
Innate and Adaptive Immunity during SARS-CoV-2 Infection: Biomolecular Cellular Markers and Mechanisms

 ,
,  , , , , , and
, , , , , and
Abstract
:
1. Introduction
1.1. Overview
1.2. Current SARS-CoV-2 Vaccine Immunogen Responses
1.3. Respiratory Microenvironment
1.4. Cytokine and Serum Proteins during SARS-CoV-2 Infection
1.5. Pre-2022 Laboratory Research Context
1.6. Role of Toll-like Receptors (TLR) or TLR Induced IFN Dysregulation
2. Innate Immune Systems and SARS-CoV-2 Research
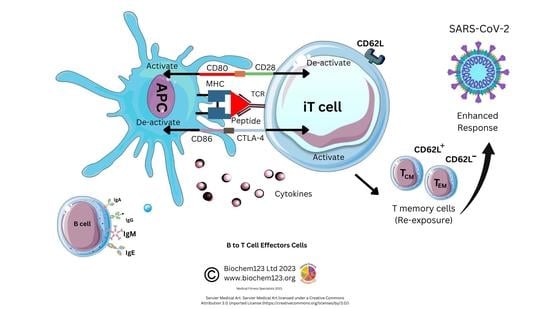
2.1. B Cell Development Dependency on T cell Activation
2.2. Antibody Isotypes and Class-Switching
2.3. Role of B Cell Markers in Current Research
2.4. B Cell Antibody Responses during Respiratory Infections
2.5. B Cells and Antibody Responses to SARS-CoV-2 Infection
2.6. Role of B Cell Markers during SARS-CoV-2 Infection and Other Conditions
3. Inflammatory Cells and Phagocytes
3.1. Neutrophil Introduction
3.2. Neutrophil Cellular Markers after Host SARS-CoV-2 Infection during COVID-19 Disease
3.3. Monocyte Cellular Development
3.4. Monocyte Cellular Markers during Host SARS-CoV-2 Infection
3.5. Macrophages Metabolism and Function
3.6. Macrophage Classification
3.7. Macrophage Metabolism Role during Polarization and SARS-CoV-2 Infection
3.8. Macrophage Phenotypes, Cytokines and Chemokines during SARS-CoV-2 Infection
4. Dendritic Cells
4.1. Dendritic Cell Overview
4.2. Role of Dendritic Cells and Cellular Markers in Disease
4.3. Dendritic Cell Role in Immune Responses to host SARS-CoV-2 Infection
5. Natural Killer Cells
5.1. Natural Killer Cell Overview
5.2. Natural Killer Cell and the Immune Response to Host SARS-CoV-2 Infection
6. T Cells and the Adaptive Immune System in SARS-CoV-2 Research
6.1. Overview to T Cells and the Adaptive Immune System
6.2. Background to T Cells in Coronaviruses and Host SARS-CoV-2 Infection
6.3. Helper T Cell Role during the Adaptive Immune Response to SARS-CoV-2
6.4. Cytotoxic T Cell Role during the Adaptive Immune Response to SARS-CoV-2
6.5. Regulatory T Cell Overview during the Adaptive Immune Response
6.6. TH17 Cell Overview during the Adaptive Immune Response
6.7. γδ T Cell Overview during the Adaptive Immune Response
7. Autoantibodies in SARS-CoV-2 Research
8. Limitations
9. Discussion
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Krishnan, U.M. The Emergence of COVID-19 as a Global Pandemic: Understanding the Epidemiology, Immune Response and Potential Therapeutic Targets of SARS-CoV-2. Biochimie 2020, 179, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.A.; Kellogg, C.; Equils, O. Neutralizing and Cross-Reacting Antibodies: Implications for Immunotherapy and SARS-CoV-2 Vaccine Development. Hum. Vaccines Immunother. 2021, 17, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Vivanti, A.J.; Vauloup-Fellous, C.; Prevot, S.; Zupan, V.; Suffee, C.; do Cao, J.; Benachi, A.; de Luca, D. Transplacental Transmission of SARS-CoV-2 Infection. Nat. Commun. 2020, 11, 3572. [Google Scholar] [CrossRef] [PubMed]
- Mahendra, M.; Nuchin, A.; Kumar, R.; Shreedhar, S.; Mahesh, P.A. Predictors of Mortality in Patients with Severe COVID-19 Pneumonia—A Retrospective Study. Adv. Respir. Med. 2021, 89, 135–144. [Google Scholar] [CrossRef]
- Zandi, M.; Shafaati, M.; Kalantar-Neyestanaki, D.; Pourghadamyari, H.; Fani, M.; Soltani, S.; Kaleji, H.; Abbasi, S. The Role of SARS-CoV-2 Accessory Proteins in Immune Evasion. Biomed. Pharmacother. 2022, 156, 113889. [Google Scholar] [CrossRef]
- Li, R.; Qin, C. Expression Pattern and Function of SARS-CoV-2 Receptor ACE2. Biosaf. Health 2021, 3, 312–318. [Google Scholar] [CrossRef]
