Enhanced Anti-Mycobacterium tuberculosis Immunity over Time with Combined Drug and Immunotherapy Treatment

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
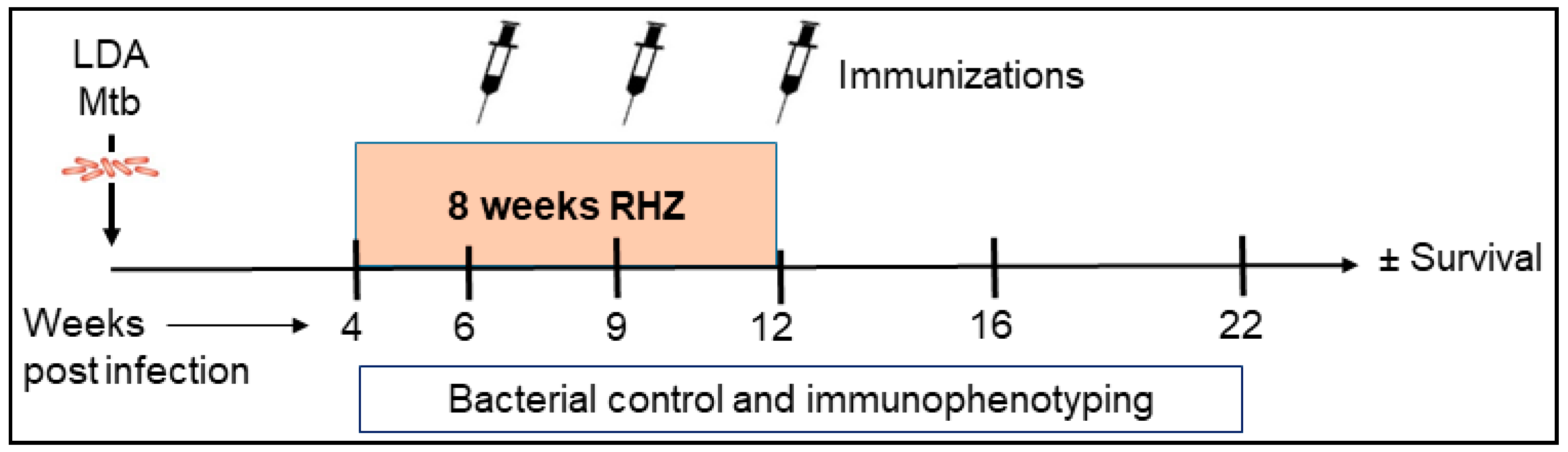
2. Materials and Methods
3. Results
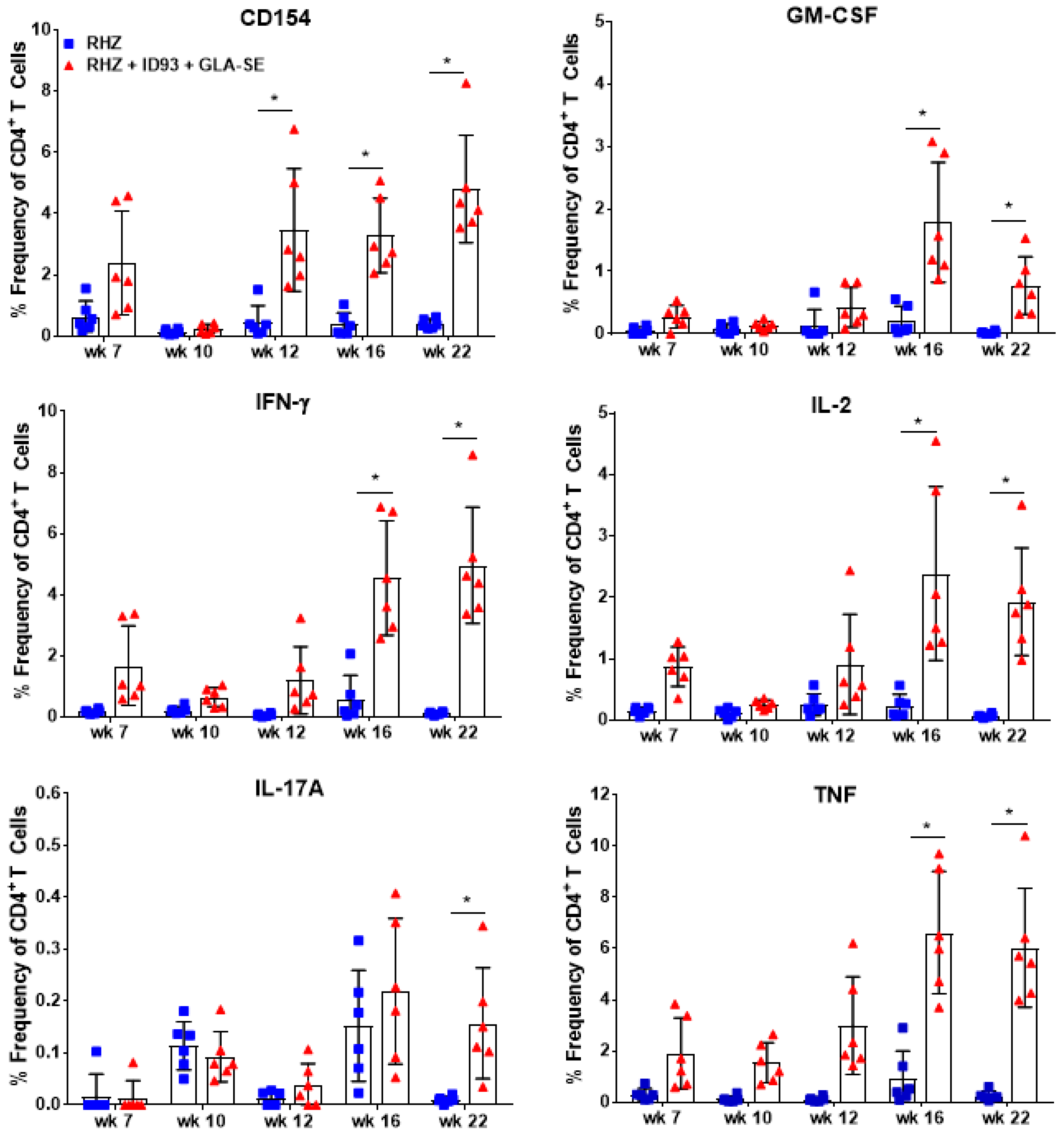
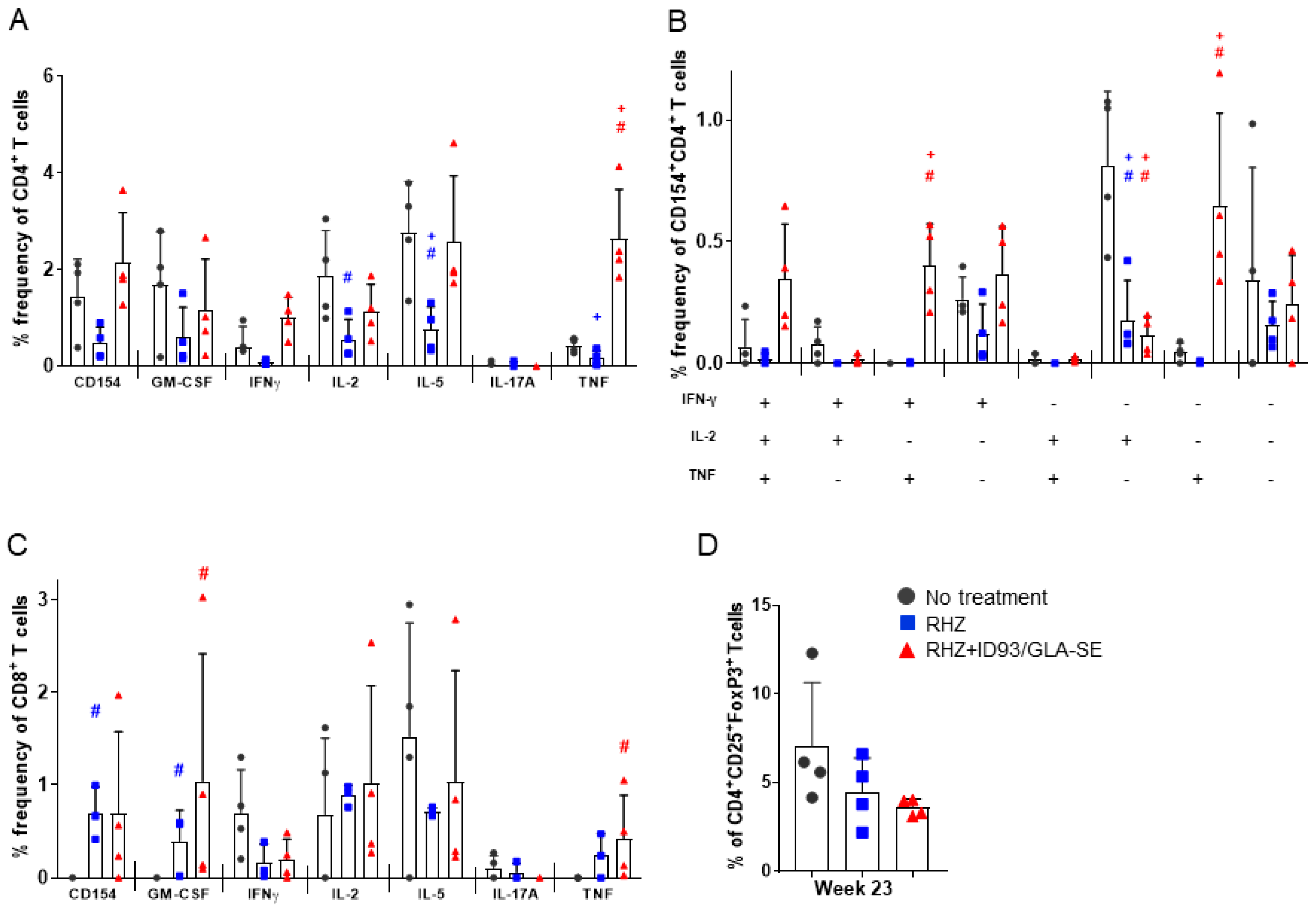
3.1. Strong CD4+ TH1 T Cell Inflammatory Responses Elicited over Time in Groups Receiving RHZ and Immunotherapy
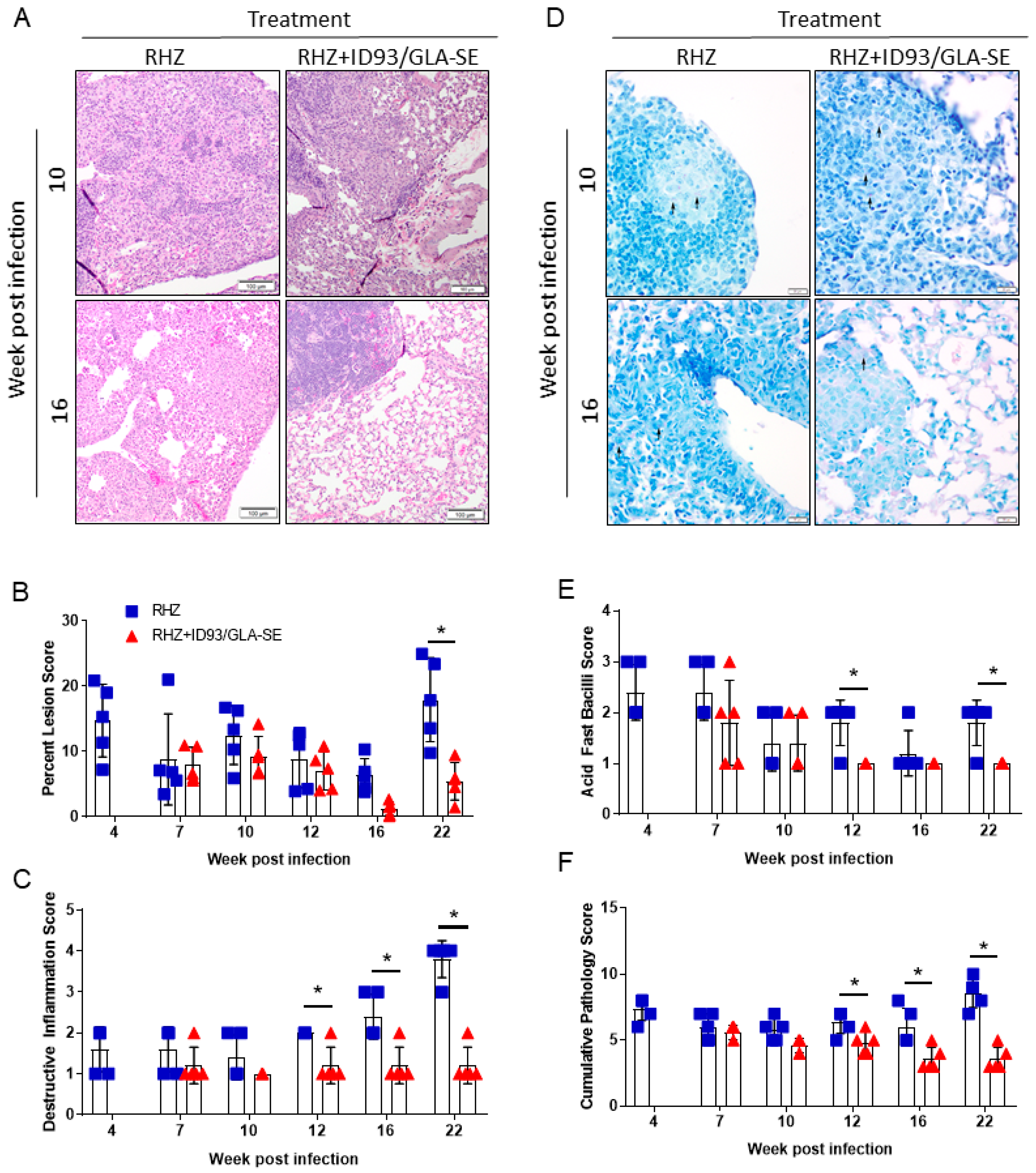
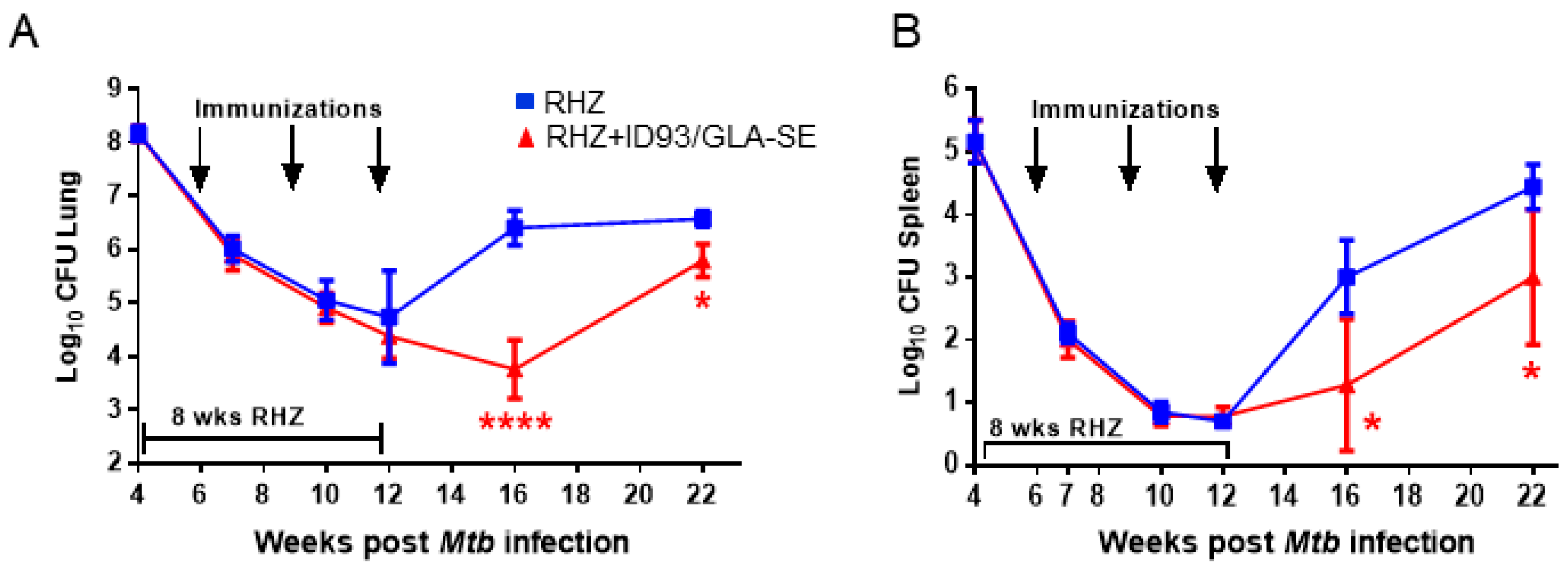
3.2. Therapeutic Treatment with RHZ+ID93/GLA-SE Results in Significantly Reduced Pathology and Bacterial Burden Compared to Drugs Alone
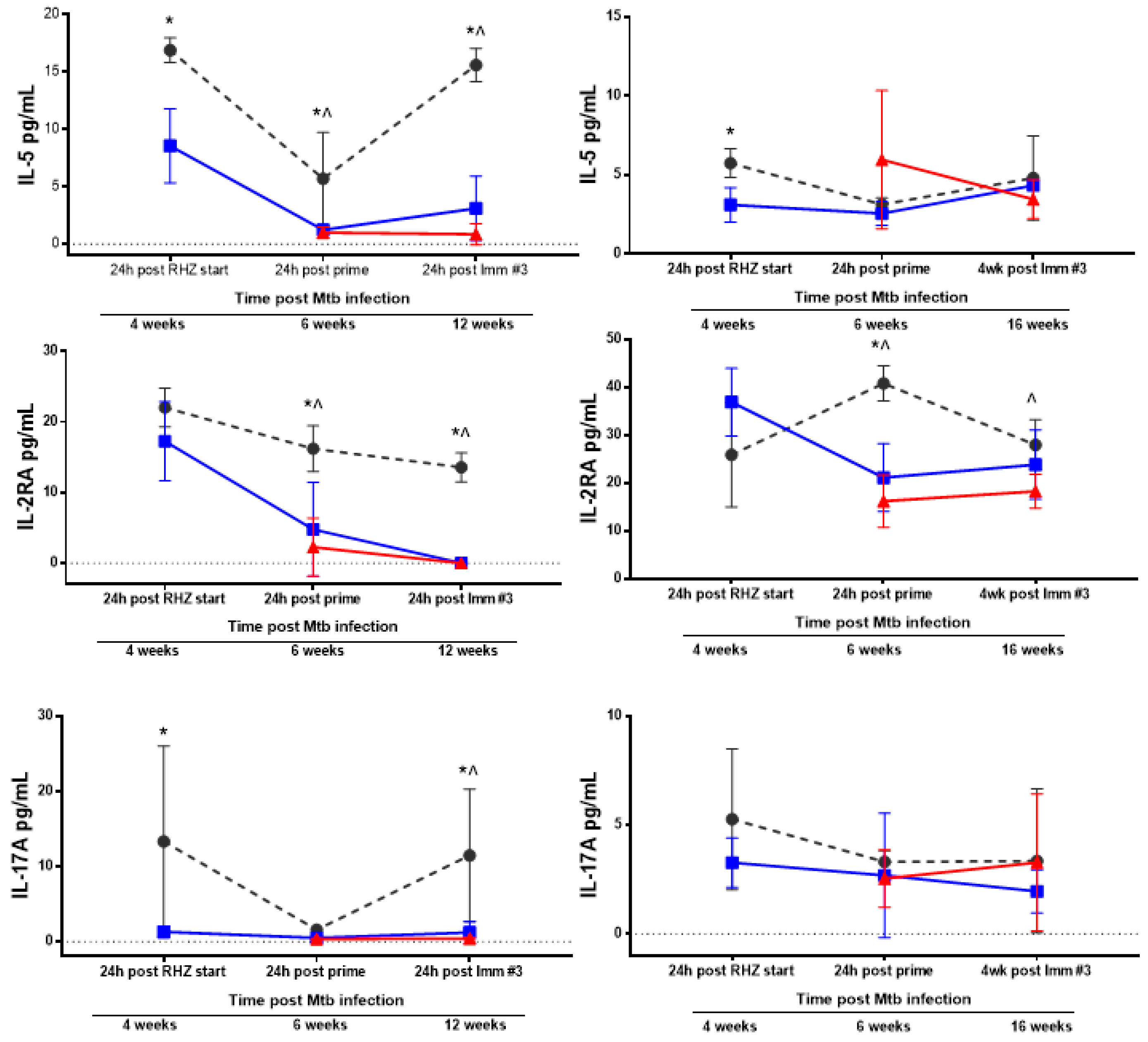
3.3. Antibiotics and Immunotherapy Reduce the Overall Inflammatory Status of an Mtb-Challenged Host
3.4. The Percentage of Regulatory T Cells in the Lung Are Not Increased Due to Immunotherapy in the Context of Drug Treatment
3.5. Immunization Provides a Significant Additive Survival Advantage over That of Antibiotics Alone
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Rodrigues, L.C.; Mangtani, P.; Abubakar, I. How does the level of BCG vaccine protection against tuberculosis fall over time? BMJ 2011, 343, d5974. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Treatment of tuberculosis: Guidelines; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Gillard, P.; Yang, P.-C.; Danilovits, M.; Su, W.-J.; Cheng, S.-L.; Pehme, L.; Bollaerts, A.; Jongert, E.; Moris, P.; Ofori-Anyinam, O.; et al. Safety and immunogenicity of the M72/AS01e candidate tuberculosis vaccine in adults with tuberculosis: A phase II randomised study. Tuberculosis 2016, 100, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Dockrell, H.M.; Ottenhoff, T.H.M.; Evans, T.G.; Zhang, Y. Tuberculosis vaccines: Opportunities and challenges. Respirology 2018. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, H.A.; Schrager, L. TB vaccine development and the end TB strategy: Importance and current status. Trans. R. Soc. Trop. Med. Hyg. 2016, 110, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Kauffman, K.D.; Sallin, M.A.; Sharpe, A.H.; Young, H.A.; Ganusov, V.V.; Barber, D.L. CD4 T cell-derived IFN-γ plays a minimal role in control of pulmonary mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog. 2016, 12, e1005667. