


Genomic Analysis of Infectious Bursal Disease Virus in Nigeria: Identification of Unique Mutations of Yet Unknown Biological Functions in Both Segments A and B

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sources of Samples
2.2. IBD Virus Antigen Detection
2.3. Sequencing
2.4. Bioinformatic Analysis
2.5. Phylogenetic Analysis
3. Results
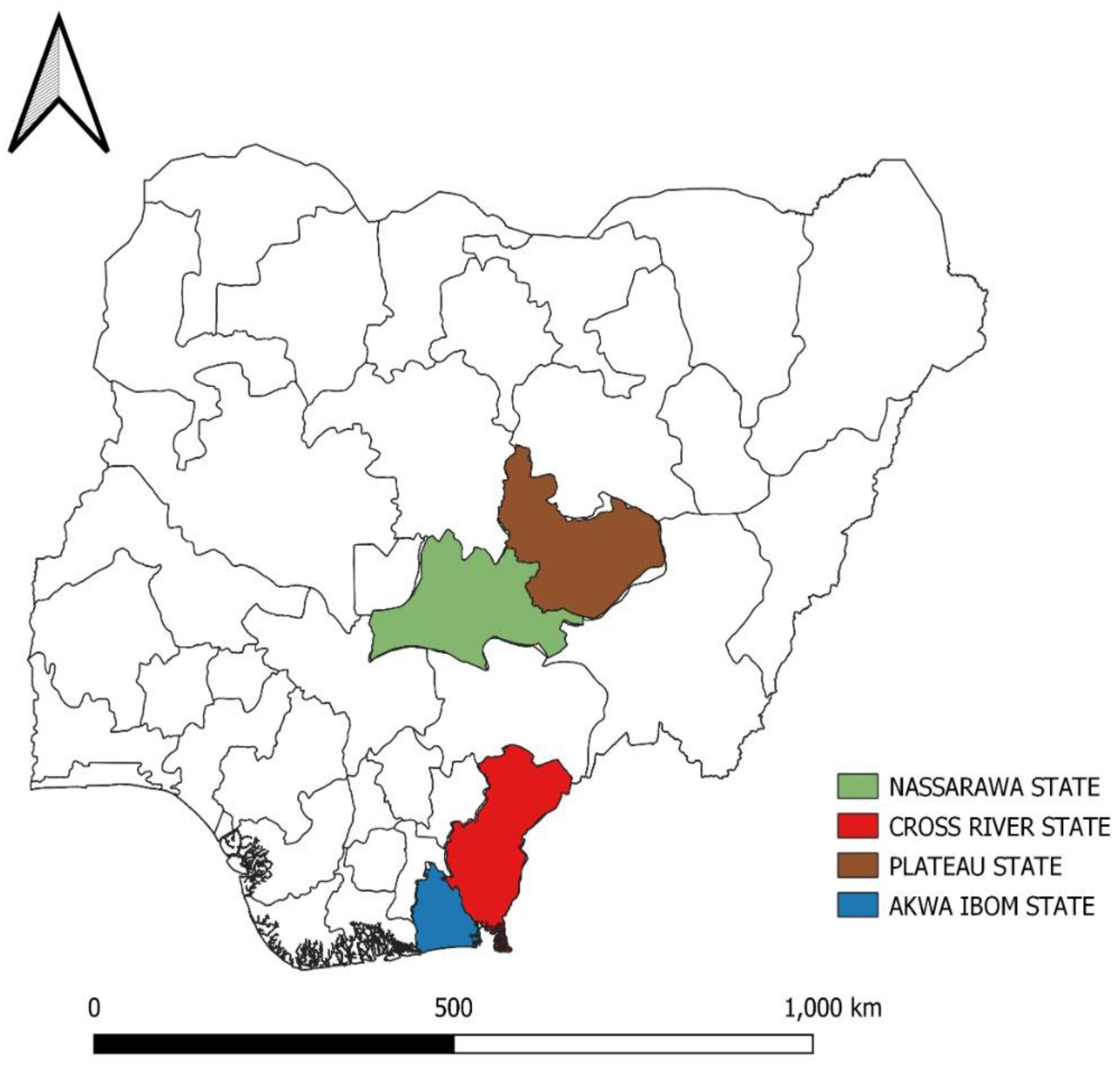
3.1. Flock History
3.2. Detection of IBD Viruses
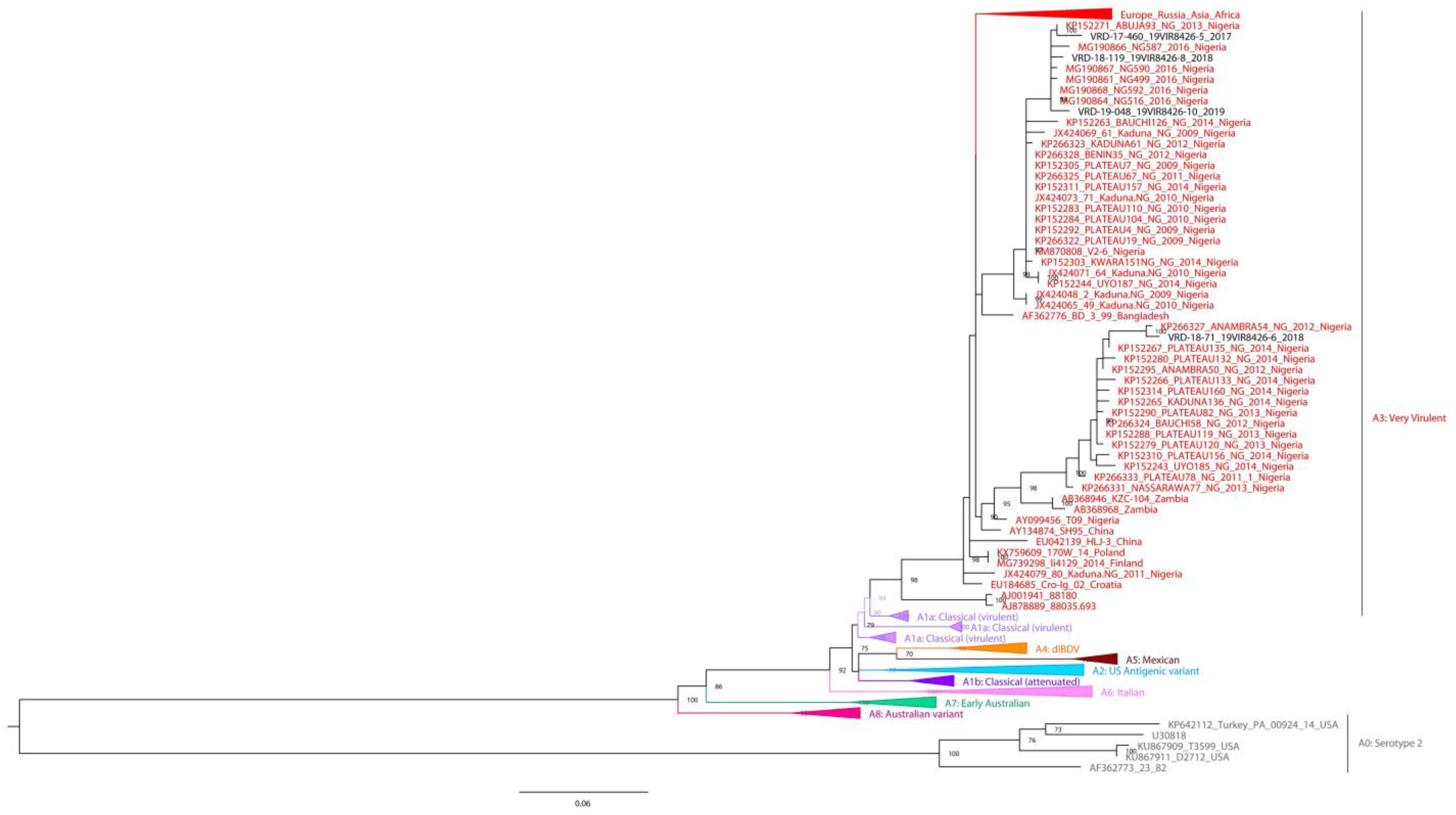
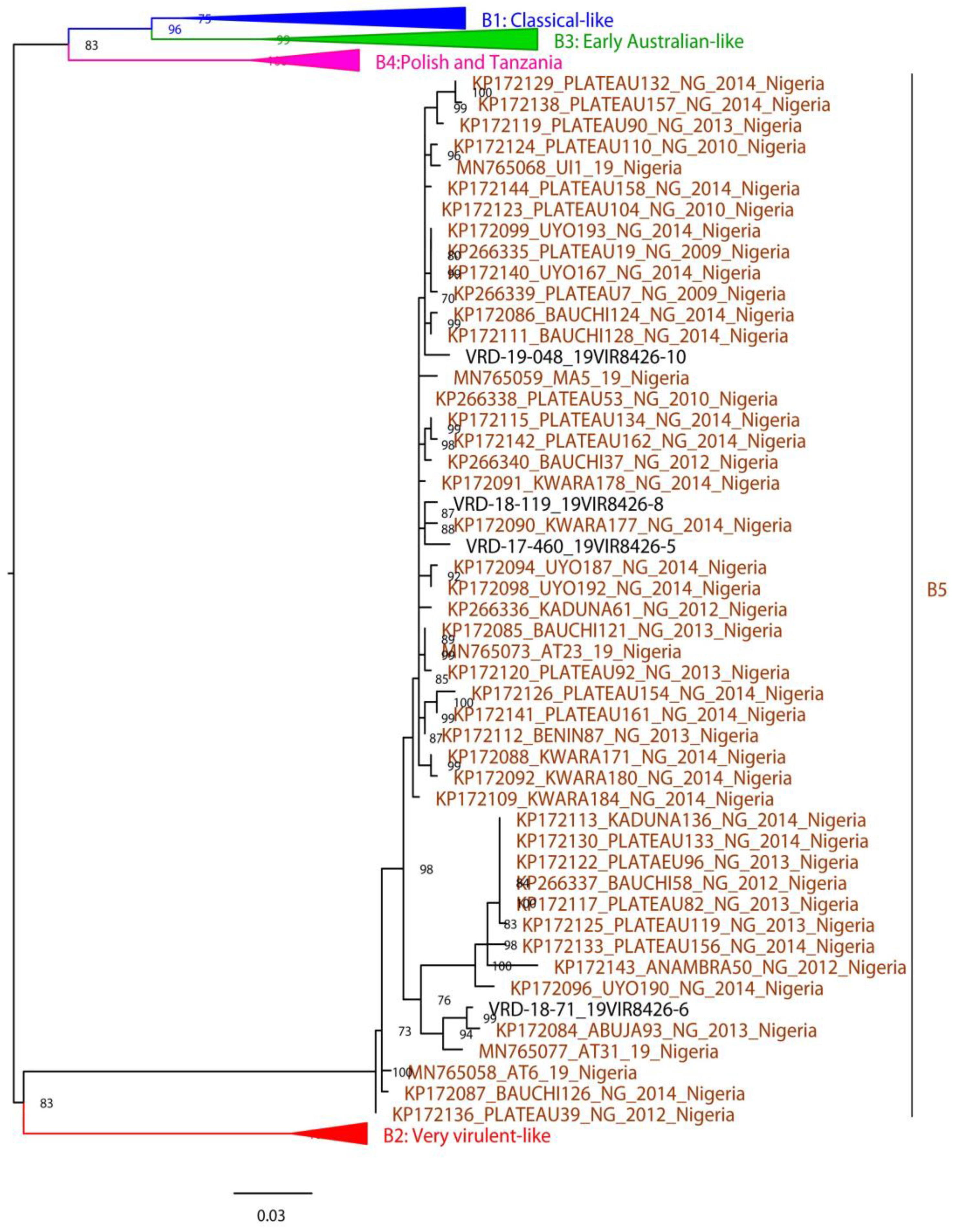
3.3. Phylogenetic Analysis
3.4. Nucleotide Sequence Analysis of Segments A and B
3.5. Amino Acid Sequence Analysis of Segments A and B
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rautenschlein, S.; Alkie, T.N. Infectious bursal disease virus in poultry: Current status and future prospects. Vet. Med. Res. Rep. 2016, 7, 9. [Google Scholar] [CrossRef]
- Eterradossi, N.; Saif, Y.M. Infectious bursal disease. In Diseases of Poultry; Swayne, D.E., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 257–283. ISBN 978-1-119-37117-5. [Google Scholar]
- Okoye, J.O.; Uzoukwu, M. An outbreak of infectious bursal disease among chickens between 16 and 20 weeks old. Avian Dis. 1981, 25, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Shekaro, A.; Josiah, I.E. Infectious bursal disease outbreak in fifteen weeks old pullets in Kaduna, Nigeria. J. Anim. Prod. Adv. 2015, 5, 636–644. [Google Scholar] [CrossRef]
- Delmas, B.; Attoui, H.; Ghosh, S.; Malik, Y.S.; Mundt, E.; Vakharia, V.N. ICTV virus taxonomy profile: Birnaviridae. J. Gen. Virol. 2019, 100, 5–6. [Google Scholar] [CrossRef]
- Müller, H.; Scholtissek, C.; Becht, H. The genome of infectious bursal disease virus consists of two segments of double-stranded RNA. J. Virol. 1979, 31, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Barrett, S.A.; Fahey, K.J. The characterization and molecular cloning of the double-stranded RNA genome of an Australian strain of infectious bursal disease virus. Virology 1985, 143, 35–44. [Google Scholar] [CrossRef]
- Birghan, C. A non-canonical Lon proteinase lacking the ATPase domain employs the Ser-Lys catalytic dyad to exercise broad control over the life cycle of a double-stranded RNA virus. EMBO J. 2000, 19, 114–123. [Google Scholar] [CrossRef]