- Wettstein, L.; Kirchhoff, F.; Münch, J. The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment. Int. J. Mol. Sci. 2022, 23, 1351. [Google Scholar] [CrossRef]
- Hoffmann, M.; Pöhlmann, S. Novel SARS-CoV-2 receptors: ASGR1 and KREMEN1. Cell Res. 2022, 32, 1–2. [Google Scholar] [CrossRef]
- Mayi, B.S.; Leibowitz, J.A.; Woods, A.T.; Ammon, K.A.; Liu, A.E.; Raja, A. The Role of Neuropilin-1 in COVID-19. PLoS Pathog. 2021, 17, e1009153. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Aleya, L.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Bungau, S. CD147-spike protein interaction in COVID-19: Get the ball rolling with a novel receptor and therapeutic target. Sci. Total Environ. 2022, 808, 152072. [Google Scholar] [CrossRef]
- Kalina, T.; Fišer, K.; Pérez-Andrés, M.; Kužílková, D.; Cuenca, M.; Bartol, S.J.W.; Blanco, E.; Engel, P.; van Zelm, M.C. CD Maps—Dynamic Profiling of CD1–CD100 Surface Expression on Human Leukocyte and Lymphocyte Subsets. Front. Immunol. 2019, 10, 2434. [Google Scholar] [CrossRef]
- Arrindell, J.; Abou Atmeh, P.; Jayet, L.; Sereme, Y.; Mege, J.-L.; Desnues, B. Vimentin Is an Important ACE2 Co-Receptor for SARS-CoV-2 in Epithelial Cells. iScience 2022, 25, 105463. [Google Scholar] [CrossRef]
- Mourier, T.; Shuaib, M.; Hala, S.; Mfarrej, S.; Alofi, F.; Naeem, R.; Alsomali, A.; Jorgensen, D.; Subudhi, A.K.; ben Rached, F.; et al. SARS-CoV-2 Genomes from Saudi Arabia Implicate Nucleocapsid Mutations in Host Response and Increased Viral Load. Nat. Commun. 2022, 13, 601. [Google Scholar] [CrossRef]
- Zhang, Z.; Nomura, N.; Muramoto, Y.; Ekimoto, T.; Uemura, T.; Liu, K.; Yui, M.; Kono, N.; Aoki, J.; Ikeguchi, M.; et al. Structure of SARS-CoV-2 Membrane Protein Essential for Virus Assembly. Nat. Commun. 2022, 13, 4399. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-Cell RNA Sequencing to Explore Immune Cell Heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef]
- Gadalla, R.; Noamani, B.; MacLeod, B.L.; Dickson, R.J.; Guo, M.; Xu, W.; Lukhele, S.; Elsaesser, H.J.; Razak, A.R.A.; Hirano, N.; et al. Validation of CyTOF Against Flow Cytometry for Immunological Studies and Monitoring of Human Cancer Clinical Trials. Front. Oncol. 2019, 9, 415. [Google Scholar] [CrossRef]
- Gil-Manso, S.; Miguens Blanco, I.; López-Esteban, R.; Carbonell, D.; López-Fernández, L.A.; West, L.; Correa-Rocha, R.; Pion, M. Comprehensive Flow Cytometry Profiling of the Immune System in COVID-19 Convalescent Individuals. Front. Immunol. 2022, 12, 5734. [Google Scholar] [CrossRef]
- Blanchard, L.; Vina, E.; Asrir, A.; Tardiveau, C.; Coudert, J.; Laffont, R.; Tarroux, D.; Bettini, S.; Veerman, K.; Lafouresse, F.; et al. Flow Cytometry Analysis of Endothelial Cells and Subsets of Exhausted CD8+ T Cells in Murine Tumor Models. STAR Protoc. 2022, 3, 101444. [Google Scholar] [CrossRef]
- Filchakova, O.; Dossym, D.; Ilyas, A.; Kuanysheva, T.; Abdizhamil, A.; Bukasov, R. Review of COVID-19 Testing and Diagnostic Methods. Talanta 2022, 244, 123409. [Google Scholar] [CrossRef]
- Kristiansen, P.A.; Page, M.; Bernasconi, V.; Mattiuzzo, G.; Dull, P.; Makar, K.; Plotkin, S.; Knezevic, I. WHO International Standard for Anti-SARS-CoV-2 Immunoglobulin. Lancet 2021, 397, 1347–1348. [Google Scholar] [CrossRef]