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Windish, H.P.; Beebe, E.A.; Argilla, D.; Huang, P.-W.D.; Reese, V.A.; Reed, S.G.; Coler, R.N. Interferon γ and tumor necrosis factor are not essential parameters of CD4(+) T-cell responses for vaccine control of tuberculosis. J. Infect. Dis. 2015, 212, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.; Urdahl, K.B. TB vaccines; promoting rapid and durable protection in the lung. Curr. Opin. Immunol. 2015, 35, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Moguche, A.O.; Shafiani, S.; Clemons, C.; Larson, R.P.; Dinh, C.; Higdon, L.E.; Cambier, C.J.; Sissons, J.R.; Gallegos, A.M.; Fink, P.J.; et al. ICOS and BCL6-dependent pathways maintain a CD4 T cell population with memory-like properties during tuberculosis. J. Exp. Med. 2015, 212, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Ireton, G.C.; Beebe, E.A.; Huang, P.-W.D.; Reese, V.A.; Argilla, D.; Coler, R.N.; Reed, S.G. Immune subdominant antigens as vaccine candidates against mycobacterium tuberculosis. J. Immunol. 2014, 193, 2911–2918. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Beebe, E.A.; Hudson, T.E.; Moon, J.J.; Fox, C.B.; Reed, S.G.; Coler, R.N. A dual TLR agonist adjuvant enhances the immunogenicity and protective efficacy of the tuberculosis vaccine antigen ID93. PLoS ONE 2014, 9, e83884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertholet, S.; Ireton, G.C.; Ordway, D.J.; Windish, H.P.; Pine, S.O.; Kahn, M.; Phan, T.; Orme, I.M.; Vedvick, T.S.; Baldwin, S.L.; et al. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug resistant mycobacterium tuberculosis. Sci. Transl. Med. 2010, 2, 53ra74. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.L.; Reese, V.A.; Huang, P.