
Immune Imprinting and Implications for COVID-19

{kind=link}
Abstract
:1. Introduction
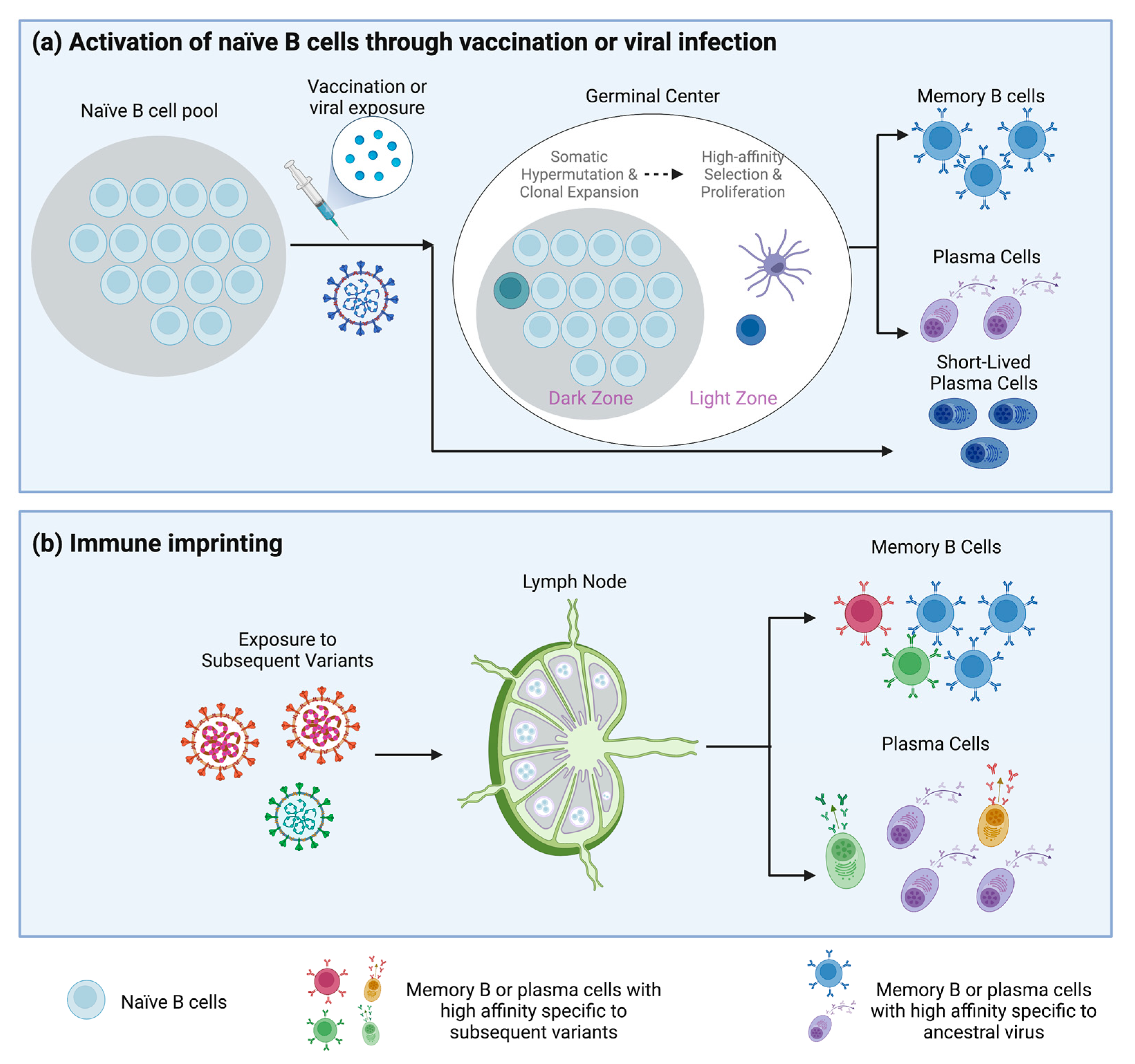
2. B Cell Memory Induced by SARS-CoV-2
3. Immune Imprinting in COVID-19 Pandemic
3.1. Immune Imprinting
3.2. Immunological Imprinting in SARS-CoV-2 Infection
3.3. Impact of Immune Imprinting on COVID-19 Vaccine
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. COVID-19—Navigating the Uncharted. N. Engl. J. Med. 2020, 382, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Tregoning, J.S.; Flight, K.E.; Higham, S.L.; Wang, Z.; Pierce, B.F. Progress of the COVID-19 Vaccine Effort: Viruses, Vaccines and Variants versus Efficacy, Effectiveness and Escape. Nat. Rev. Immunol. 2021, 21, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Kurhade, C.; Zou, J.; Xia, H.; Liu, M.; Chang, H.C.; Ren, P.; Xie, X.; Shi, P. Low Neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1, and XBB.1 by Parental MRNA Vaccine or a BA.5-Bivalent Booster. Nat. Med. 2022. [Google Scholar] [CrossRef]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of COVID-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.-M.; et al. COVID-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Shinde, V.; Bhikha, S.; Hoosain, Z.; Archary, M.; Bhorat, Q.; Fairlie, L.; Lalloo, U.; Masilela, M.S.L.; Moodley, D.; Hanley, S. Efficacy of NVX-CoV2373 COVID-19 Vaccine against the B. 1.351 Variant. N. Engl. J. Med. 2021, 384, 1899–1909. [Google Scholar] [CrossRef]
- Callaway, E. Heavily Mutated Omicron Variant Puts Scientists on Alert. Nature 2021, 600, 21. [Google Scholar] [CrossRef]
- Callaway, E.; Ledford, H. How Bad Is Omicron? What Scientists Know so Far. Nature 2021, 600, 197–199. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 Humoral Immunity Induces Convergent Omicron RBD Evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef]
- Davis-Gardner, M.E.; Lai, L.; Wali, B.; Samaha, H.; Solis, D.; Lee, M.; Porter-Morrison, A.; Hentenaar, I.T.; Yamamoto, F.; Godbole, S.; et al. Neutralization against BA.2.75.2, BQ.1.1, and XBB from MRNA Bivalent Booster. N. Engl. J. Med. 2022, 388, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Gagne, M.; Moliva, J.I.; Foulds, K.E.; Andrew, S.F.; Flynn, B.J.; Werner, A.P.; Wagner, D.A.; Teng, I.-T.; Lin, B.C.; Moore, C.; et al. MRNA-1273 or MRNA-Omicron Boost in Vaccinated Macaques Elicits Similar B Cell Expansion, Neutralizing Responses, and Protection from Omicron. Cell 2022, 185, 1556–1571.e18. [Google Scholar] [CrossRef] [PubMed]
- Wrammert, J.; Koutsonanos, D.; Li, G.-M.; Edupuganti, S.; Sui, J.; Morrissey, M.; McCausland, M.; Skountzou, I.; Hornig, M.; Lipkin, W.I.; et al. Broadly Cross-Reactive Antibodies Dominate the Human B Cell Response against 2009 Pandemic H1N1 Influenza Virus Infection. J. Exp. Med. 2011, 208, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Ellebedy, A.H. The Germinal Centre B Cell Response to SARS-CoV-2. Nat. Rev. Immunol. 2022, 22, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Williams, C.M.; Wijesundara, D.K.; Furuya, Y. Impact of Pre-Existing Immunity to Influenza on Live-Attenuated Influenza Vaccine (LAIV) Immunogenicity. Vaccines 2020, 8, 683. [Google Scholar] [CrossRef] [PubMed]