- Al-Sheboul, S.A.; Brown, B.; Shboul, Y.; Fricke, I.; Imarogbe, C.; Alzoubi, K.H. An Immunological Review of SARS-CoV-2 Infection and Vaccine Serology: Innate and Adaptive Responses to MRNA, Adenovirus, Inactivated and Protein Subunit Vaccines. Vaccines 2022, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Kellam, P.; Barclay, W. The Dynamics of Humoral Immune Responses Following SARS-CoV-2 Infection and the Potential for Reinfection. J. Gen. Virol. 2020, 101, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.-M.; Padera, R.F.; Solomon, I.H.; Kanjilal, S.; Hammer, M.M.; Hornick, J.L.; Sholl, L.M. In Situ Detection of SARS-CoV-2 in Lungs and Airways of Patients with COVID-19. Mod. Pathol. 2020, 33, 2104–2114. [Google Scholar] [CrossRef]
- Zeng, Y.; Qiu, Y.; Jiang, W.; Fu, B.M. Glycocalyx Acts as a Central Player in the Development of Tumor Microenvironment by Extracellular Vesicles for Angiogenesis and Metastasis. Cancers 2022, 14, 5415. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Derangement of the endothelial glycocalyx in sepsis. J. Thromb. Haemost. 2019, 17, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Ninham, B.; Reines, B.; Battye, M.; Thomas, P. Pulmonary Surfactant and COVID-19: A New Synthesis. QRB Discov. 2022, 3, e6. [Google Scholar] [CrossRef]
- Han, S.; Mallampalli, R.K. The Role of Surfactant in Lung Disease and Host Defense against Pulmonary Infections. Ann. Am. Thorac. Soc. 2015, 12, 765–774. [Google Scholar] [CrossRef]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A Molecular Single-Cell Lung Atlas of Lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef]
- Zhang, Z.; Newton, K.; Kummerfeld, S.K.; Webster, J.; Kirkpatrick, D.S.; Phu, L.; Eastham-Anderson, J.; Liu, J.; Lee, W.P.; Wu, J.; et al. Transcription Factor Etv5 Is Essential for the Maintenance of Alveolar Type II Cells. Proc. Natl. Acad. Sci. USA 2017, 114, 3903–3908. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 Infection: The Role of Cytokines in COVID-19 Disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling Serum Cytokines in COVID-19 Patients Reveals IL-6 and IL-10 Are Disease Severity Predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef]
- Can, F.K.; Özkurt, Z.; Öztürk, N.; Sezen, S. Effect of IL-6, IL-8/CXCL8, IP-10/CXCL 10 Levels on the Severity in COVID-19 Infection. Int. J. Clin. Pract. 2021, 75. [Google Scholar] [CrossRef]
- Valente, M.; Dölen, Y.; van Dinther, E.; Vimeux, L.; Fallet, M.; Feuillet, V.; Figdor, C.G. Cross-Talk between INKT Cells and CD8 T Cells in the Spleen Requires the IL-4/CCL17 Axis for the Generation of Short-Lived Effector Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 25816–25827. [Google Scholar] [CrossRef]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- Khalil, B.A.; Shakartalla, S.B.; Goel, S.; Madkhana, B.; Halwani, R.; Maghazachi, A.A.; AlSafar, H.; Al-Omari, B.; al Bataineh, M.T. Immune Profiling of COVID-19 in Correlation with SARS and MERS. Viruses 2022, 14, 164. [Google Scholar] [CrossRef]
- Zhang, N.; Zhao, Y.D.; Wang, X.M. CXCL10 an important chemokine associated with cytokine storm in COVID-19 infected patients. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7497–7505. [Google Scholar] [CrossRef] [PubMed]
- Başar, E.Z.; Sönmez, H.E.; Uzuner, H.; Karadenizli, A.; Güngör, H.S.; Akgün, G.; Yetimakman, A.F.; Öncel, S.; Babaoğlu, K. CXCL10/IP10 as a Biomarker Linking Multisystem Inflammatory Syndrome and Left Ventricular Dysfunction in Children with SARS-CoV-2. J. Clin. Med. 2022, 11, 1416. [Google Scholar] [CrossRef]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-Associated Coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, L.; Schaller, T.; Zwiebel, M.; Karayel, Ö.; Müller-Reif, J.B.; Zeng, W.-F.; Dintner, S.; Hirschbühl, K.; Märkl, B.; Claus, R.; et al. Quantitative Multi-Organ Proteomics of Fatal COVID-19 Uncovers Tissue-Specific Effects beyond Inflammation. medxRiv, 2022; pre-print. [Google Scholar] [CrossRef]