-W.D.; Beebe, E.A.; Podell, B.K.; Reed, S.G.; Coler, R.N. Protection and long-lived immunity induced by the ID93/GLA-se vaccine candidate against a clinical mycobacterium tuberculosis isolate. Clin. Vaccine Immunol. CVI 2016, 23, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Coler, R.N.; Bertholet, S.; Moutaftsi, M.; Guderian, J.A.; Windish, H.P.; Baldwin, S.L.; Laughlin, E.M.; Duthie, M.S.; Fox, C.B.; Carter, D.; et al. Development and characterization of synthetic glucopyranosyl lipid adjuvant system as a vaccine adjuvant. PLoS ONE 2011, 6, e16333. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.L.; Bertholet, S.; Reese, V.A.; Ching, L.K.; Reed, S.G.; Coler, R.N. The importance of adjuvant formulation in the development of a tuberculosis vaccine. J. Immunol. 2012, 188, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Coler, R.N.; Bertholet, S.; Pine, S.O.; Orr, M.T.; Reese, V.; Windish, H.P.; Davis, C.; Kahn, M.; Baldwin, S.L.; Reed, S.G. Therapeutic immunization against mycobacterium tuberculosis is an effective adjunct to antibiotic treatment. J. Infect. Dis. 2013, 207, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Duthie, M.S.; Windish, H.P.; Lucas, E.A.; Guderian, J.A.; Hudson, T.E.; Shaverdian, N.; O’Donnell, J.; Desbien, A.L.; Reed, S.G.; et al. Myd88 and TRIF synergistic interaction is required for Th1-cell polarization with a synthetic TLR4 agonist adjuvant. Eur. J. Immunol. 2013, 43, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Fox, C.B.; Baldwin, S.L.; Sivananthan, S.J.; Lucas, E.; Lin, S.; Phan, T.; Moon, J.J.; Vedvick, T.S.; Reed, S.G.; et al. Adjuvant formulation structure and composition are critical for the development of an effective vaccine against tuberculosis. J. Control Release 2013, 172, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.B.; Kim, W.S.; Kim, J.S.; Kim, H.; Kwon, K.W.; Han, S.J.; Cho, S.N.; Coler, R.N.; Reed, S.G.; Shin, S.J. Pulmonary immunity and durable protection induced by the ID93/GLA-se vaccine candidate against the hyper-virulent korean beijing mycobacterium tuberculosis strain k. Vaccine 2016, 34, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Penn-Nicholson, A.; Tameris, M.; Smit, E.; Day, T.A.; Musvosvi, M.; Jayashankar, L.; Vergara, J.; Mabwe, S.; Bilek, N.; Geldenhuys, H.; et al. Safety and immunogenicity of the novel tuberculosis vaccine ID93/GLA-se in BCG-vaccinated healthy adults in south Africa: A randomised, double-blind, placebo-controlled phase 1 trial. Lancet Respir. Med. 2018, 6, 287–298. [Google Scholar] [CrossRef]