- Palm, A.-K.E.; Henry, C. Remembrance of Things Past: Long-Term B Cell Memory after Infection and Vaccination. Front. Immunol. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Treanor, B. B-cell Receptor: From Resting State to Activate. Immunology 2012, 136, 21–27. [Google Scholar] [CrossRef]
- Huang, C. Germinal Center Reaction. B Cells Immun. Toler. 2020, 1254, 47–53. [Google Scholar]
- Tomohiro, K.; Wataru, I. Memory B Cells. Nat. Rev. Immunol. 2015, 15, 149–159. [Google Scholar]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The Generation of Antibody-Secreting Plasma Cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- Lee, J.H.; Sutton, H.J.; Cottrell, C.A.; Phung, I.; Ozorowski, G.; Sewall, L.M.; Nedellec, R.; Nakao, C.; Silva, M.; Richey, S.T. Long-Primed Germinal Centres with Enduring Affinity Maturation and Clonal Migration. Nature 2022, 609, 998–1004. [Google Scholar] [CrossRef]
- Gitlin, A.D.; Shulman, Z.; Nussenzweig, M.C. Clonal Selection in the Germinal Centre by Regulated Proliferation and Hypermutation. Nature 2014, 509, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Viant, C.; Weymar, G.H.J.; Escolano, A.; Chen, S.; Hartweger, H.; Cipolla, M.; Gazumyan, A.; Nussenzweig, M.C. Antibody Affinity Shapes the Choice between Memory and Germinal Center B Cell Fates. Cell 2020, 183, 1298–1311. [Google Scholar] [CrossRef] [PubMed]
- Amanna, I.J.; Slifka, M.K. Mechanisms That Determine Plasma Cell Lifespan and the Duration of Humoral Immunity. Immunol. Rev. 2010, 236, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Hartley, G.E.; Edwards, E.S.J.; Aui, P.M.; Varese, N.; Stojanovic, S.; McMahon, J.; Peleg, A.Y.; Boo, I.; Drummer, H.E.; Hogarth, P.M. Rapid Generation of Durable B Cell Memory to SARS-CoV-2 Spike and Nucleocapsid Proteins in COVID-19 and Convalescence. Sci. Immunol. 2020, 5, eabf8891. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.S.; Kim, W.; Kalaidina, E.; Goss, C.W.; Rauseo, A.M.; Schmitz, A.J.; Hansen, L.; Haile, A.; Klebert, M.K.; Pusic, I. SARS-CoV-2 Infection Induces Long-Lived Bone Marrow Plasma Cells in Humans. Nature 2021, 595, 421–425. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, M.; Peng, Y.; Liang, Y.; Wei, J.; Xing, L.; Guo, L.; Li, X.; Li, J.; Wang, J.; et al. Longitudinal Analysis of Antibody Dynamics in COVID-19 Convalescents Reveals Neutralizing Responses up to 16 Months after Infection. Nat. Microbiol. 2022, 7, 423–433. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y. Evolution of Antibody Immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Aid, M.; Chandrashekar, A.; Yu, J.; McMahan, K.; Wegmann, F.; Jacob-Dolan, C.; Maron, J.S.; Atyeo, C.; Wan, H. A Homologous or Variant Booster Vaccine after Ad26. COV2. S Immunization Enhances SARS-CoV-2–Specific Immune Responses in Rhesus Macaques. Sci. Transl. Med. 2022, 14, eabm4996. [Google Scholar] [CrossRef]
