Peptide Vaccine: Progress and Challenges


Abstract
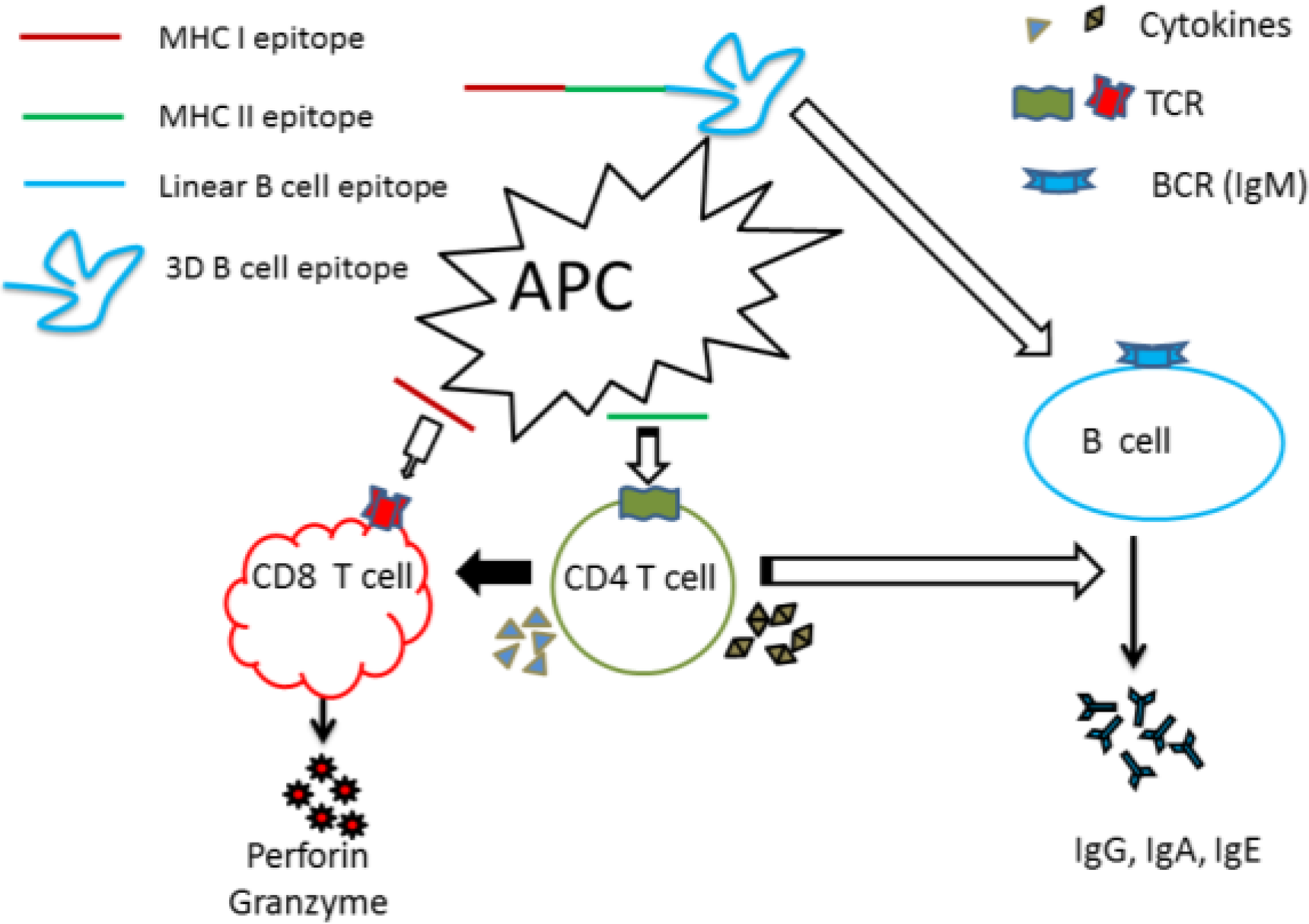
:1. Introduction

{kind=link}
| S.N. | Peptide Vaccines Under Development | Clinical Indications for Candidate Peptide Vaccines Under Development | |
|---|---|---|---|
| 1 | No. of Phase I Studies | 270 | Anticancer studies, Malaria, Falciparum Malaria, Anti-Plasmodium vivax, Influenza, Alzheimer’s disease, Insulin dependent diabetes mellitus, Hand foot and mouth disease, anti HIV, HCV, HBV, CMV, Diabetes Mellitus, Type One, Cat allergy, Allergy |
| 2 | No. of Phase II Studies | 224 | Anticancer studies, anti HIV, HCV, HBV, CMV, Pneumococcal, genital Herpes—Herpes Simplex Type II, Tuberculosis, Diabetes, Diabetes Mellitus, Type One, Cat allergy, Ragweed allergy, Grass allergy, Ashtma, House dust mites - Rhinoconjuctivitis, maximum studies–anticancer |
| 3 | No. of Phase III Studies | 12 | Anti-cancer studies |
| 4 | No. of Phase IV Studies | NIL | No peptide vaccine reached market yet |
2. Considerations and Methods for the Design of Peptide Vaccines
- (1)
- Structural resolution of desired antigen and its monoclonal antibody complex using nuclear magnetic resonance and X-ray crystallography to identify interactions at atomic level [29];
- (2)
- Mass Spectrometry for the identification of monoclonal antibody binding antigenic epitope, and then using in silico techniques mapping them on the whole antigen to describe structure and sequence of the epitope [30]. Such computational analysis is usually done by first excluding antigen non-binding regions, and subsequently mapping the amino acid residues of the antigen identified by mass spectrophotometry analysis and the crystal structure;
- (3)
- “Mimotopes” are peptides mimicking antigenic conformational structures that are recognized by paratope antibody. This is usually achieved by first generating a specific phage display library [31]. The identified peptides are then aligned to antigen sequence and subsequently superimposed to its 3D structure using in silico tools. An alternative approach is to express antigenic peptide from recombinant cDNA library and then screen for binding to specific monoclonal antibody. Using in silico tools, the selected peptide antigens can be further sequenced and aligned with antigen sequence, and if available, 3D structures can be superimposed. Some algorithms that can be of use are MimoPro, Mimox, Pepitope, MimoDB 2.0 [32];
- (4)
- (5)
- Usage of databases containing known T cell epitopes or peptides including information of their respective MHC binding and affinity of binding, the antigens involved in various clinical conditions, HLA restriction, host specificity, primary sequence of antigen etc. In case of development of peptide vaccines, selecting the correct target MHC or HLA is very critical, as the vaccine candidate should bind majority of HLAs in the population [35]. IMGT HLA database, which gives information of MHC alleles and its polymorphism and distribution in the community, is useful for this purpose [36]. A variety of structure-based algorithms also are available for in silico prediction of T cell epitopes. They are roughly classified as homology modeling, protein threading, and protein-protein docking. Prediction of conformational epitopes can be done using sequence, structure based, or binding matrices in silico algorithms such as DiscoTope, CEP, EPCES, PEPITO, SEPPA, EPSVR, ElliPro, BLAST-MODELLER, Epitpopia, CBTOPE, BEEPro, IEDB, SYFPEITHI, BIMAS, SMM, ANN, HMMs, SVMs, PROPRED, NetChop-3.0, etc. [36,37,38,39].