- Lombardo, E.; Maraver, A.; Espinosa, I.; Fernández-Arias, A.; Rodriguez, J.F. VP5, the Nonstructural Polypeptide of Infectious Bursal Disease Virus, Accumulates within the Host Plasma Membrane and Induces Cell Lysis. Virology 2000, 277, 345–357. [Google Scholar] [CrossRef]
- Méndez, F.; de Garay, T.; Rodríguez, D.; Rodríguez, J.F. Infectious Bursal Disease Virus VP5 Polypeptide: A Phosphoinositide-Binding Protein Required for Efficient Cell-to-Cell Virus Dissemination. PLoS ONE 2015, 10, e0123470. [Google Scholar] [CrossRef]
- Shwed, P.S.; Dobos, P.; Cameron, L.A.; Vakharia, V.N.; Duncan, R. Birnavirus VP1 Proteins Form a Distinct Subgroup of RNA-Dependent RNA Polymerases Lacking a GDD Motif. Virology 2002, 296, 241–250. [Google Scholar] [CrossRef]
- McFerran, J.B.; McNulty, M.S.; McKillop, E.R.; Connor, T.J.; McCracken, R.M.; Collins, D.S.; Allan, G.M. Isolation and serological studies with infectious bursal disease viruses from fowl, turkeys and ducks: Demonstration of a second serotype. Avian Pathol. 1980, 9, 395–404. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, T.P. Acute infectious bursal disease in poultry: A review. Avian Pathol. 2000, 29, 175–194. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.; Islam, M.R.; Raue, R. Research on infectious bursal disease—The past, the present and the future. Vet. Microbiol. 2003, 97, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Nouën, C.L.; Rivallan, G.; Toquin, D.; Darlu, P.; Morin, Y.; Beven, V.; de Boisseson, C.; Cazaban, C.; Comte, S.; Gardin, Y.; et al. Very virulent infectious bursal disease virus: Reduced pathogenicity in a rare natural segment-B-reassorted isolate. J. Gen. Virol. 2006, 87, 209–216. [Google Scholar] [CrossRef]
- He, X.; Xiong, Z.; Yang, L.; Guan, D.; Yang, X.; Wei, P. Molecular epidemiology studies on partial sequences of both genome segments reveal that reassortant infectious bursal disease viruses were dominantly prevalent in southern China during 2000–2012. Arch. Virol. 2014, 159, 3279–3292. [Google Scholar] [CrossRef]
- Michel, L.O.; Jackwood, D.J. Classification of infectious bursal disease virus into genogroups. Arch. Virol. 2017, 162, 3661–3670. [Google Scholar] [CrossRef]