- Sette, A.; Crotty, S. Adaptive Immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef]
- Kasturi, S.P.; Skountzou, I.; Albrecht, R.A.; Koutsonanos, D.; Hua, T.; Nakaya, H.I.; Ravindran, R.; Stewart, S.; Alam, M.; Kwissa, M. Programming the Magnitude and Persistence of Antibody Responses with Innate Immunity. Nature 2011, 470, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Barouch, D.H. T Cell Immunity to COVID-19 Vaccines. Science 2022, 377, 821–822. [Google Scholar] [CrossRef] [PubMed]
- Mahrokhian, S.H.; Tostanoski, L.H.; Jacob-Dolan, C.; Zahn, R.C.; Wegmann, F.; McMahan, K.; Yu, J.; Gebre, M.S.; Bondzie, E.A.; Wan, H. Durability and Expansion of Neutralizing Antibody Breadth Following Ad26. COV2. S Vaccination of Mice. NPJ Vaccines 2022, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological 432 Memory to SARS-CoV-2 Assessed for up to 8 Months after Infection. Science 2021, 371, 6529. [Google Scholar] [CrossRef] [PubMed]
- Purtha, W.E.; Tedder, T.F.; Johnson, S.; Bhattacharya, D.; Diamond, M.S. Memory B Cells, but Not Long-Lived Plasma Cells, Possess Antigen Specificities for Viral Escape Mutants. J. Exp. Med. 2011, 208, 2599–2606. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F. SARS-CoV-2 and HIV-1—A Tale of Two Vaccines. Nat. Rev. Immunol. 2021, 21, 543–544. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Gaebler, C.; Muecksch, F.; Lorenzi, J.C.C.; Wang, Z.; Cho, A.; Agudelo, M.; Barnes, C.O.; Gazumyan, A.; Finkin, S. Convergent Antibody Responses to SARS-CoV-2 in Convalescent Individuals. Nature 2020, 584, 437–442. [Google Scholar] [CrossRef]
- Cao, Y.; Su, B.; Guo, X.; Sun, W.; Deng, Y.; Bao, L.; Zhu, Q.; Zhang, X.; Zheng, Y.; Geng, C. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell 2020, 182, 73–84. [Google Scholar] [CrossRef]
- Wang, K.; Jia, Z.; Bao, L.; Wang, L.; Cao, L.; Chi, H.; Hu, Y.; Li, Q.; Zhou, Y.; Jiang, Y. Memory B Cell Repertoire from Triple Vaccinees against Diverse SARS-CoV-2 Variants. Nature 2022, 603, 919–925. [Google Scholar] [CrossRef]
- He, B.; Liu, S.; Wang, Y.; Xu, M.; Cai, W.; Liu, J.; Bai, W.; Ye, S.; Ma, Y.; Hu, H. Rapid Isolation and Immune Profiling of SARS-CoV-2 Specific Memory B Cell in Convalescent COVID-19 Patients via LIBRA-Seq. Signal Transduct. Target. Ther. 2021, 6, 195. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, P.; Muecksch, F.; Cho, A.; Ben Tanfous, T.; Canis, M.; Witte, L.; Johnson, B.; Raspe, R.; Schmidt, F. Memory B Cell Responses to Omicron Subvariants after SARS-CoV-2 MRNA Breakthrough Infection in Humans. J. Exp. Med. 2022, 219. [Google Scholar] [CrossRef] [PubMed]