3. Induction of Protective T-Cell and B-Cell Mediated Immunity by Peptide Vaccines

4. Particulate Peptide Vaccine Delivery Methods
4.1. Emulsions
| Example of Emulsion Based Adjuvant | Composition | Features | Encapsulated Peptide Antigen | Ref. |
|---|---|---|---|---|
| Freunds complete adjuvant | w/o emulsion of a mineral oil, paraffin and killed mycobacteria | Capable of generating high immunization titers Strong adverse reactions | Human papillomaviruses (HPV) E5 | [65] |
| Montanide™ ISA 720 and 51 | w/o emulsion with squalene as the oil and mono-oleate as the surfactant | Associated with adverse drug reactions; extensive and costly emulsification is needed | Human papillomaviruses (HPV) E6, E7 | [65,66] |
| MF59™ | o/w emulsion with squalene oil dispersed with the help of surfactants viz. polysorbate 80 and sorbitan oleate | Great safety profile, able to activate immune cells directly | Melanoma Peptides | [67] |
| AS02™ | Composed of squalene and two hydrophobic immune adjuvants viz. MPL1™, a synthetic derivative of lipid A and QS-21, a purified saponin extract | Capable of inducing both humoral and cellular response | A recombinantly produced fusion of circumsporozoite protein (CS) and hepatitis B surface antigen (HBsAg), called RTS,S | [68] |
4.2. Liposomes
4.3. Virosomes, ISCOMS and Related Particles
4.4. Polymeric Particles
4.5. Other Particulate Systems
4.6. Adjuvants Approved for Human Use
4.7. Safety Issues with Particulate Delivery Strategies
5. Summary
Acknowledgements
Authors Contributions
Conflicts of Interest
References
- Bachler, B.C.; Humbert, M.; Palikuqi, B.; Siddappa, N.B.; Lakhashe, S.K.; Rasmussen, R.A.; Ruprecht, R.M. Novel biopanning strategy to identify epitopes associated with vaccine protection. J. Virol. 2013, 87, 4403–4416. [Google Scholar] [CrossRef]
- Perrie, Y.; Kirby, D.; Bramwell, V.W.; Mohammed, A.R. Recent developments in particulate-based vaccines. Recent Pat. Drug Deliv. Formul. 2007, 1, 117–129. [Google Scholar] [CrossRef]
- Black, M.; Trent, A.; Tirrell, M.; Olive, C. Advances in the design and delivery of peptide subunit vaccines with a focus on toll-like receptor agonists. Expert Rev. Vaccines 2010, 9, 157–173. [Google Scholar] [CrossRef]
- Thompson, A.L.; Staats, H.F. Cytokines: The future of intranasal vaccine adjuvants. Clin. Dev. Immunol. 2011, 2011. [Google Scholar] [CrossRef]
- Petrovsky, N.; Aguilar, J.C. Vaccine adjuvants: Current state and future trends. Immunol. Cell Biol. 2004, 82, 488–496. [Google Scholar] [CrossRef]
- Sesardic, D. Synthetic peptide vaccines. J. Med. Microbiol. 1993, 39, 241–242. [Google Scholar] [CrossRef]
- Bijker, M.S.; Melief, C.J.; Offringa, R.; van der Burg, S.H. Design and development of synthetic peptide vaccines: Past, present and future. Expert Rev. Vaccines 2007, 6, 591–603. [Google Scholar] [CrossRef]
- Lin, S.Y.; Cheng, C.W.; Su, E.C. Prediction of B-cell epitopes using evolutionary information and propensity scales. BMC Bioinform. 2013, 14. [Google Scholar] [CrossRef]
- Purcell, A.W.; McCluskey, J.; Rossjohn, J. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 2007, 6, 404–414. [Google Scholar] [CrossRef]
- Aguilar, J.C.; Rodriguez, E.G. Vaccine adjuvants revisited. Vaccine 2007, 25, 3752–3762. [Google Scholar] [CrossRef]
- Liu, Y.; McNevin, J.; Zhao, H.; Tebit, D.M.; Troyer, R.M.; McSweyn, M.; Ghosh, A.K.; Shriner, D.; Arts, E.J.; McElrath, M.J.; et al. Evolution of human immunodeficiency virus type 1 cytotoxic T-lymphocyte epitopes: Fitness-balanced escape. J. Virol. 2007, 81, 12179–12188. [Google Scholar] [CrossRef]
- Kolesanova, E.F.; Sanzhakov, M.A.; Kharybin, O.N. Development of the schedule for multiple parallel “difficult” Peptide synthesis on pins. Int. J. Pept. 2013, 2013. [Google Scholar] [CrossRef]
- Epstein, J.E.; Giersing, B.; Mullen, G.; Moorthy, V.; Richie, T.L. Malaria vaccines: Are we getting closer? Curr. Opin. Mol. Ther. 2007, 9, 12–24. [Google Scholar]
- Volpina, O.M.; Gelfanov, V.M.; Yarov, A.V.; Surovoy, A.Y.; Chepurkin, A.V.; Ivanov, V.T. New virus-specific T-helper epitopes of foot-and-mouth disease viral VP1 protein. FEBS Lett. 1993, 333, 175–178. [Google Scholar] [CrossRef]
- Tarradas, J.; Monso, M.; Munoz, M.; Rosell, R.; Fraile, L.; Frías, M.T.; Domingo, M.; Andreu, D.; Sobrino, F.; Ganges, L. Partial protection against classical swine fever virus elicited by dendrimeric vaccine-candidate peptides in domestic pigs. Vaccine 2011, 29, 4422–4429. [Google Scholar] [CrossRef]