- Islam, M.R.; Nooruzzaman, M.; Rahman, T.; Mumu, T.T.; Rahman, M.M.; Chowdhury, E.H.; Eterradossi, N.; Müller, H. A unified genotypic classification of infectious bursal disease virus based on both genome segments. Avian Pathol. 2021, 50, 190–206. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, L.; Jiang, N.; Gao, L.; Li, K.; Gao, Y.; Liu, C.; Cui, H.; Pan, Q.; Zhang, Y.; et al. An improved scheme for infectious bursal disease virus genotype classification based on both genome-segments A and B. J. Integr. Agric. 2021, 20, 1372–1381. [Google Scholar] [CrossRef]
- Owolodun, O.A.; Yakubu, B.; Jambol, A.R.; Audu, B.J.; Dogonyaro, B.B.; Luka, P.D. Further evidence for very virulent infectious bursal disease virus in vaccinated chickens in Nigeria. Trop. Anim. Health Prod. 2015, 47, 1437–1441. [Google Scholar] [CrossRef]
- Nwagbo, I.O.; Shittu, I.; Nwosuh, C.I.; Ezeifeka, G.O.; Odibo, F.J.C.; Michel, L.O.; Jackwood, D.J. Molecular characterization of field infectious bursal disease virus isolates from Nigeria. Vet. World 2016, 9, 1420–1428. [Google Scholar] [CrossRef]
- Owoade, A.A.; Mulders, M.N.; Kohnen, J.; Ammerlaan, W.; Muller, C.P. High sequence diversity in infectious bursal disease virus serotype 1 in poultry and turkey suggests West-African origin of very virulent strains. Arch. Virol. 2004, 149, 653–672. [Google Scholar] [CrossRef] [PubMed]
- Adamu, J.; Owoade, A.A.; Abdu, P.A.; Kazeem, H.M.; Fatihu, M.Y. Characterization of field and vaccine infectious bursal disease viruses from Nigeria revealing possible virulence and regional markers in the VP2 minor hydrophilic peaks. Avian Pathol. 2013, 42, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Luka, P.D.; Yakubu, B.; Jambol, A.R.; Audu, B.J.; Dogonyaro, B.B.; Owolodun, O.A. Detection and differentiation of infectious bursal disease virus from the outbreaks in two layer farms by PCR-RFLP in Jos, Nigeria. Vet. World 2014, 7, 30–33. [Google Scholar] [CrossRef]
- Nwagbo, I.O.; Shittu, I.; Qasim, A.M.M.; Joannis, T.M. Continuous circulation and mutation at the hypervariable region of the VP2 gene of very virulent infectious bursal disease virus in Nigeria. Niger. Vet. J. 2018, 39, 143. [Google Scholar] [CrossRef]