- Ooi, Y.M.; Colten, H.R. Genetic defect in secretion of complement c5 in mice. Nature 1979, 282, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Turner, O.C.; Keefe, R.G.; Sugawara, I.; Yamada, H.; Orme, I.M. SWR mice are highly susceptible to pulmonary infection with mycobacterium tuberculosis. Infect. Immun. 2003, 71, 5266–5272. [Google Scholar] [CrossRef] [PubMed]
- Turner, O.C.; Sugawara; Yamada, H.; Cummings, B.; Orme, I.M. Crystalloid inclusions in the cytoplasm of alveolar macrophages of the SWR/J mouse. A possible cause of susceptibility to mycobacterium tuberculosis? J. Submicrosc. Cytol. Pathol. 2001, 33, 217–219. [Google Scholar] [PubMed]
- Tonaco, M.M.; Moreira, J.D.; Nunes, F.F.C.; Loures, C.M.G.; Souza, L.R.; Martins, J.M.; Silva, H.R.; Porto, A.H.R.; Toledo, V.; Miranda, S.S.; et al. Evaluation of profile and functionality of memory t cells in pulmonary tuberculosis. Immunol. Lett. 2017, 192, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Kramnik, I.; Beamer, G. Mouse models of human tb pathology: Roles in the analysis of necrosis and the development of host-directed therapies. Semin. Immunopathol. 2016, 38, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Apt, A.; Kramnik, I. Man and mouse tb: Contradictions and solutions. Tuberculosis 2009, 89, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Santucci, N.; D’Attilio, L.; Kovalevski, L.; Bozza, V.; Besedovsky, H.; del Rey, A.; Bay, M.L.; Bottasso, O. A multifaceted analysis of immune-endocrine-metabolic alterations in patients with pulmonary tuberculosis. PLoS ONE 2011, 6, e26363. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Clark, K.; Smith, C.; Hopwood, J.; Lynard, O.; Toolan, M.; Creer, D.; Barker, J.; Breen, R.; Brown, T.; et al. Variation in C-reactive protein response according to host and mycobacterial characteristics in active tuberculosis. BMC Infect. Dis. 2016, 16, 265. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.; Vittinghoff, E.; Segal, M.R.; MacKenzie, W.R.; Johnson, J.L.; Gitta, P.; Saukkonen, J.; Anderson, J.; Weiner, M.; Engle, M.; et al. Serum biomarkers of treatment response within a randomized clinical trial for pulmonary tuberculosis. Tuberculosis 2015, 95, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Díaz, A.; Bongiovanni, B.; D’Attilio, L.; Santucci, N.; Dídoli, G.; Fernández, R.D.V.; Kovalevski, L.; Lioi, S.; Gardeñez, W.; Brandan, N.; et al. The clinical recovery of tuberculosis patients undergoing specific treatment is associated with changes in the immune and neuroendocrine responses. Pathog. Dis. 2017, 75, ftx087. [Google Scholar] [CrossRef] [PubMed]
- Niazi, M.K.K.; Dhulekar, N.; Schmidt, D.; Major, S.; Cooper, R.; Abeijon, C.; Gatti, D.M.; Kramnik, I.; Yener, B.; Gurcan, M.; et al. Lung necrosis and neutrophils reflect common pathways of susceptibility to mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis. Models Mech. 2015, 8, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Nouailles, G.; Dorhoi, A.; Koch, M.; Zerrahn, J.; Weiner, J.; Faé, K.C.; Arrey, F.; Kuhlmann, S.; Bandermann, S.; Loewe, D.; et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J. Clin. Investig. 2014, 124, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Juarrero, M.; Hattle, J.M.; Izzo, A.; Junqueira-Kipnis, A.P.; Shim, T.S.; Trapnell, B.C.; Cooper, A.M.; Orme, I.M. Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control mycobacterium tuberculosis infection. J. Leukoc. Biol. 2005, 77, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Won, E.-J.; Kee, S.-J. Biomarkers for discrimination between latent tuberculosis infection and active tuberculosis disease. J. Infect. 2017, 74, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Djouma, F.N.; Noubom, M.; Ngomba, A.V.; Donfack, H.; Kouomboua, P.S.M.; Saah, M.A.F. Determinants of death among tuberculosis patients in a semi urban diagnostic and treatment centre of Bafoussam, West Cameroon: A retrospective case-control study. Pan Afr. Med. J. 2015, 22, 253. [Google Scholar] [CrossRef] [PubMed]
