
T-ALL Cells as Tool Cells for CAR T Therapy

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. DNA Constructs and Lentivirus Production
2.3. Flow Cytometry
2.4. Apoptosis Assays
3. Results
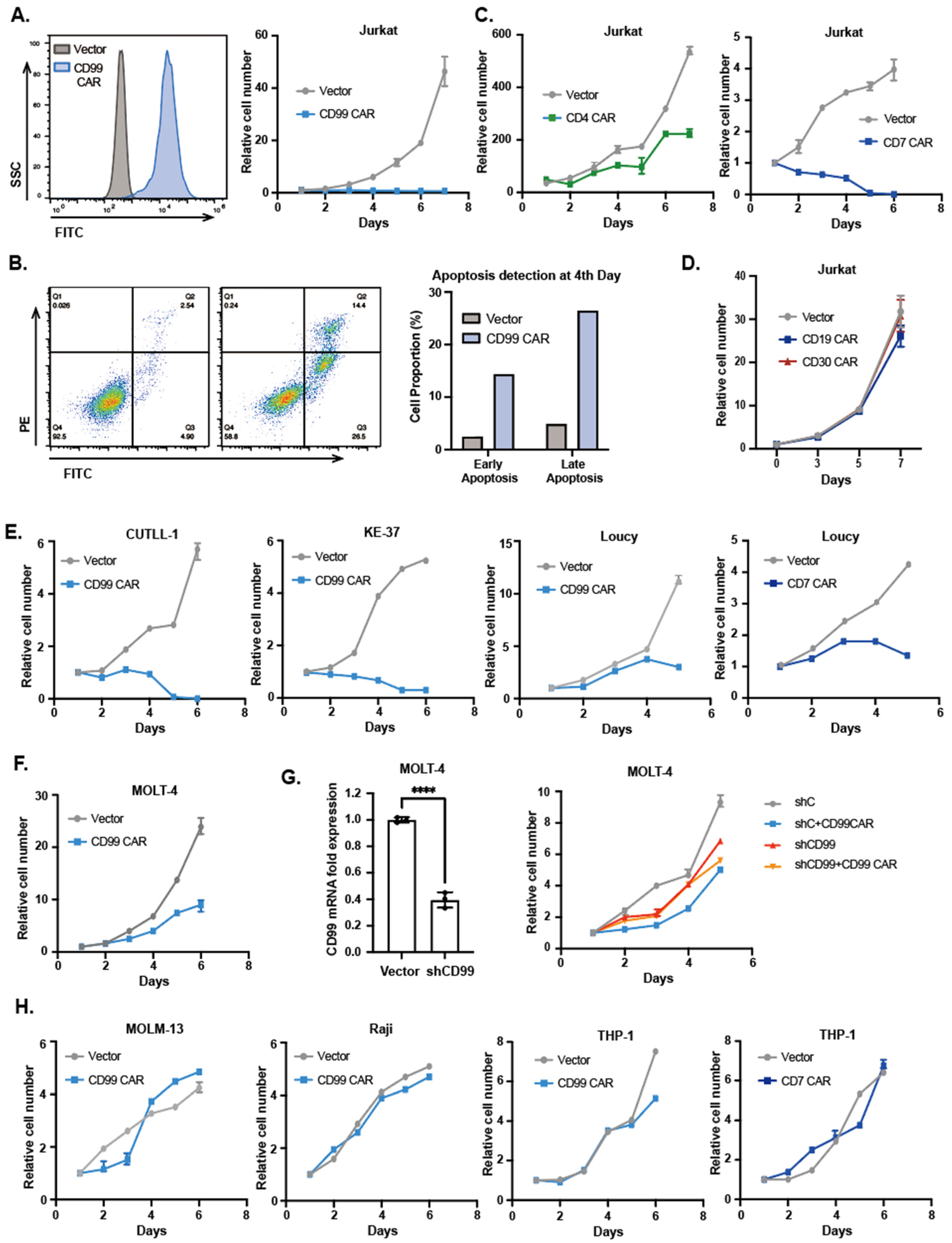
3.1. T-ALL Cell Lines Transduced with CAR Has Similar Cytotoxicity to That of CAR T Cells
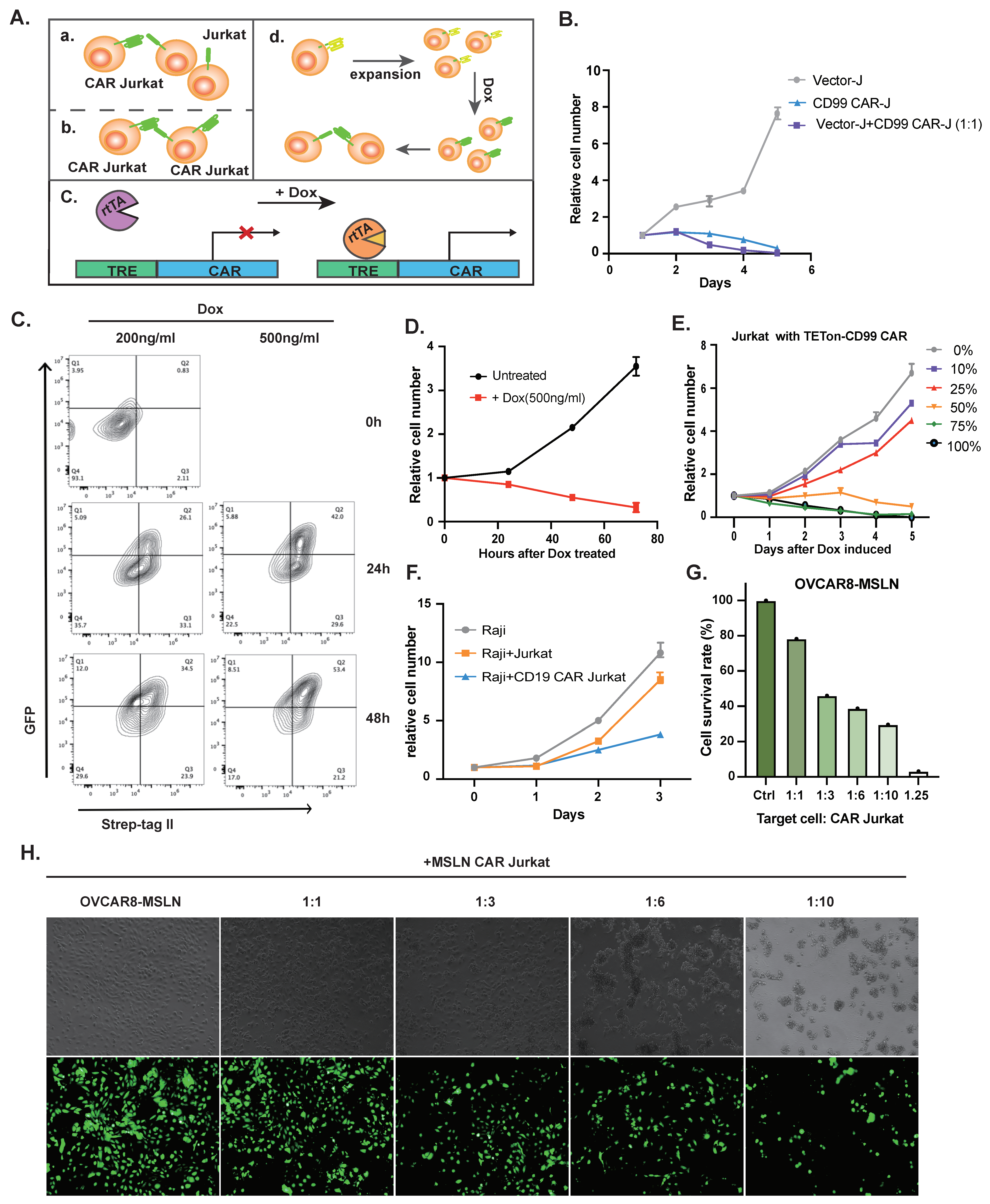
3.2. Application of CAR Jurkat Cell Lines Treating T-ALL or Non-T-ALL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiaretti, S.; Foà, R. T-cell acute lymphoblastic leukemia. Haematologica 2009, 94, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Raetz, E.A.; Teachey, D.T. T-cell acute lymphoblastic leukemia. Hematology 2016, 2016, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 33, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.; Kaminskiy, Y.; Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Rakhmatullina, A.; Petukhov, A.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Knowns and Unknowns about CAR-T Cell Dysfunction. Cancers 2022, 14, 1078. [Google Scholar] [CrossRef]
- Fleischer, L.C.; Spencer, H.T.; Raikar, S.S. Targeting T cell malignancies using CAR-based immunotherapy: Challenges and potential solutions. J. Hematol. Oncol. 2019, 12, 141. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, M.; Tesio, M.; June, C.H.; Houot, R. CAR T-cells for T-cell malignancies: Challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia 2018, 32, 2307–2315. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Fröschl, G.; Printz, D.; De Zen, L.; Gaipa, G.; Ratei, R.; Basso, G.; Biondi, A.; Ludwig, W.-D.; Gadner, H.; et al. CD99 expression in T-lineage ALL: Implications for flow cytometric detection of minimal residual disease. Leukemia 2004, 18, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhang, Z.; Cen, H.; Wu, H.; Zhang, S.; Liu, J.; Leng, Y.; Ren, A.; Liu, X.; Zhang, Z.; et al. CAR T cells targeting CD99 as an approach to eradicate T-cell acute lymphoblastic leukemia without normal blood cells toxicity. J. Hematol. Oncol. 2021, 14, 162. [Google Scholar] [CrossRef]