- Captur, G.; Moon, J.C.; Topriceanu, C.-C.; Joy, G.; Swadling, L.; Hallqvist, J.; Doykov, I.; Patel, N.; Spiewak, J.; Baldwin, T.; et al. Plasma Proteomic Signature Predicts Who Will Get Persistent Symptoms Following SARS-CoV-2 Infection. eBioMedicine 2022, 85, 104293. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Flerlage, T.; Boyd, D.F.; Meliopoulos, V.; Thomas, P.G.; Schultz-Cherry, S. Influenza virus and SARS-CoV-2: Pathogenesis and host responses in the respiratory tract. Nat. Rev. Microbiol. 2021, 19, 425–441. [Google Scholar] [CrossRef]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; van der Werf, S.; et al. Syncytia Formation by SARS-CoV-2-infected Cells. EMBO J. 2021, 40, e107405. [Google Scholar] [CrossRef]
- Norlander, A.E.; Peebles, R.S., Jr. Innate Type 2 Responses to Respiratory Syncytial Virus Infection. Viruses 2020, 12, 521. [Google Scholar] [CrossRef]
- Lin, L.; Li, Q.; Wang, Y.; Shi, Y. Syncytia Formation during SARS-CoV-2 Lung Infection: A Disastrous Unity to Eliminate Lymphocytes. Cell Death Differ. 2021, 28, 2019–2021. [Google Scholar] [CrossRef]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeño, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs That Inhibit TMEM16 Proteins Block SARS-CoV-2 Spike-Induced Syncytia. Nature 2021, 594, 88–93. [Google Scholar] [CrossRef]
- Ludington, J.G.; Ansari, S.A.; Schmaier, A.A.; Enjyoji, K.; Nilsson-Payant, B.E.; Bram, Y.; Chandar, V.; Borczuk, A.; tenOever, B.R.; Schwartz, R.E.; et al. SARS-CoV-2 Ion Channel ORF3a Enables TMEM16F-Dependent Phosphatidylserine Externalization to Augment Procoagulant Activity of the Tenase and Prothrombinase Complexes. Blood 2021, 138, 1. [Google Scholar] [CrossRef]
- Wu, N.; Cernysiov, V.; Davidson, D.; Song, H.; Tang, J.; Luo, S.; Lu, Y.; Qian, J.; Gyurova, I.E.; Waggoner, S.N.; et al. Critical Role of Lipid Scramblase TMEM16F in Phosphatidylserine Exposure and Repair of Plasma Membrane after Pore Formation. Cell Rep. 2020, 30, 1129–1140.e5. [Google Scholar] [CrossRef]
- Lind, S.E. Phosphatidylserine Is an Overlooked Mediator of COVID-19 Thromboinflammation. Heliyon 2021, 7, e06033. [Google Scholar] [CrossRef]
- Sheng, Y.H.; Hasnain, S.Z. Mucus and Mucins: The Underappreciated Host Defence System. Front. Cell Infect. Microbiol. 2022, 12, 744. [Google Scholar] [CrossRef]
- Ballester, B.; Milara, J.; Cortijo, J. The role of mucin 1 in respiratory diseases. Eur. Respir. Rev. 2021, 30, 200149, PMCID:PMC9488590. [Google Scholar] [CrossRef] [PubMed]
- Huang, B. Mucins Produced by Type II Pneumocyte: Culprits in SARS-CoV-2 Pathogenesis. Cell Mol. Immunol. 2021, 18, 1823–1825. [Google Scholar] [CrossRef]
- Lu, W.; Liu, X.; Wang, T.; Liu, F.; Zhu, A.; Lin, Y.; Luo, J.; Ye, F.; He, J.; Zhao, J.; et al. Elevated MUC1 and MUC5AC mucin protein levels in airway mucus of critical ill COVID-19 patients. J. Med. Virol. 2021, 93, 582–584. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through down-Regulating MHC-I. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV-2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci. 2021, 30, 1114–1130. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I Response Timing Relative to Virus Replication Determines MERS Coronavirus Infection Outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Hijano, D.R.; Vu, L.D.; Kauvar, L.M.; Tripp, R.A.; Polack, F.P.; Cormier, S.A. Role of Type I Interferon (IFN) in the Respiratory Syncytial Virus (RSV) Immune Response and Disease Severity. Front. Immunol. 2019, 10, 566. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Liu, N.; Qin, Z.; Wang, Y. Mitochondrial-Derived Damage-Associated Molecular Patterns Amplify Neuroinflammation in Neurodegenerative Diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qin, L.; Zhang, P.; Li, K.; Liang, L.; Sun, J.; Xu, B.; Dai, Y.; Li, X.; Zhang, C.; et al. Longitudinal COVID-19 Profiling Associates IL-1RA and IL-10 with Disease Severity and RANTES with Mild Disease. JCI Insight 2020, 5, e139834. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, D.; Gentili, V.; Rizzo, S.; Schiuma, G.; Beltrami, S.; Strazzabosco, G.; Fernandez, M.; Caccuri, F.; Caruso, A.; Rizzo, R. TLR3 and TLR7 RNA Sensor Activation during SARS-CoV-2 Infection. Microorganisms 2021, 9, 1820. [Google Scholar] [CrossRef]
- Solanich, X.; Vargas-Parra, G.; van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; Antolí, A.; del Valle, J.; Rocamora-Blanch, G.; Setién, F.; Esteller, M.; et al. Genetic Screening for TLR7 Variants in Young and Previously Healthy Men With Severe COVID-19. Front. Immunol. 2021, 12, 2965. [Google Scholar] [CrossRef]
- Rahman, A.H.; Taylor, D.K.; Turka, L.A. The Contribution of Direct TLR Signaling to T Cell Responses. Immunol. Res. 2009, 45, 25–36. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 Trafficking and Its Influence on LPS-Induced pro-Inflammatory Signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Tang, L.; Wang, F.; Huang, S.; Liu, S.; Lei, Y.; Wang, S.; Xie, Z.; Wang, W.; et al. TLR4 Regulates RORγt+ Regulatory T-Cell Responses and Susceptibility to Colon Inflammation through Interaction with Akkermansia Muciniphila. Microbiome 2022, 10, 98. [Google Scholar] [CrossRef]
- Visan, I. RORγt+ Treg Cells. Nat. Immunol. 2015, 16, 906. [Google Scholar] [CrossRef]
- Sugitharini, V.; Shahana, P.; Prema, A.; Berla Thangam, E. TLR2 and TLR4 Co-Activation Utilizes Distinct Signaling Pathways for the Production of Th1/Th2/Th17 Cytokines in Neonatal Immune Cells. Cytokine 2016, 85, 191–200. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 Spike Protein Interacts with and Activates TLR41. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Liu, L.; Aron, C.Z.; Grable, C.M.; Robles, A.; Liu, X.; Liu, Y.; Fatheree, N.Y.; Rhoads, J.M.; Alcorn, J.L. Surfactant Protein A Reduces TLR4 and Inflammatory Cytokine MRNA Levels in Neonatal Mouse Ileum. Sci. Rep. 2021, 11, 2593. [Google Scholar] [CrossRef]
- Schattner, M. Platelet TLR4 at the Crossroads of Thrombosis and the Innate Immune Response. J. Leukoc. Biol. 2019, 105, 873–880. [Google Scholar] [CrossRef]
- Becker, R.C.; Sexton, T.; Smyth, S. COVID-19 and Biomarkers of Thrombosis: Focus on von Willebrand Factor and Extracellular Vesicles. J. Thromb. Thrombolysis 2021, 52, 1010–1019. [Google Scholar] [CrossRef]
- Xue, W.; Ding, C.; Qian, K.; Liao, Y. The Interplay Between Coronavirus and Type I IFN Response. Front. Microbiol. 2022, 12, 805472. [Google Scholar] [CrossRef]
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral Activities of Type I Interferons to SARS-CoV-2 Infection. Antivir. Res. 2020, 179, 104811. [Google Scholar] [CrossRef]
- Klouda, T.; Hao, Y.; Kim, H.; Kim, J.; Olejnik, J.; Hume, A.J.; Ayyappan, S.; Hong, X.; Melero-Martin, J.; Fang, Y.; et al. Interferon-Alpha or -Beta Facilitates SARS-CoV-2 Pulmonary Vascular Infection by Inducing ACE2. Angiogenesis 2022, 25, 225–240. [Google Scholar] [CrossRef]
- Fukuda, Y.; Homma, T.; Inoue, H.; Goto, Y.; Sato, Y.; Ikeda, H.; Onitsuka, C.; Sato, H.; Akimoto, K.; Ebato, T.; et al. Serum IL-28A/IFN-Λ2 Is Linked to Disease Severity of COVID-19. Sci. Rep. 2022, 12, 5458. [Google Scholar] [CrossRef]