- Hachmann, N.P.; Miller, J.; Collier, A.Y.; Ventura, J.D.; Yu, J.; Rowe, M.; Bondzie, E.A.; Powers, O.; Surve, N.; Hall, K.; et al. Neutralization Escape by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4, and BA.5. N. Engl. J. Med. 2022, 387, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Pinto, D.; Walls, A.C.; Liu, Z.; De Marco, A.; Benigni, F.; Zatta, F.; Silacci-Fregni, C.; Bassi, J.; Sprouse, K.R. Imprinted Antibody Responses against SARS-CoV-2 Omicron Sublineages. Science 2022, 378, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.J.; Pade, C.; Gibbons, J.M.; Otter, A.D.; Lin, K.-M.; Muñoz Sandoval, D.; Pieper, F.P.; Butler, D.K.; Liu, S.; Joy, G.; et al. Immune Boosting by B.1.1.529 (Omicron) Depends on Previous SARS-CoV-2 Exposure. Science 2022, 377, eabq1841. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Francis, T. On the Doctrine of Original Antigenic Sin. Proc. Am. Philos. Soc. 1960, 104, 572–578. [Google Scholar]
- Ndifon, W. A Simple Mechanistic Explanation for Original Antigenic Sin and Its Alleviation by Adjuvants. J. R. Soc. Interface 2015, 12, 20150627. [Google Scholar] [CrossRef]
- Henry, C.; Palm, A.-K.E.; Krammer, F.; Wilson, P.C. From Original Antigenic Sin to the Universal Influenza Virus Vaccine. Trends Immunol. 2018, 39, 70–79. [Google Scholar] [CrossRef]
- Zhang, A.; Stacey, H.D.; Mullarkey, C.E.; Miller, M.S. Original Antigenic Sin: How First Exposure Shapes Lifelong Anti–Influenza Virus Immune Responses. J. Immunol. 2019, 202, 335–340. [Google Scholar] [CrossRef]
- Cortina-Ceballos, B.; Godoy-Lozano, E.E.; Téllez-Sosa, J.; Ovilla-Muñoz, M.; Sámano-Sánchez, H.; Aguilar-Salgado, A.; Gómez-Barreto, R.E.; Valdovinos-Torres, H.; López-Martínez, I.; Aparicio-Antonio, R.; et al. Longitudinal Analysis of the Peripheral B Cell Repertoire Reveals Unique Effects of Immunization with a New Influenza Virus Strain. Genome Med. 2015, 7, 124. [Google Scholar] [CrossRef]
- Tan, Y.-C.; Blum, L.K.; Kongpachith, S.; Ju, C.-H.; Cai, X.; Lindstrom, T.M.; Sokolove, J.; Robinson, W.H. High-Throughput Sequencing of Natively Paired Antibody Chains Provides Evidence for Original Antigenic Sin Shaping the Antibody Response to Influenza Vaccination. Clin. Immunol. 2014, 151, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.F.; Huang, Y.; Kaur, K.; Popova, L.I.; Ho, I.Y.; Pauli, N.T.; Dunand, C.J.H.; Taylor, W.M.; Lim, S.; Huang, M.; et al. Immune History Profoundly Affects Broadly Protective B Cell Responses to Influenza. Sci. Transl. Med. 2015, 7, 316ra192. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B.; Rojanasuphot, S.; Sangkawibha, N. Original Antigenic Sin in Dengue. Am. J. Trop. Med. Hyg. 1983, 32, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Midgley, C.M.; Bajwa-Joseph, M.; Vasanawathana, S.; Limpitikul, W.; Wills, B.; Flanagan, A.; Waiyaiya, E.; Tran, H.B.; Cowper, A.E.; Chotiyarnwon, P.; et al. An In-Depth Analysis of Original Antigenic Sin in Dengue Virus Infection. J. Virol. 2011, 85, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, N.I.; Fisher, P.B. Surface-Epitope Masking (SEM) BT-Cancer Genomics and Proteomics: Methods and Protocols; Fisher, P.B., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 245–258. ISBN 978-1-59745-335-6. [Google Scholar]
