg twice | neutralizing titers | [56] |
| LNP (Arcturus) | self-amplifying | mouse | 2 µg once | neutralizing titers and protection | [58] |
| LNP (Acuitas) | self-amplifying | mouse | 0.1 µg twice | neutralizing titers | [60] |
| LNP (Acuitas) | non-modified | Syrian Hamster | 10 µg twice | protection | [53] |
| LNP (MC3) | nucleoside-modified | ferret | 50 µg once | neutralizing titers | [97] |
| Cationic Nanoemulsion | self-amplifying | non-human primate | 75 µg twice | neutralizing titers | [70] |
| LNP (MC3) | nucleoside-modified | non-human primate | 200 µg twice | neutralizing titers | [97] |
| LNP (MC3 or Moderna Lipid H) | nucleoside-modified | non-human primate | 5 µg twice | neutralizing titers | [42] |
| LNP (Acuitas) | nucleoside-modified | non-human primate | 30 µg twice | neutralizing titers | [49] |
| LNP (Moderna) | nucleoside-modified | non-human primate | 100 µg twice | neutralizing titers | [45] |
| LNP (Translate Bio) | non-modified | non-human primate | 15 µg twice | neutralizing titers | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buschmann, M.D.; Carrasco, M.J.; Alishetty, S.; Paige, M.; Alameh, M.G.; Weissman, D. Nanomaterial Delivery Systems for mRNA Vaccines. Vaccines 2021, 9, 65. https://doi.org/10.3390/vaccines9010065
Buschmann MD, Carrasco MJ, Alishetty S, Paige M, Alameh MG, Weissman D. Nanomaterial Delivery Systems for mRNA Vaccines. Vaccines. 2021; 9(1):65. https://doi.org/10.3390/vaccines9010065
Chicago/Turabian StyleBuschmann, Michael D., Manuel J. Carrasco, Suman Alishetty, Mikell Paige, Mohamad Gabriel Alameh, and Drew Weissman. 2021. "Nanomaterial Delivery Systems for mRNA Vaccines" Vaccines 9, no. 1: 65. https://doi.org/10.3390/vaccines9010065