- Kart, L.; Buyukoglan, H.; Tekin, I.O.; Altin, R.; Senturk, Z.; Gulmez, I.; Demir, R.; Ozesmi, M. Correlation of serum tumor necrosis factor-alpha, interleukin-4 and soluble interleukin-2 receptor levels with radiologic and clinical manifestations in active pulmonary tuberculosis. Med. Inflamm. 2003, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Shitrit, D.; Izbicki, G.; Shitrit, A.B.-G.; Raz, M.; Sulkes, J.; Kramer, M.R. Role of soluble interleukin-2 receptor levels in patients with latent tuberculosis. Lung 2006, 184, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Skogstrand, K.; Thysen, A.H.; Jørgensen, C.S.; Rasmussen, E.M.; Andersen, Å.B.; Lillebaek, T.; Hougaard, D.M.; Houen, G. Antigen-induced cytokine and chemokine release test for tuberculosis infection using adsorption of stimulated whole blood on filter paper and multiplex analysis. Scand. J. Clin. Lab. Investig. 2012, 72, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Zambuzi, F.A.; Cardoso-Silva, P.M.; Espindola, M.S.; Soares, L.S.; Galvão-Lima, L.J.; Brauer, V.S.; Gomes, M.S.; Amaral, L.R.; Schaller, M.; Bollela, V.R.; et al. Identification of promising plasma immune biomarkers to differentiate active pulmonary tuberculosis. Cytokine 2016, 88, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Sigal, G.B.; Segal, M.R.; Mathew, A.; Jarlsberg, L.; Wang, M.; Barbero, S.; Small, N.; Haynesworth, K.; Davis, J.L.; Weiner, M.; et al. Biomarkers of tuberculosis severity and treatment effect: A directed screen of 70 host markers in a randomized clinical trial. EBioMedicine 2017, 25, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, Z.; Yu, L.; Li, H.; Lin, J.; Guo, H.; Wang, W.; Chen, L.; Zhang, X.; Wang, Y.; et al. Profiling the human immune response to mycobacterium tuberculosis by human cytokine array. Tuberculosis 2016, 97, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Frahm, M.; Goswami, N.D.; Owzar, K.; Hecker, E.; Mosher, A.; Cadogan, E.; Nahid, P.; Ferrari, G.; Stout, J.E. Discriminating