- Arowolo, O.A.; George, U.E.; Luka, P.D.; Maurice, N.A.; Atuman, Y.J.; Shallmizhili, J.J.; Shittu, I.; Oluwayelu, D.O. Infectious bursal disease in Nigeria: Continuous circulation of reassortant viruses. Trop. Anim. Health Prod. 2021, 53, 271. [Google Scholar] [CrossRef]
- Dey, S.; Pathak, D.; Ramamurthy, N.; Maity, H.K.; Chellappa, M.M. Infectious bursal disease virus in chickens: Prevalence, impact, and management strategies. Vet. Med. Res. Rep. 2019, 10, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.J.; Sreedevi, B.; LeFever, L.J.; Sommer-Wagner, S.E. Studies on naturally occurring infectious bursal disease viruses suggest that a single amino acid substitution at position 253 in VP2 increases pathogenicity. Virology 2008, 377, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Escaffre, O.; Le Nouën, C.; Amelot, M.; Ambroggio, X.; Ogden, K.M.; Guionie, O.; Toquin, D.; Müller, H.; Islam, M.R.; Eterradossi, N. Both Genome Segments Contribute to the Pathogenicity of Very Virulent Infectious Bursal Disease Virus. J. Virol. 2013, 87, 2767–2780. [Google Scholar] [CrossRef] [PubMed]
- Shehata, A.A.; Sultan, H.; Halami, M.Y.; Talaat, S.; Vahlenkamp, T.W. Molecular characterization of very virulent infectious bursal disease virus strains circulating in Egypt from 2003 to 2014. Arch. Virol. 2017, 162, 3803–3815. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, H.; Chen, G.; Liu, H.; Wang, S.; Xia, D.; Yu, Y.; Zhang, Y.; Jiang, J.; Ma, J.; et al. Identification and assessment of pathogenicity of a naturally reassorted infectious bursal disease virus from Henan, China. Poult. Sci. 2019, 98, 6433–6444. [Google Scholar] [CrossRef]
- Hussain, A.; Wu, T.; Li, H.; Fan, L.; Li, K.; Gao, L.; Wang, Y.; Gao, Y.; Liu, C.; Cui, H.; et al. Pathogenic Characterization and Full Length Genome Sequence of a Reassortant Infectious Bursal Disease Virus Newly Isolated in Pakistan. Virol. Sin. 2019, 34, 102–105. [Google Scholar] [CrossRef]
- Chen, F.; Liu, J.; Yan, Z.; Liu, D.; Ji, J.; Qin, J.; Li, H.; Ma, J.; Bi, Y.; Xie, Q. Complete Genome Sequence Analysis of a Natural Reassortant Infectious Bursal Disease Virus in China. J. Virol. 2012, 86, 11942–11943. [Google Scholar] [CrossRef] [PubMed]