- Heuberger, J.; Trimpert, J.; Vladimirova, D.; Goosmann, C.; Lin, M.; Schmuck, R.; Mollenkopf, H.; Brinkmann, V.; Tacke, F.; Osterrieder, N.; et al. Epithelial Response to IFN-γ Promotes SARS-CoV-2 Infection. EMBO Mol. Med. 2021, 13, e13191. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Rivera, A.; Parker, D.; Durbin, J.E. Type III IFNs: Beyond Antiviral Protection. Semin. Immunol. 2019, 43, 101303. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Benitez, M.J.; Strich, J.R.; Alehashemi, S.; Stein, S.; Rastegar, A.; Almeida De Jesus, A.; Bhuyan, F.; Ramelli, S.; Babyak, A.; Perez-Valencia, L.; et al. Antiviral Innate Immunity Is Diminished in the Upper Respiratory Tract of Severe COVID-19 Patients. medxRiv, 2022; pre-print. [Google Scholar] [CrossRef]
- Xu, G.; Qi, F.; Wang, H.; Liu, Y.; Wang, X.; Zou, R.; Yuan, J.; Liao, X.; Liu, Y.; Zhang, S.; et al. The Transient IFN Response and the Delay of Adaptive Immunity Feature the Severity of COVID-19. Front. Immunol. 2022, 12, 5846. [Google Scholar] [CrossRef]
- Bondet, V.; Rodero, M.P.; Posseme, C.; Bost, P.; Decalf, J.; Haljasmägi, L.; Bekaddour, N.; Rice, G.I.; Upasani, V.; Herbeuval, J.-P.; et al. Differential Levels of IFNα Subtypes in Autoimmunity and Viral Infection. Cytokine 2021, 144, 155533. [Google Scholar] [CrossRef]
- Lam, J.H.; Baumgarth, N. The Multifaceted B Cell Response to Influenza Virus. J. Immunol. 2019, 202, 351–359. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Gaebler, C.; Muecksch, F.; Lorenzi, J.C.C.; Wang, Z.; Cho, A.; Agudelo, M.; Barnes, C.O.; Gazumyan, A.; Finkin, S.; et al. Convergent Antibody Responses to SARS-CoV-2 in Convalescent Individuals. Nature 2020, 584, 437–442. [Google Scholar] [CrossRef]
- Rosser, E.C.; Mauri, C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef]
- Mishra, D.; Singh, S.; Narayan, G. Role of B Cell Development Marker CD10 in Cancer Progression and Prognosis. Mol. Biol. Int. 2016, 2016, 432869. [Google Scholar] [CrossRef]
- Röltgen, K.; Boyd, S.D. Antibody and B Cell Responses to SARS-CoV-2 Infection and Vaccination. Cell Host Microbe 2021, 29, 1063–1075. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Hayes, J.; Wormald, M.; Rudd, P.; Davey, G. Fc Gamma Receptors: Glycobiology and Therapeutic Prospects. J. Inflamm. Res. 2016, 9, 209–219. [Google Scholar] [CrossRef]
- Junker, F.; Gordon, J.; Qureshi, O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol. 2020, 11, 1393. [Google Scholar] [CrossRef]
- Woof, J.M.; Kerr, M.A. The Function of Immunoglobulin A in Immunity. J. Pathol. 2006, 208, 270–282. [Google Scholar] [CrossRef]
- Woof, J.M.; Russell, M.W. Structure and Function Relationships in IgA. Mucosal. Immunol. 2011, 4, 590–597. [Google Scholar] [CrossRef]
- Monteiro, R.C.; van de Winkel, J.G.J. IgA Fc Receptors. Annu. Rev. Immunol. 2003, 21, 177–204. [Google Scholar] [CrossRef]
- De Tymowski, C.; Heming, N.; Correia, M.D.T.; Abbad, L.; Chavarot, N.; le Stang, M.-B.; Flament, H.; Bex, J.; Boedec, E.; Bounaix, C.; et al. CD89 Is a Potent Innate Receptor for Bacteria and Mediates Host Protection from Sepsis. Cell Rep. 2019, 27, 762–775.e5. [Google Scholar] [CrossRef]
- Van der Poll, T.; Shankar-Hari, M.; Wiersinga, W.J. The immunology of sepsis. Immunity 2021, 54, 2450–2464. [Google Scholar] [CrossRef]
- Amendt, T.; el Ayoubi, O.; Linder, A.T.; Allies, G.; Young, M.; Setz, C.S.; Jumaa, H. Primary Immune Responses and Affinity Maturation Are Controlled by IgD. Front. Immunol. 2021, 12, 709240. [Google Scholar] [CrossRef]