- Stanekova, Z.; Kiraly, J.; Stropkovska, A.; Mikušková, T.; Mucha, V.; Kostolanský, F.; Varečková, E. Heterosubtypic protective immunity against influenza a virus induced by fusion peptide of the hemagglutinin in comparison to ectodomain of M2 protein. Acta Virol. 2011, 55, 61–67. [Google Scholar] [CrossRef]
- Oscherwitz, J.; Yu, F.; Cease, K.B. A synthetic peptide vaccine directed against the 2ss2–2ss3 loop of domain 2 of protective antigen protects rabbits from inhalation anthrax. J. Immunol. 2010, 185, 3661–3668. [Google Scholar] [CrossRef]
- Solares, A.M.; Baladron, I.; Ramos, T.; Valenzuela, C.; Borbon, Z.; Fanjull, S.; Gonzalez, L.; Castillo, D.; Esmir, J.; Granadillo, M.; et al. Safety and immunogenicity of a human papillomavirus peptide vaccine (CIGB-228) in women with high-grade cervical intraepithelial neoplasia: first-in-human, proof-of-concept trial. ISRN Obstet. Gynecol. 2011, 2011. [Google Scholar] [CrossRef]
- Bernhardt, S.L.; Gjertsen, M.K.; Trachsel, S.; Møller, M.; Eriksen, J.A.; Meo, M.; Buanes, T.; Gaudernack, G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: A dose escalating phase I/II study. Br. J. Cancer 2006, 95, 1474–1482. [Google Scholar] [CrossRef]
- Brunsvig, P.F.; Aamdal, S.; Gjertsen, M.K.; Kvalheim, G.; Markowski-Grimsrud, C.J.; Sve, I.; Dyrhaug, M.; Trachsel, S.; Møller, M.; Eriksen, J.A.; et al. Telomerase peptide vaccination: A phase I/II study in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2006, 55, 1553–1564. [Google Scholar] [CrossRef]
- Brunsvig, P.F.; Kyte, J.A.; Kersten, C.; Sundstrøm, S.; Møller, M.; Nyakas, M.; Hansen, G.L.; Gaudernack, G.; Aamdal, S. Telomerase peptide vaccination in NSCLC: A phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin. Cancer Res. 2011, 17, 6847–6857. [Google Scholar] [CrossRef]
- Kyte, J.A.; Gaudernack, G.; Dueland, S.; Trachsel, S.; Julsrud, L.; Aamdal, S. Telomerase peptide vaccination combined with temozolomide: A clinical trial in stage IV melanoma patients. Clin. Cancer Res. 2011, 17, 4568–4580. [Google Scholar]
- Greten, T.F.; Forner, A.; Korangy, F.; N’Kontchou, G.; Barget, N.; Ayuso, C.; Ormandy, L.A.; Manns, M.P.; Beaugrand, M.; Bruix, J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 2010, 10, e209. [Google Scholar] [CrossRef]
- Kyte, J.A.; Trachsel, S.; Risberg, B.; Thor, S.P.; Lislerud, K.; Gaudernack, G. Unconventional cytokine profiles and development of T cell memory in long-term survivors after cancer vaccination. Cancer Immunol. Immunother. 2009, 58, 1609–1626. [Google Scholar] [CrossRef]
- Demento, S.L.; Siefert, A.L.; Bandyopadhyay, A.; Sharp, F.A.; Fahmy, T.M. Pathogen-associated molecular patterns on biomaterials: A paradigm for engineering new vaccines. Trends Biotechnol. 2011, 29, 294–306. [Google Scholar] [CrossRef]
- Burioni, R.; Perotti, M.; Mancini, N.; Clementi, M. Perspectives for the utilization of neutralizing human monoclonal antibodies as anti-HCV drugs. J. Hepatol. 2008, 49, 299–300. [Google Scholar] [CrossRef]
- Testa, J.S.; Philip, R. Role of T-cell epitope-based vaccine in prophylactic and therapeutic applications. Future Virol. 2012, 7, 1077–1088. [Google Scholar] [CrossRef]
- Lanier, J.G.; Newman, M.J.; Lee, E.M.; Sette, A.; Ahmed, R. Peptide vaccination using nonionic block copolymers induces protective anti-viral CTL responses. Vaccine 1999, 18, 549–557. [Google Scholar] [CrossRef]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef]
- Hager-Braun, C.; Tomer, K.B. Determination of protein-derived epitopes by mass spectrometry. Expert Rev. Proteomics 2005, 2, 745–756. [Google Scholar] [CrossRef]
- Panina-Bordignon, P.; Demotz, S.; Corradin, G.; Lanzavecchia, A. Study on the immunogenicity of human class-II-restricted T-cell epitopes: Processing constraints, degenerate binding, and promiscuous recognition. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 445–451. [Google Scholar] [CrossRef]
- Huang, J.; Ru, B.; Zhu, P.; Nie, F.; Yang, J.; Wang, X.; Dai, P.; Lin, H.; Guo, F.B.; Rao, N. MimoDB 2.0: A mimotope database and beyond. Nucleic Acids Res. 2012, 40, D271–D277. [Google Scholar] [CrossRef]
- Gershoni, J.M.; Roitburd-Berman, A.; Siman-Tov, D.D.; Tarnovitski, F.N.; Weiss, Y. Epitope mapping: The first step in developing epitope-based vaccines. BioDrugs 2007, 21, 145–156. [Google Scholar] [CrossRef]