- Raja, P.; Senthilkumar, T.M.A.; Parthiban, M.; Thangavelu, A.; Gowri, A.M.; Palanisammi, A.; Kumanan, K. Complete Genome Sequence Analysis of a Naturally Reassorted Infectious Bursal Disease Virus from India. Genome Announc. 2016, 4, e00709-16. [Google Scholar] [CrossRef] [PubMed]
- Drissi Touzani, C.; Fellahi, S.; Fassi Fihri, O.; Gaboun, F.; Khayi, S.; Mentag, R.; Lico, C.; Baschieri, S.; El Houadfi, M.; Ducatez, M. Complete genome analysis and time scale evolution of very virulent infectious bursal disease viruses isolated from recent outbreaks in Morocco. Infect. Genet. Evol. 2020, 77, 104097. [Google Scholar] [CrossRef]
- Eterradossi, N.; Arnauld, C.; Toquin, D.; Rivallan, G. Critical amino acid changes in VP2 variable domain are associated with typical and atypical antigenicity in very virulent infectious bursal disease viruses. Arch. Virol. 1998, 143, 1627–1636. [Google Scholar] [CrossRef]
- Jackwood, D.J.; Sommer-Wagner, S.E. Amino acids contributing to antigenic drift in the infectious bursal disease Birnavirus (IBDV). Virology 2011, 409, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Soubies, S.; Courtillon, C.; Briand, F.-X.; Allée, C.; Amelot, M.; De Boisseson, C.; Lucas, P.; Blanchard, Y.; Belahouel, A.; et al. Infectious bursal disease virus in Algeria: Detection of highly pathogenic reassortant viruses. Infect. Genet. Evol. 2018, 60, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Lachheb, J.; Jbenyeni, A.; Nsiri, J.; Larbi, I.; Ammouna, F.; El Behi, I.; Ghram, A. Full-length genome sequencing of a very virulent infectious bursal disease virus isolated in Tunisia. Poult. Sci. 2021, 100, 496–506. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, L.; Chen, Y.; Lu, Z.; Gao, L.; Wang, Y.; Gao, Y.; Gao, H.; Cui, H.; Li, K.; et al. Cyclophilin A Interacts with Viral VP4 and Inhibits the Replication of Infectious Bursal Disease Virus. Biomed Res. Int. 2015, 2015, 719454. [Google Scholar] [CrossRef]
- Rudd, M.F.; Heine, H.G.; Sapats, S.I.; Parede, L.; Ignjatovic, J. Characterisation of an Indonesian very virulent strain of infectious bursal disease virus. Arch. Virol. 2002, 147, 1303–1322. [Google Scholar] [CrossRef]