between latent and active tuberculosis with multiple biomarker responses. Tuberculosis 2011, 91, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Pollock, K.M.; Whitworth, H.S.; Montamat-Sicotte, D.J.; Grass, L.; Cooke, G.S.; Kapembwa, M.S.; Kon, O.M.; Sampson, R.D.; Taylor, G.P.; Lalvani, A. T-cell immunophenotyping distinguishes active from latent tuberculosis. J. Infect. Dis. 2013, 208, 952–968. [Google Scholar] [CrossRef] [PubMed]
- Keicho, N.; Matsushita, I.; Tanaka, T.; Shimbo, T.; Hang, N.T.L.; Sakurada, S.; Kobayashi, N.; Hijikata, M.; Huu Thuong, P.; Thi Lien, L. Circulating levels of adiponectin, leptin, fetuin-a and retinol-binding protein in patients with tuberculosis: Markers of metabolism and inflammation. PLoS ONE 2012, 7, e38703. [Google Scholar] [CrossRef] [PubMed]
- Perreau, M.; Rozot, V.; Welles, H.C.; Belluti-Enders, F.; Vigano, S.; Maillard, M.; Dorta, G.; Mazza-Stalder, J.; Bart, P.-A.; Roger, T.; et al. Lack of mycobacterium tuberculosis–specific interleukin-17a–producing cd4+ t cells in active disease. Eur. J. Immunol. 2013, 43, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Billeskov, R.; Lindenstrøm, T.; Woodworth, J.; Vilaplana, C.; Cardona, P.-J.; Cassidy, J.P.; Mortensen, R.; Agger, E.M.; Andersen, P. High antigen dose is detrimental to post-exposure vaccine protection against tuberculosis. Front. Immunol. 2017, 8, 1973. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, S.; Kannanganat, S.; Posey, J.E.; Amara, R.R.; Sable, S.B. Attrition of t-cell functions and simultaneous upregulation of inhibitory markers correspond with the waning of bcg-induced protection against tuberculosis in mice. PLoS ONE 2014, 9, e113951. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.G.; Du, Y.; Malherbe, S.T.; Shankar, S.; Braun, J.; Valvo, J.; Ronacher, K.; Tromp, G.; Tabb, D.L.; Alland, D.; et al. Host blood rna signatures predict the outcome of tuberculosis treatment. Tuberculosis 2017, 107, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Zak, D.E.; Penn-Nicholson, A.; Scriba, T.J.; Thompson, E.; Suliman, S.; Amon, L.M.; Mahomed, H.; Erasmus, M.; Whatney, W.; Hussey, G.D.; et al. A blood rna signature for tuberculosis disease risk: A prospective cohort study. Lancet 2016, 387, 2312–2322. [Google Scholar] [CrossRef]