- Zarnitsyna, V.I.; Ellebedy, A.H.; Davis, C.; Jacob, J.; Ahmed, R.; Antia, R. Masking of Antigenic Epitopes by Antibodies Shapes the Humoral Immune Response to Influenza. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140248. [Google Scholar] [CrossRef]
- Bowen, J.E.; Addetia, A.; Dang, H.V.; Stewart, C.; Brown, J.T.; Sharkey, W.K.; Sprouse, K.R.; Walls, A.C.; Mazzitelli, I.G.; Logue, J.K.; et al. Omicron Spike Function and Neutralizing Activity Elicited by a Comprehensive Panel of Vaccines. Science 2022, 377, 890–894. [Google Scholar] [CrossRef]
- Röltgen, K.; Nielsen, S.C.A.; Silva, O.; Younes, S.F.; Zaslavsky, M.; Costales, C.; Yang, F.; Wirz, O.F.; Solis, D.; Hoh, R.A.; et al. Immune Imprinting, Breadth of Variant Recognition, and Germinal Center Response in Human SARS-CoV-2 Infection and Vaccination. Cell 2022, 185, 1025–1040.e14. [Google Scholar] [CrossRef]
- Aydillo, T.; Rombauts, A.; Stadlbauer, D.; Aslam, S.; Abelenda-Alonso, G.; Escalera, A.; Amanat, F.; Jiang, K.; Krammer, F.; Carratala, J.; et al. Immunological Imprinting of the Antibody Response in COVID-19 Patients. Nat. Commun. 2021, 12, 3781. [Google Scholar] [CrossRef]
- El Sahly, H.M.; Baden, L.R.; Essink, B.; Doblecki-Lewis, S.; Martin, J.M.; Anderson, E.J.; Campbell, T.B.; Clark, J.; Jackson, L.A.; Fichtenbaum, C.J. Efficacy of the MRNA-1273 SARS-CoV-2 Vaccine at Completion of Blinded Phase. N. Engl. J. Med. 2021, 385, 1774–1785. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C. Safety and Efficacy of the BNT162b2 MRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S. Safety and Efficacy of an RAd26 and RAd5 Vector-Based Heterologous Prime-Boost COVID-19 Vaccine: An Interim Analysis of a Randomised Controlled Phase 3 Trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, M.N.; Minassian, A.M.; Ewer, K.J.; Flaxman, A.L.; Folegatti, P.M.; Owens, D.R.; Voysey, M.; Aley, P.K.; Angus, B.; Babbage, G. Safety and Immunogenicity of ChAdOx1 NCoV-19 Vaccine Administered in a Prime-Boost Regimen in Young and Old Adults (COV002): A Single-Blind, Randomised, Controlled, Phase 2/3 Trial. Lancet 2020, 396, 1979–1993. [Google Scholar] [CrossRef] [PubMed]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B. Safety and Efficacy of Single-Dose Ad26. COV2. S Vaccine against COVID-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhang, Y.; Wang, Y.; Wang, H.; Yang, Y.; Gao, G.F.; Tan, W.; Wu, G.; Xu, M.; Lou, Z. Safety and Immunogenicity of an Inactivated SARS-CoV-2 Vaccine, BBIBP-CorV: A Randomised, Double-Blind, Placebo-Controlled, Phase 1/2 Trial. Lancet Infect. Dis. 2021, 21, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zeng, G.; Pan, H.; Li, C.; Hu, Y.; Chu, K.; Han, W.; Chen, Z.; Tang, R.; Yin, W. Safety, Tolerability, and Immunogenicity of an Inactivated SARS-CoV-2 Vaccine in Healthy Adults Aged 18–59 Years: A Randomised, Double-Blind, Placebo-Controlled, Phase 1/2 Clinical Trial. Lancet Infect. Dis. 2021, 21, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Al Kaabi, N.; Zhang, Y.; Xia, S.; Yang, Y.; Al Qahtani, M.M.; Abdulrazzaq, N.; Al Nusair, M.; Hassany, M.; Jawad, J.S.; Abdalla, J. Effect of 2 Inactivated SARS-CoV-2 Vaccines on Symptomatic COVID-19 Infection in Adults: A Randomized Clinical Trial. JAMA 2021, 326, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, L.M.; Kotloff, K.L.; Gay, C.L.; Áñez, G.; Adelglass, J.M.; Barrat Hernández, A.Q.; Harper, W.L.; Duncanson, D.M.; McArthur, M.A.; Florescu, D.F. Efficacy and Safety of NVX-CoV2373 in Adults in the United States and Mexico. N. Engl. J. Med. 2022, 386, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Keech, C.; Albert, G.; Cho, I.; Robertson, A.; Reed, P.; Neal, S.; Plested, J.S.; Zhu, M.; Cloney-Clark, S.; Zhou, H. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N. Engl. J. Med. 2020, 383, 2320–2332. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Choi, A.; Koch, M.; Wu, K.; Chu, L.; Ma, L.; Hill, A.; Nunna, N.; Huang, W.; Oestreicher, J.; Colpitts, T. Safety and Immunogenicity of SARS-CoV-2 Variant MRNA Vaccine Boosters in Healthy Adults: An Interim Analysis. Nat. Med. 2021, 27, 2025–2031. [Google Scholar] [CrossRef]
- Khoury, D.S.; Docken, S.S.; Subbarao, K.; Kent, S.J.; Davenport, M.P.; Cromer, D. Predicting the Efficacy of Variant-Modified COVID-19 Vaccine Boosters. medRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Chalkias, S.; Harper, C.; Vrbicky, K.; Walsh, S.R.; Essink, B.; Brosz, A.; McGhee, N.; Tomassini, J.E.; Chen, X.; Chang, Y. A Bivalent Omicron-Containing Booster Vaccine against COVID-19. N. Engl. J. Med. 2022, 387, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Collier, A.Y.; Miller, J.; Hachmann, N.P.; McMahan, K.; Liu, J.; Bondzie, E.A.; Gallup, L.; Rowe, M.; Schonberg, E.; Thai, S.; et al. Immunogenicity of BA.5 Bivalent MRNA Vaccine Boosters. N. Engl. J. Med. 2023, 388, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bowen, A.; Valdez, R.; Gherasim, C.; Gordon, A.; Liu, L.; Ho, D.D. Antibody Response to Omicron BA.4–BA.5 Bivalent Booster. N. Engl. J. Med. 2023, 388, 567–569. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Barrett, J.; He, X. Immune Imprinting and Implications for COVID-19. Vaccines 2023, 11, 875. https://doi.org/10.3390/vaccines11040875
Zhou Z, Barrett J, He X. Immune Imprinting and Implications for COVID-19. Vaccines. 2023; 11(4):875. https://doi.org/10.3390/vaccines11040875
Chicago/Turabian StyleZhou, Zhiqian, Julia Barrett, and Xuan He. 2023. "Immune Imprinting and Implications for COVID-19" Vaccines 11, no. 4: 875. https://doi.org/10.3390/vaccines11040875