- Ye, C.; Wang, Y.; Zhang, E.; Han, X.; Yu, Z.; Liu, H. VP1 and VP3 Are Required and Sufficient for Translation Initiation of Uncapped Infectious Bursal Disease Virus Genomic Double-Stranded RNA. J. Virol. 2018, 92, e01345-17. [Google Scholar] [CrossRef]
- Li, R.; Wang, H.; Huang, G.; Zhang, M. Residues of 862, 921 of VP3 are associated with virulence in infectious bursal disease virus. Nat. Sci. 2010, 02, 718–725. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, X.; Kang, Z.; Yu, F.; Qin, L.; Gao, H.; Gao, Y.; Wang, X. A single amino acid in the C-terminus of VP3 protein influences the replication of attenuated infectious bursal disease virus in vitro and in vivo. Antivir. Res. 2010, 87, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.; Villegas, P.; Hernández, D.; Banda, A.; Maya, L.; Romero, V.; Tomás, G.; Pérez, R. Sequence variability and evolution of the terminal overlapping VP5 gene of the infectious bursal disease virus. Virus Genes 2010, 41, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.L.; Omar, A.R.; Hair-Bejo, M.; Aini, I.; Seow, H.F. Comparative analysis of viral RNA and apoptotic cells in bursae following infection with infectious bursal disease virus. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 433–443. [Google Scholar] [CrossRef]
- Yao, K.; Goodwin, M.A.; Vakharia, V.N. Generation of a Mutant Infectious Bursal Disease Virus That Does Not Cause Bursal Lesions. J. Virol. 1998, 72, 2647–2654. [Google Scholar] [CrossRef] [PubMed]
- Le Nouën, C.; Toquin, D.; Müller, H.; Raue, R.; Kean, K.M.; Langlois, P.; Cherbonnel, M.; Eterradossi, N. Different Domains of the RNA Polymerase of Infectious Bursal Disease Virus Contribute to Virulence. PLoS ONE 2012, 7, e28064. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Ma, S.-J.; Zhang, Y.-G.; Li, X.-S.; Gao, X.-Y.; Cui, B.-A.; Chen, H.-Y. Genomic sequence analysis of a new reassortant infectious bursal disease virus from commercial broiler flocks in central China. Arch. Virol. 2013, 158, 1973–1978. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Virus Id | Year | Flock Size | Outbreak Location | Specie | Vaccination Status |
|---|---|---|---|---|---|