- Thompson, E.G.; Shankar, S.; Gideon, H.P.; Braun, J.; Valvo, J.; Skinner, J.; Aderem, A.; Flynn, J.L.; Lin, P.L.; Zak, D.E. Blood RNA signatures prospectively discriminate controllers from progressors early after low-dose m. Tuberculosis infection of cynomolgus macaques. J. Infect. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Burel, J.G.; Lindestam Arlehamn, C.S.; Khan, N.; Seumois, G.; Greenbaum, J.A.; Taplitz, R.; Gilman, R.H.; Saito, M.; Vijayanand, P.; Sette, A.; et al. Transcriptomic analysis of CD4+ T cells reveals novel immune signatures of latent tuberculosis. J. Immunol. 2018, 200, 3283–3290. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Tasneen, R.; Tyagi, S.; Soni, H.; Converse, P.J.; Mdluli, K.; Nuermberger, E.L. Bactericidal and sterilizing activity of a novel regimen with bedaquiline, pretomanid, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00913-17. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larsen, S.E.; Baldwin, S.L.; Orr, M.T.; Reese, V.A.; Pecor, T.; Granger, B.; Dubois Cauwelaert, N.; Podell, B.K.; Coler, R.N. Enhanced Anti-Mycobacterium tuberculosis Immunity over Time with Combined Drug and Immunotherapy Treatment. Vaccines 2018, 6, 30. https://doi.org/10.3390/vaccines6020030
Larsen SE, Baldwin SL, Orr MT, Reese VA, Pecor T, Granger B, Dubois Cauwelaert N, Podell BK, Coler RN. Enhanced Anti-Mycobacterium tuberculosis Immunity over Time with Combined Drug and Immunotherapy Treatment. Vaccines. 2018; 6(2):30. https://doi.org/10.3390/vaccines6020030
Chicago/Turabian StyleLarsen, Sasha E., Susan L. Baldwin, Mark T. Orr, Valerie A. Reese, Tiffany Pecor, Brian Granger, Natasha Dubois Cauwelaert, Brendan K. Podell, and Rhea N. Coler. 2018. "Enhanced Anti-Mycobacterium tuberculosis Immunity over Time with Combined Drug and Immunotherapy Treatment" Vaccines 6, no. 2: 30. https://doi.org/10.3390/vaccines6020030