- Langeveld, J.P.; Casal, J.I.; Osterhaus, A.D.; Cortés, E.; de Swart, R.; Vela, C.; Dalsgaard, K.; Puijk, W.C.; Schaaper, W.M.; Meloen, R.H. First peptdie vaccine providing protection against viral infection in the target animal; studies of canine parvovirus in dogs. J. Virol. 1994, 68, 4506–4513. [Google Scholar]
- Kupriianova, M.A.; Zhmak, M.N.; Koroev, D.O.; Chepurkin, A.V.; Vol’pina, O.M.; Ivanov, V.T. Synthetic peptide designs based on immunoactive fragments of the VP1 protein of the foot-and-mouth disease virus strain A22. Bioorg. Khimiia 2000, 26, 926–932. [Google Scholar]
- Robinson, J.; Waller, M.J.; Fail, S.C.; Mcwilliam, H.; Lopez, R.; Parham, P.; Marsh, S.G.E. The IMGT/HLA database. Nucleic Acids Res. 2009, 37, D1013–D1017. [Google Scholar] [CrossRef]
- Schuler, M.M.; Nastke, M.D.; Stevanovikc, S. SYFPEITHI: Database for searching and T-cell epitope prediction. Methods Mol. Biol. 2007, 409, 75–93. [Google Scholar]
- Michielin, O.; Luescher, I.; Karplus, M. Modeling of the TCR-MHC-peptide complex. J. Mol. Biol. 2000, 300, 1205–1235. [Google Scholar] [CrossRef]
- Lin, H.H.; Ray, S.; Tongchusak, S.; Reinherz, E.L.; Brusic, V. Evaluation of MHC class I peptide binding prediction servers: Applications for vaccine research. BMC Immunol. 2008, 9. [Google Scholar] [CrossRef]
- Townsend, A.R.; Rothbard, J.; Gotch, F.M.; Bahadur, G.; Wraith, D.; McMichael, A.J. The epitopes of influenza nucleoprotein recognized by cytotoxic T lymphocytes can be defined with short synthetic peptides. Cell 1986, 44, 959–968. [Google Scholar] [CrossRef]
- Townsend, A.R.; McMichael, A.J. Specificity of cytotoxic T lymphocytes stimulated with influenza virus. Studies in mice and humans. Prog. Allergy 1985, 36, 10–43. [Google Scholar]
- Townsend, A.R.; Skehel, J.J.; Taylor, P.M.; Palese, P. Recognition of influenza A virus nucleoprotein by an H-2-restricted cytotoxic T-cell clone. Virology 1984, 133, 456–459. [Google Scholar] [CrossRef]
- Aichele, P.; Brduscha-Riem, K.; Zinkernagel, R.M.; Hengartner, H.; Pircher, H. T cell priming versus T cell tolerance induced by synthetic peptides. J. Exp. Med. 1995, 182, 261–266. [Google Scholar] [CrossRef]
- Aichele, P.; Hengartner, H.; Zinkernagel, R.M.; Schulz, M. Antiviral cytotoxic T cell response induced by in vivo priming with a free synthetic peptide. J. Exp. Med. 1990, 171, 1815–1820. [Google Scholar] [CrossRef]
- Azizi, A.; Anderson, D.E.; Torres, J.V.; Ogrel, A.; Ghorbani, M.; Soare, C.; Sandstrom, P.; Fournier, J.; Diaz-Mitoma, F. Induction of broad cross-subtype-specific HIV-1 immune responses by a novel multivalent HIV-1 peptide vaccine in cynomolgus macaques. J. Immunol. 2008, 180, 2174–2186. [Google Scholar] [CrossRef]
- Kast, W.M.; Roux, L.; Curren, J.; Blom, H.J.; Voordouw, A.C.; Meloen, R.H.; Kolakofsky, D.; Melief, C.J. Protection against lethal Sendai virus infection by in vivo priming of virus-specific cytotoxic T lymphocytes with a free synthetic peptide. Proc. Natl. Acad. Sci. USA 1991, 88, 2283–2287. [Google Scholar] [CrossRef]
- Wahala, W.M.; Silva, A.M. The human antibody response to dengue virus infection. Viruses 2011, 3, 2374–2395. [Google Scholar] [CrossRef]
- Joshi, M.D.; Unger, W.J.; Storm, G.; van, K.Y.; Mastrobattista, E. Targeting tumor antigens to dendritic cells using particulate carriers. J. Control Release 2012, 161, 25–37. [Google Scholar] [CrossRef]
- Xu, Z.; Ramishetti, S.; Tseng, Y.C.; Guo, S.; Wang, Y.; Huang, L. Multifunctional nanoparticles co-delivering Trp2 peptide and CpG adjuvant induce potent cytotoxic T-lymphocyte response against melanoma and its lung metastasis. J. Control Release 2013, 172, 259–265. [Google Scholar] [CrossRef]
- Quakkelaar, E.D.; Melief, C.J. Experience with synthetic vaccines for cancer and persistent virus infections in nonhuman primates and patients. Adv. Immunol. 2012, 114, 77–106. [Google Scholar] [CrossRef]
- Quakkelaar, E.D.; Bunnik, E.M.; van Alphen, F.P.; Boeser-Nunnink, B.D.; van Nuenen, A.C.; Schuitemaker, H. Escape of human immunodeficiency virus type 1 from broadly neutralizing antibodies is not associated with a reduction of viral replicative capacity in vitro. Virology 2007, 363, 447–453. [Google Scholar] [CrossRef]
- Moisa, A.A.; Kolesanova, E.F. Synthetic peptide vaccines. Biomed. Khimiia 2011, 57, 14–30. [Google Scholar]