| VRD/17/460_19VIR8426-5 | 2017 | 350 | Plateau | Chicken | Not vaccinated |
| VRD/18/71_19VIR8426-6 | 2018 | 30,000 | Cross-River | Chicken | IBDV twice |
| VRD/18/119_19VIR8426-8 | 2018 | 5000 | Nasarawa | Chicken | IBDV twice |
| VRD/19/048_19VIR8426-10 | 2019 | 3000 | Akwa-Ibom | Chicken | IBDV twice |
| Virus ID | Segment | Accession Number | % Similarity | Description | Accession Number | Country |
|---|---|---|---|---|---|---|
| VRD/17/460_19VIR8426-5 | A | OP311683 | 98.08 | IBDV 80/Kaduna.NG/2011 | JX424079.1 | Nigeria |
| B | OP311687 | 90.09 | IBDV strain 150133/3.2 | MF969112.1 | Algeria | |
| VRD/18/71_19VIR8426-6 | A | OP311684 | 96.61 | IBDV strain T09 | AY099456.1 | Nigeria |
| B | OP311688 | 90.43 | IBDV HuB-1 | GQ449693.1 | China | |
| VRD/18/119_19VIR8426-8 | A | OP311685 | 98.08 | IBDV 80/Kaduna.NG/2011 | JX424079.1 | Nigeria |
| B | OP311689 | 90.20 | IBDV strain 150133/3.2 | MF969112.1 | Algeria | |
| VRD/19/048_19VIR8426-10 | A | OP311682 | 98.16 | IBDV 80/Kaduna.NG/2011 | JX424079.1 | Nigeria |
| B | OP311686 | 92.91 | IBDV strain 150133/3.2 | MF969112.1 | Algeria |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nwagbo, I.; Milani, A.; Salviato, A.; Zamperin, G.; Sulaiman, L.; Maurice, N.; Meseko, C.; Fusaro, A.; Shittu, I. Genomic Analysis of Infectious Bursal Disease Virus in Nigeria: Identification of Unique Mutations of Yet Unknown Biological Functions in Both Segments A and B. Vaccines 2023, 11, 867. https://doi.org/10.3390/vaccines11040867
Nwagbo I, Milani A, Salviato A, Zamperin G, Sulaiman L, Maurice N, Meseko C, Fusaro A, Shittu I. Genomic Analysis of Infectious Bursal Disease Virus in Nigeria: Identification of Unique Mutations of Yet Unknown Biological Functions in Both Segments A and B. Vaccines. 2023; 11(4):867. https://doi.org/10.3390/vaccines11040867
Chicago/Turabian StyleNwagbo, Ijeoma, Adelaide Milani, Annalisa Salviato, Gianpiero Zamperin, Lanre Sulaiman, Nanven Maurice, Clement Meseko, Alice Fusaro, and Ismaila Shittu. 2023. "Genomic Analysis of Infectious Bursal Disease Virus in Nigeria: Identification of Unique Mutations of Yet Unknown Biological Functions in Both Segments A and B" Vaccines 11, no. 4: 867. https://doi.org/10.3390/vaccines11040867