- Rabinowich, H.; Pricop, L.; Herberman, R.B.; Whiteside, T.L. Expression and function of CD7 molecule on human natural killer cells. J. Immunol. 1994, 152, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Png, Y.T.; Vinanica, N.; Kamiya, T.; Shimasaki, N.; Coustan-Smith, E.; Campana, D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017, 1, 2348–2360. [Google Scholar] [CrossRef] [PubMed]
- Manara, M.C.; Pasello, M.; Scotlandi, K. CD99: A Cell Surface Protein with an Oncojanus Role in Tumors. Genes 2018, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Eng, W.S.; Hu, W.; Khalaj, M.; Garrett-Bakelman, F.E.; Tavakkoli, M.; Levine, R.L.; Carroll, M.; Klimek, V.M.; Melnick, A.M.; et al. CD99 is a therapeutic target on disease stem cells in myeloid malignancies. Sci. Transl. Med. 2017, 9, eaaj2025. [Google Scholar] [CrossRef]
- Terada, T. TDT (−), KIT (+), CD34 (+), CD99 (+) precursor T lymphoblastic leukemia/lymphoma. Int. J. Clin. Exp. Pathol. 2012, 5, 167–170. [Google Scholar]
- Milanezi, F.; Pereira, E.M.; Ferreira, F.V.; Leitão, D.; Schmitt, F.C. CD99/MIC-2 surface protein expression in breast carcinomas. Histopathology 2001, 39, 578–583. [Google Scholar] [CrossRef]
- Rocchi, A.; Manara, M.C.; Sciandra, M.; Zambelli, D.; Nardi, F.; Nicoletti, G.; Garofalo, C.; Meschini, S.; Astolfi, A.; Colombo, M.P.; et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J. Clin. Investig. 2010, 120, 668–680. [Google Scholar] [CrossRef]
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On Systems For Doxycycline-inducible Gene Expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef]