- Fayolle, C.; bdel-Motal, U.M.; Berg, L.; Deriaud, E.; Jondal, M.; Leclerc, C. Induction of cytotoxic T-cell response by optimal-length peptides does not require CD4+ T-cell help. Immunology 1996, 89, 41–45. [Google Scholar]
- Fayolle, C.; Sebo, P.; Ladant, D.; Ullmann, A.; Leclerc, C. In vivo induction of CTL responses by recombinant adenylate cyclase of Bordetella pertussis carrying viral CD8+ T cell epitopes. J. Immunol. 1996, 156, 4697–4706. [Google Scholar]
- Zwaveling, S.; Ferreira Mota, S.C.; Nouta, J.; Johnson, M.; Lipford, G.B.; Offringa, R.; van der Burg, S.H.; Melief, C.J. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J. Immunol. 2002, 169, 350–358. [Google Scholar] [CrossRef]
- Welters, M.J.; Kenter, G.G.; Piersma, S.J.; Vloon, A.P.; Löwik, M.J.; Berends-van der Meer, D.M.; Drijfhout, J.W.; Valentijn, A.R.; Wafelman, A.R.; Oostendorp, J.; et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin. Cancer Res. 2008, 14, 178–187. [Google Scholar]
- Welters, M.J.; van der, L.P.; van den Eeden, S.J.; Kwappenberg, K.M.C.; Drijfhout, J.W.; Fleuren, G.J.; Kenter, G.G.; Melief, C.J.M.; van der Burg, S.H.; Offringa, R. Detection of human papillomavirus type 18 E6 and E7-specific CD4+ T-helper 1 immunity in relation to health versus disease. Int. J. Cancer 2006, 118, 950–956. [Google Scholar]
- Welters, M.J.; Kenter, G.G.; de Vos van Steenwijk, P.J.; Löwik, M.J.; Berends-van der Meer, D.M.; Essahsah, F.; Stynenbosch, L.F.; Vloon, A.P.; Ramwadhdoebe, T.H.; Piersma, S.J.; et al. Success or failure of vaccination for HPV16-positive vulvar lesions correlates with kinetics and phenotype of induced T-cell responses. Proc. Natl. Acad. Sci. USA 2010, 107, 11895–11899. [Google Scholar]
- Panina-Bordignon, P.; Tan, A.; Termijtelen, A.; Demotz, S.; Corradin, G.; Lanzavecchia, A. Universally immunogenic T cell epitopes: Promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur. J. Immunol. 1989, 19, 2237–2242. [Google Scholar] [CrossRef]
- Agadjanyan, M.G.; Ghochikyan, A.; Petrushina, I.; Vasilevko, V.; Movsesyan, N.; Mkrtichyan, M.; Saing, T.; Cribbs, D.H. Prototype Alzheimer’s disease vaccine using the immunodominant B cell epitope from beta-amyloid and promiscuous T cell epitope pan HLA DR-binding peptide. J. Immunol. 2005, 174, 1580–1586. [Google Scholar]
- Chen, W.; Ede, N.J.; Jackson, D.C.; McCluskey, J.; Purcell, A.W. CTL recognition of an altered peptide associated with asparagine bond rearrangement. Implications for immunity and vaccine design. J. Immunol. 1996, 157, 1000–1005. [Google Scholar]
- Jackson, D.C.; Purcell, A.W.; Fitzmaurice, C.J.; Zeng, W.; Hart, D.N. The central role played by peptides in the immune response and the design of peptide-based vaccines against infectious diseases and cancer. Curr. Drug Targets 2002, 3, 175–196. [Google Scholar]
- Koh, Y.T.; Higgins, S.A.; Weber, J.S.; Kast, W.M. Immunological consequences of using three different clinical/laboratory techniques of emulsifying peptide-based vaccines in incomplete Freund’s adjuvant. J. Transl. Med. 2006, 4. [Google Scholar] [CrossRef]
- Kalariya, M.; Ganta, S.; Amiji, M. Multi-compartmental vaccine delivery system for enhanced immune response to gp100 peptide antigen in melanoma immunotherapy. Pharm. Res. 2012, 29, 3393–3403. [Google Scholar] [CrossRef]
- Chen, Y.F.; Lin, C.W.; Tsao, Y.P.; Chen, S.L. Cytotoxic-T-lymphocyte human papillomavirus type 16 E5 peptide with CpG-oligodeoxynucleotide can eliminate tumor growth in C57BL/6 mice. J. Virol. 2004, 78, 1333–1343. [Google Scholar] [CrossRef]
- Van Driel, W.J.; Ressing, M.E.; Kenter, G.G.; Brandt, R.M.; Krul, E.J.; van Rossum, A.B.; Schuuring, E.; Offringa, R.; Bauknecht, T.; Tamm-Hermelink, A.; et al. Vaccination with HPV16 peptides of patients with advanced cervical carcinoma: Clinical evaluation of a phase I–II trial. Eur. J. Cancer 1999, 35, 946–952. [Google Scholar] [CrossRef]
- Slingluff, C.L., Jr.; Petroni, G.R.; Yamshchikov, G.V.; Barnd, D.L.; Eastham, S.; Galavotti, H.; Patterson, J.W.; Deacon, D.H.; Hibbitts, S.; Teates, D.; et al. Clinical and immunologic results of a randomized phase II trial of vaccination using four melanoma peptides either administered in granulocyte-macrophage colony-stimulating factor in adjuvant or pulsed on dendritic cells. J. Clin. Oncol. 2003, 21, 4016–4026. [Google Scholar] [CrossRef]