- Roche, P.A.; Furuta, K. The Ins and Outs of MHC Class II-Mediated Antigen Processing and Presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Ishina, I.A.; Zakharova, M.Y.; Kurbatskaia, I.N.; Mamedov, A.E.; Belogurov, A.A., Jr.; Gabibov, A.G. MHC Class II Presentation in Autoimmunity. Cells 2023, 12, 314. [Google Scholar] [CrossRef] [PubMed]
- Katikaneni, D.S.; Jin, L. B Cell MHC Class II Signaling: A Story of Life and Death. Hum. Immunol. 2019, 80, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.J.R.; Watkins, D.I. Impact of MHC Class I Diversity on Immune Control of Immunodeficiency Virus Replication. Nat. Rev. Immunol. 2008, 8, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A Biomarker for B Cell Development, Lymphoma Diagnosis and Therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739.e6. [Google Scholar] [CrossRef]
- Wei, C.; Anolik, J.; Cappione, A.; Zheng, B.; Pugh-Bernard, A.; Brooks, J.; Lee, E.-H.; Milner, E.C.B.; Sanz, I. A New Population of Cells Lacking Expression of CD27 Represents a Notable Component of the B Cell Memory Compartment in Systemic Lupus Erythematosus. J. Immunol. 2007, 178, 6624–6633. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Hu, F. Double-Negative (DN) B Cells: An under-Recognized Effector Memory B Cell Subset in Autoimmunity. Clin. Exp. Immunol. 2021, 205, 119–127. [Google Scholar] [CrossRef]
- Hu, F.; Zhang, W.; Shi, L.; Liu, X.; Jia, Y.; Xu, L.; Zhu, H.; Li, Y.; Xu, D.; Lu, L.; et al. Impaired CD27+IgD+ B Cells With Altered Gene Signature in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 626. [Google Scholar] [CrossRef]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From Activation Marker to Metabolic Gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Rincon-Arevalo, H.; Wiedemann, A.; Stefanski, A.-L.; Lettau, M.; Szelinski, F.; Fuchs, S.; Frei, A.P.; Steinberg, M.; Kam-Thong, T.; Hatje, K.; et al. Deep Phenotyping of CD11c+ B Cells in Systemic Autoimmunity and Controls. Front. Immunol. 2021, 12, 635615. [Google Scholar] [CrossRef]
- Levack, R.C.; Newell, K.L.; Cabrera-Martinez, B.; Cox, J.; Perl, A.; Bastacky, S.I.; Winslow, G.M. Adenosine Receptor 2a Agonists Target Mouse CD11c+T-Bet+ B Cells in Infection and Autoimmunity. Nat. Commun. 2022, 13, 452. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, S.; Qian, J.; Wang, Y.; Yu, X.; Dai, D.; Dai, M.; Wu, L.; Liao, Z.; Xue, Z.; et al. T-Bet+CD11c+ B Cells Are Critical for Antichromatin Immunoglobulin G Production in the Development of Lupus. Arthritis Res. Ther. 2017, 19, 225. [Google Scholar] [CrossRef]
- Agematsu, K. Memory B cells and CD27. Histol. Histopathol. 2000, 15, 573–576. [Google Scholar] [CrossRef]
- Hendriks, J.; Gravestein, L.A.; Tesselaar, K.; van Lier, R.A.W.; Schumacher, T.N.M.; Borst, J. CD27 Is Required for Generation and Long-Term Maintenance of T Cell Immunity. Nat. Immunol. 2000, 1, 433–440. [Google Scholar] [CrossRef]
- Thorarinsdottir, K.; Camponeschi, A.; Gjertsson, I.; Mårtensson, I.-L. CD21 B Cells: A Snapshot of a Unique B Cell Subset in Health and Disease. Scand. J. Immunol. 2015, 82, 254–261. [Google Scholar] [CrossRef]
- Bernard, N.J. Double-Negative B Cells. Nat. Rev. Rheumatol. 2018, 14, 684. [Google Scholar] [CrossRef]
- Golinski, M.-L.; Demeules, M.; Derambure, C.; Riou, G.; Maho-Vaillant, M.; Boyer, O.; Joly, P.; Calbo, S. CD11c+ B Cells Are Mainly Memory Cells, Precursors of Antibody Secreting Cells in Healthy Donors. Front. Immunol. 2020, 11, 32. [Google Scholar] [CrossRef]