- O’Dwyer, K.M. The challenge to further improvements in survival of patients with T-ALL: Current treatments and new insights from disease pathogenesis. Semin. Hematol. 2020, 57, 149–156. [Google Scholar] [CrossRef]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell. Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar] [PubMed]
- Liu, Q.; Xu, Y.; Mou, J.; Tang, K.; Fu, X.; Li, Y.; Xing, Y.; Rao, Q.; Xing, H.; Tian, Z.; et al. Irradiated chimeric antigen receptor engineered NK-92MI cells show effective cytotoxicity against CD19+ malignancy in a mouse model. Cytotherapy 2020, 22, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Fabian, K.P.; Hodge, J.W. The emerging role of off-the-shelf engineered natural killer cells in targeted cancer immunotherapy. Mol. Ther. Oncolytics 2021, 23, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Cui, H.; Caligiuri, M.A.; Yu, J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 168. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, W.; Huang, B.; Xu, S.; Li, Y.; Zhu, L.; Yiqiao, H.; Wu, Z.; Wu, X. AAV-mediated in vivo CAR gene therapy for targeting human T-cell leukemia. Blood Cancer J. 2021, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Park, J.; Du, Y.; Kim, H.R.; Wang, G.; Errami, Y.; Chen, S. One-step generation of modular CAR-T cells with AAV–Cpf1. Nat. Methods 2019, 16, 247–254. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, A.; Zhao, Y.; Zhu, H. T-ALL Cells as Tool Cells for CAR T Therapy. Vaccines 2023, 11, 854. https://doi.org/10.3390/vaccines11040854
Ren A, Zhao Y, Zhu H. T-ALL Cells as Tool Cells for CAR T Therapy. Vaccines. 2023; 11(4):854. https://doi.org/10.3390/vaccines11040854
Chicago/Turabian StyleRen, Anqi, Yuan Zhao, and Haichuan Zhu. 2023. "T-ALL Cells as Tool Cells for CAR T Therapy" Vaccines 11, no. 4: 854. https://doi.org/10.3390/vaccines11040854