- Pinder, M.; Reece, W.H.; Plebanski, M.; Akinwunmi, P.; Flanagan, K.L.; Lee, E.A.; Doherty, T.; Milligan, P.; Jaye, A.; Tornieporth, N.; et al. Cellular immunity induced by the recombinant Plasmodium falciparum malaria vaccine, RTS,S/AS02, in semi-immune adults in The Gambia. Clin. Exp. Immunol. 2004, 135, 286–293. [Google Scholar] [CrossRef]
- Beebe, M.; Qin, M.; Moi, M.; Wu, S.; Heiati, H.; Walker, L.; Newman, M.; Fikes, J.; Ishioka, G.Y. Formulation and characterization of a ten-peptide single-vial vaccine, EP-2101, designed to induce cytotoxic T-lymphocyte responses for cancer immunotherapy. Hum. Vaccines 2008, 4, 210–218. [Google Scholar] [CrossRef]
- Iseki, K.; Matsunaga, H.; Komatsu, N.; Suekane, S.; Noguchi, M.; Itoh, K.; Yamada, A. Evaluation of a new oil adjuvant for use in peptide-based cancer vaccination. Cancer Sci. 2010, 101, 2110–2114. [Google Scholar] [CrossRef]
- Liang, M.T.; Davies, N.M.; Blanchfield, J.T.; Toth, I. Particulate systems as adjuvants and carriers for peptide and protein antigens. Curr. Drug Deliv. 2006, 3, 379–388. [Google Scholar] [CrossRef]
- Nagata, T.; Toyota, T.; Ishigaki, H.; Ichihashi, T.; Kajino, K.; Kashima, Y.; Itoh, Y.; Mori, M.; Oda, H.; Yamamura, H.; et al. Peptides coupled to the surface of a kind of liposome protect infection of influenza viruses. Vaccine 2007, 25, 4914–4921. [Google Scholar] [CrossRef]
- Taneichi, M.; Tanaka, Y.; Kakiuchi, T.; Uchida, T. Liposome-coupled peptides induce long-lived memory CD8 T cells without CD4 T cells. PLoS One 2010, 5, e15091. [Google Scholar]
- Kohyama, S.; Ohno, S.; Suda, T.; Taneichi, M.; Yokoyama, S.; Mori, M.; Kobayashi, A.; Hayashi, H.; Uchida, T.; Matsui, M. Efficient induction of cytotoxic T lymphocytes specific for severe acute respiratory syndrome (SARS)-associated coronavirus by immunization with surface-linked liposomal peptides derived from a non-structural polyprotein 1a. Antivir. Res. 2009, 84, 168–177. [Google Scholar] [CrossRef]
- Takagi, A.; Matsui, M.; Ohno, S.; Duan, H.; Moriya, O.; Kobayashi, N.; Oda, H.; Mori, M.; Kobayashi, A.; Taneichi, M.; et al. Highly efficient antiviral CD8+ T-cell induction by peptides coupled to the surfaces of liposomes. Clin. Vaccine Immunol. 2009, 16, 1383–1392. [Google Scholar] [CrossRef]
- Guan, H.H.; Budzynski, W.; Koganty, R.R.; Krantz, M.J.; Reddish, M.A.; Rogers, J.A.; Longenecker, B.M.; Samuel, J. Liposomal formulations of synthetic MUC1 peptides: Effects of encapsulation versus surface display of peptides on immune responses. Bioconjug. Chem. 1998, 9, 451–458. [Google Scholar]
- Cortesi, R.; Argnani, R.; Esposito, E.; Dalpiaz, A.; Scatturin, A.; Bortolotti, F.; Lufino, M.; Guerrini, R.; Cavicchioni, G.; Incorvaia, C.; et al. Cationic liposomes as potential carriers for ocular administration of peptides with anti-herpetic activity. Int. J. Pharm. 2006, 317, 90–100. [Google Scholar] [CrossRef]
- Henriksen-Lacey, M.; Korsholm, K.S.; Andersen, P.; Perrie, Y.; Christensen, D. Liposomal vaccine delivery systems. Expert Opin. Drug Deliv. 2011, 8, 505–519. [Google Scholar] [CrossRef]
- Sugita, T.; Yoshikawa, T.; Gao, J.Q.; Shimokawa, M.; Oda, A.; Niwa, T.; Akashi, M.; Tsutsumi, Y.; Mayumi, T.; Nakagawa, S. Fusogenic liposome can be used as an effective vaccine carrier for peptide vaccination to induce cytotoxic T lymphocyte (CTL) response. Biol. Pharm. Bull. 2005, 28, 192–193. [Google Scholar] [CrossRef]
- Chang, J.S.; Choi, M.J.; Cheong, H.S.; Kim, K. Development of Th1-mediated CD8+ effector T cells by vaccination with epitope peptides encapsulated in pH-sensitive liposomes. Vaccine 2001, 19, 3608–3614. [Google Scholar] [CrossRef]
- Chang, J.S.; Choi, M.J.; Kim, T.Y.; Cho, S.Y.; Cheong, H.S. Immunogenicity of synthetic HIV-1 V3 loop peptides by MPL adjuvanted pH-sensitive liposomes. Vaccine 1999, 17, 1540–1548. [Google Scholar] [CrossRef]
- Hayashi, A.; Wakita, H.; Yoshikawa, T.; Nakanishi, T.; Tsutsumi, Y.; Mayumie, T.; Mukai, Y.; Yoshioka, Y.; Okada, N.; Nakagawa, S.; et al. A strategy for efficient cross-presentation of CTL-epitope peptides leading to enhanced induction of in vivo tumor immunity. J. Control Release 2007, 117, 11–19. [Google Scholar] [CrossRef]
- Awate, S.; Babiuk, L.A.; Mutwiri, G. Mechanisms of action of adjuvants. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef]
- Okitsu, S.L.; Boato, F.; Mueller, M.S.; Li, D.B.; Vogel, D.; Westerfeld, N.; Zurbriggen, R.; Robinson, J.A.; Pluschke, G. Antibodies elicited by a virosomally formulated Plasmodium falciparum serine repeat antigen-5 derived peptide detect the processed 47 kDa fragment both in sporozoites and merozoites. Peptides 2007, 28, 2051–2060. [Google Scholar] [CrossRef]
- Cryz, S.J.; Que, J.U.; Gluck, R. A virosome vaccine antigen delivery system does not stimulate an antiphospholipid antibody response in humans. Vaccine 1996, 14, 1381–1383. [Google Scholar] [CrossRef]
- Westerfeld, N.; Pluschke, G.; Zurbriggen, R. Optimized Malaria-antigens delivered by immunostimulating reconstituted influenza virosomes. Wien. klinische Wochenschr. 2006, 118, 50–57. [Google Scholar] [CrossRef]
- Herzog, C.; Hartmann, K.; Kunzi, V.; Kürsteiner, O.; Mischler, R.; Lazar, H.; Glück, R. Eleven years of inflexal V-a virosomal adjuvanted influenza vaccine. Vaccine 2009, 27, 4381–4387. [Google Scholar] [CrossRef]
- Sun, H.X.; Xie, Y.; Ye, Y.P. ISCOMs and ISCOMATRIX. Vaccine 2009, 27, 4388–4401. [Google Scholar] [CrossRef]
- Morelli, A.B.; Becher, D.; Koernig, S.; Silva, A.; Drane, D.; Maraskovsky, E. ISCOMATRIX: A novel adjuvant for use in prophylactic and therapeutic vaccines against infectious diseases. J. Med. Microbiol. 2012, 61, 935–943. [Google Scholar] [CrossRef]
- Duewell, P.; Kisser, U.; Heckelsmiller, K.; Hoves, S.; Stoitzner, P.; Koernig, S.; Morelli, A.B.; Clausen, B.E.; Dauer, M.; Eigler, A.; et al. ISCOMATRIX adjuvant combines immune activation with antigen delivery to dendritic cells in vivo leading to effective cross-priming of CD8+ T cells. J. Immunol. 2011, 187, 55–63. [Google Scholar] [CrossRef]
- Agrawal, L.; Haq, W.; Hanson, C.V.; Rao, D.N. Generating neutralizing antibodies, Th1 response and MHC non restricted immunogenicity of HIV-I env and gag peptides in liposomes and ISCOMs with in-built adjuvanticity. J. Immune Based Ther. Vaccines 2003, 1. [Google Scholar] [CrossRef] [Green Version]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef]
- Kang, N.; Perron, M.E.; Prud’homme, R.E.; Zhang, Y.; Gaucher, G.; Leroux, J.C. Stereocomplex block copolymer micelles: Core-shell nanostructures with enhanced stability. Nano Lett. 2005, 5, 315–319. [Google Scholar]
- Correia-Pinto, J.F.; Csaba, N.; Alonso, M.J. Vaccine delivery carriers: Insights and future perspectives. Int. J. Pharm. 2013, 440, 27–38. [Google Scholar] [CrossRef]
- Buyuktimkin, B.; Wang, Q.; Kiptoo, P.; Stewart, J.M.; Berkland, C.; Siahaan, T.J. Vaccine-like controlled-release delivery of an immunomodulating peptide to treat experimental autoimmune encephalomyelitis. Mol. Pharm. 2012, 9, 979–985. [Google Scholar] [CrossRef]
- Heffernan, M.J.; Zaharoff, D.A.; Fallon, J.K.; Schlom, J.; Greiner, J.W. In vivo efficacy of a chitosan/IL-12 adjuvant system for protein-based vaccines. Biomaterials 2011, 32, 926–932. [Google Scholar]
- Saenz, L.; Neira-Carrillo, A.; Paredes, R.; Cortes, M.; Bucarey, S.; Arias, J.L. Chitosan formulations improve the immunogenicity of a GnRH-I peptide-based vaccine. Int. J. Pharm. 2009, 369, 64–71. [Google Scholar] [CrossRef]
- Zaharoff, D.A.; Rogers, C.J.; Hance, K.W.; Schlom, J.; Greiner, J.W. Chitosan solution enhances the immunoadjuvant properties of GM-CSF. Vaccine 2007, 25, 8673–8686. [Google Scholar] [CrossRef]
- Chua, B.Y.; Al, K.M.; Zeng, W.; Mainwaring, D.; Jackson, D.C. Chitosan microparticles and nanoparticles as biocompatible delivery vehicles for peptide and protein-based immunocontraceptive vaccines. Mol. Pharm. 2012, 9, 81–90. [Google Scholar] [CrossRef]
- Gaertner, H.F.; Cerini, F.; Kamath, A.; Rochat, A.-F.; Siegrist, C.-A.; Menin, L.; Hartley, O. Efficient orthogonal bioconjugation of dendrimers for synthesis of bioactive nanoparticles. Bioconjug. Chem. 2011, 22, 1103–1114. [Google Scholar]
- Chen, Y.S.; Hung, Y.C.; Lin, L.W.; Liau, I.; Hong, M.Y.; Huang, G.S. Size-dependent impairment of cognition in mice caused by the injection of gold nanoparticles. Nanotechnology 2010, 21. [Google Scholar] [CrossRef]
- Jalali, S.A.; Sankian, M.; Tavakkol-Afshari, J.; Jaafari, M.R. Induction of tumor-specific immunity by multi-epitope rat HER2/neu-derived peptides encapsulated in LPD Nanoparticles. Nanomedicine 2012, 8, 692–701. [Google Scholar] [CrossRef]
- Daftarian, P.; Mansour, M.; Benoit, A.C.; Pohajdak, B.; Hoskin, D.W.; Brown, R.G.; Kast, W.M. Eradication of established HPV 16-expressing tumors by a single administration of a vaccine composed of a liposome-encapsulated CTL-T helper fusion peptide in a water-in-oil emulsion. Vaccine 2006, 24, 5235–5244. [Google Scholar] [CrossRef]
- Jiang, T.; Singh, B.; Li, H.S.; Kim, Y.K.; Kang, S.K.; Nah, J.W.; Choi, Y.J.; Cho, C.S. Targeted oral delivery of BmpB vaccine using porous PLGA microparticles coated with M cell homing peptide-coupled chitosan. Biomaterials 2014, 35, 2365–2373. [Google Scholar] [CrossRef]
- Fischer, S.; Schlosser, E.; Mueller, M.; Csaba, N.; Merkle, H.P.; Groettrup, M.; Gander, B. Concomitant delivery of a CTL-restricted peptide antigen and CpG ODN by PLGA microparticles induces cellular immune response. J. Drug Target. 2009, 17, 652–661. [Google Scholar] [CrossRef]
- Garlapati, S.; Garg, R.; Brownlie, R.; Latimer, L.; Simko, E.; Hancock, R.E.; Babiuk, L.A.; Gerdts, V.; Potter, A.; van Drunen Littel-van den Hurk, S. Enhanced immune responses and protection by vaccination with respiratory syncytial virus fusion protein formulated with CpG oligodeoxynucleotide and innate defense regulator peptide in polyphosphazene microparticles. Vaccine 2012, 30, 5206–5214. [Google Scholar] [CrossRef]
- Haining, W.N.; Anderson, D.G.; Little, S.R.; von Bergwelt-Baildon, M.S.; Cardoso, A.A.; Alves, P.; Kosmatopoulos, K.; Nadler, L.M.; Langer, R.; Kohane, D.S. pH-triggered microparticles for peptide vaccination. J. Immunol. 2004, 173, 2578–2585. [Google Scholar]
- Seubert, A.; Calabro, S.; Santini, L.; Galli, B.; Genovese, A.; Valentini, S.; Aprea, S.; Colaprico, A.; D’Oro, U.; Giuliani, M.M.; et al. Adjuvanticity of the oil-in-water emulsion MF59 is independent of Nlrp3 inflammasome but requires the adaptor protein MyD88. Proc. Natl. Acad. Sci. USA 2011, 108, 11169–11174. [Google Scholar] [CrossRef]
- Calabro, S.; Tortoli, M.; Baudner, B.C.; Pacitto, A.; Cortese, M.; O’Hagan, D.T.; de Gregorio, E.; Seubert, A.; Wack, A. Vaccine adjuvants alum and MF59 induce rapid recruitment of neutrophils and monocytes that participate in antigen transport to draining lymph nodes. Vaccine 2011, 29, 1812–1823. [Google Scholar] [CrossRef]
- Dupuis, M.; McDonald, D.M.; Ott, G. Distribution of adjuvant MF59 and antigen gD2 after intramuscular injection in mice. Vaccine 1999, 18, 434–439. [Google Scholar] [CrossRef]
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. 2009, 183, 6186–6197. [Google Scholar] [CrossRef]
- Baldrick, P.; Richardson, D.; Elliott, G.; Wheeler, A.W. Safety evaluation of monophosphoryl lipid A (MPL): An immunostimulatory adjuvant. Regul. Toxicol. Pharmacol. 2002, 35, 398–413. [Google Scholar] [CrossRef]
- Lawson, L.B.; Freytag, L.C.; Clements, J.D. Use of nanocarriers for transdermal vaccine delivery. Clin. Pharmacol. Ther. 2007, 82, 641–643. [Google Scholar] [CrossRef]
- Shima, F.; Akagi, T.; Akashi, M. The role of hydrophobicity in the disruption of erythrocyte membrane by nanoparticles composed of hydrophobically modified poly(gamma-glutamic acid). J. Biomater. Sci. Polym. Ed. 2014, 25, 203–210. [Google Scholar] [CrossRef]
- Yang, Y.; Neef, T.; Mittelholzer, C.; Garcia Garayoa, E.; Bläuenstein, P.; Schibli, R.; Aebi, U.; Burkhard, P. The biodistribution of self-assembling protein nanoparticles shows they are promising vaccine platforms. J. Nanobiotechnol. 2013, 11. [Google Scholar] [CrossRef]
- WHO Vaccine Standardization. Available online: http://www.who.int/biologicals/publications/nonclinical_evaluation_vaccines_nov_2003.pdf (accessed on 11 April 2014).
- Glenn, G.M.; Smith, G.; Fries, L.; Raghunandan, R.; Lu, H.; Zhou, B.; Thomas, D.N.; Hickman, S.P.; Kpamegan, E.; Boddapati, S.; et al. Safety and immunogenicity of a Sf9 insect cell-derived respiratory syncytial virus fusion protein nanoparticle vaccine. Vaccine 2013, 31, 524–532. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515-536. https://doi.org/10.3390/vaccines2030515
Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK. Peptide Vaccine: Progress and Challenges. Vaccines. 2014; 2(3):515-536. https://doi.org/10.3390/vaccines2030515
Chicago/Turabian StyleLi, Weidang, Medha D. Joshi, Smita Singhania, Kyle H. Ramsey, and Ashlesh K. Murthy. 2014. "Peptide Vaccine: Progress and Challenges" Vaccines 2, no. 3: 515-536. https://doi.org/10.3390/vaccines2030515