- Castleman, M.J.; Stumpf, M.M.; Therrien, N.R.; Smith, M.J.; Lesteberg, K.E.; Palmer, B.E.; Maloney, J.P.; Janssen, W.J.; Mould, K.J.; Beckham, J.D.; et al. Autoantibodies Elicited with SARS-CoV-2 Infection Are Linked to Alterations in Double Negative B Cells. Front. Immunol. 2022, 13, 988125. [Google Scholar] [CrossRef]
- Luo, H.; Jia, T.; Chen, J.; Zeng, S.; Qiu, Z.; Wu, S.; Li, X.; Lei, Y.; Wang, X.; Wu, W.; et al. The Characterization of Disease Severity Associated IgG Subclasses Response in COVID-19 Patients. Front. Immunol. 2021, 12, 632814. [Google Scholar] [CrossRef]
- Lee, H.; Kovacs, C.; Mattman, A.; Hollander, Z.; Chen, V.; Ng, R.; Leung, J.M.; Sin, D.D. The Impact of IgG Subclass Deficiency on the Risk of Mortality in Hospitalized Patients with COPD. Respir. Res. 2022, 23, 141. [Google Scholar] [CrossRef]
- Gorse, G.J.; Patel, G.B.; Vitale, J.N.; O’Connor, T.Z. Prevalence of Antibodies to Four Human Coronaviruses Is Lower in Nasal Secretions than in Serum. Clin. Vaccine Immunol. 2010, 17, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Liu, D.; Sun, J.; Li, F.; Zhao, J. Immune Responses to Human Respiratory Coronaviruses Infection in Mouse Models. Curr. Opin. Virol. 2022, 52, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Burlington, D.B.; Clements, M.L.; Meiklejohn, G.; Phelan, M.; Murphy, B.R. Hemagglutinin-Specific Antibody Responses in Immunoglobulin G, A, and M Isotypes as Measured by Enzyme-Linked Immunosorbent Assay after Primary or Secondary Infection of Humans with Influenza A Virus. Infect. Immun. 1983, 41, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-N.; Lin, S.-C.; Carney, P.J.; Li, J.; Liu, F.; Lu, X.; Liu, M.; Stevens, J.; Levine, M.; Katz, J.M.; et al. IgM, IgG, and IgA Antibody Responses to Influenza A(H1N1)Pdm09 Hemagglutinin in Infected Persons during the First Wave of the 2009 Pandemic in the United States. Clin. Vaccine Immunol. 2014, 21, 1054–1060. [Google Scholar] [CrossRef]
- Wu, J.; Tang, L.; Ma, Y.; Li, Y.; Zhang, D.; Li, Q.; Mei, H.; Hu, Y. Immunological Profiling of COVID-19 Patients with Pulmonary Sequelae. mBio 2021, 12, e01599-21. [Google Scholar] [CrossRef]
- Ciabattini, A.; Pastore, G.; Fiorino, F.; Polvere, J.; Lucchesi, S.; Pettini, E.; Auddino, S.; Rancan, I.; Durante, M.; Miscia, M.; et al. Evidence of SARS-CoV-2-Specific Memory B Cells Six Months After Vaccination With the BNT162b2 MRNA Vaccine. Front. Immunol. 2021, 12, 740708. [Google Scholar] [CrossRef]
- Plūme, J.; Galvanovskis, A.; Šmite, S.; Romanchikova, N.; Zayakin, P.; Linē, A. Early and Strong Antibody Responses to SARS-CoV-2 Predict Disease Severity in COVID-19 Patients. J. Transl. Med. 2022, 20, 176. [Google Scholar] [CrossRef]
- Chen, S.; Guan, F.; Candotti, F.; Benlagha, K.; Camara, N.O.S.; Herrada, A.A.; James, L.K.; Lei, J.; Miller, H.; Kubo, M.; et al. The Role of B Cells in COVID-19 Infection and Vaccination. Front. Immunol. 2022, 13, 988536. [Google Scholar] [CrossRef]
- Sosa-Hernández, V.A.; Torres-Ruíz, J.; Cervantes-Díaz, R.; Romero-Ramírez, S.; Páez-Franco, J.C.; Meza-Sánchez, D.E.; Juárez-Vega, G.; Pérez-Fragoso, A.; Ortiz-Navarrete, V.; Ponce-de-León, A.; et al. B Cell Subsets as Severity-Associated Signatures in COVID-19 Patients. Front. Immunol. 2020, 11, 611004. [Google Scholar] [CrossRef]
- Nguyen-Contant, P.; Embong, A.K.; Kanagaiah, P.; Chaves, F.A.; Yang, H.; Branche, A.R.; Topham, D.J.; Sangster, M.Y. S Protein-Reactive IgG and Memory B Cell Production after Human SARS-CoV-2 Infection Includes Broad Reactivity to the S2 Subunit. mBio 2020, 11, e01991-20. [Google Scholar] [CrossRef]
- Staats, L.A.N.; Pfeiffer, H.; Knopf, J.; Lindemann, A.; Fürst, J.; Kremer, A.E.; Hackstein, H.; Neurath, M.F.; Muñoz, L.E.; Achenbach, S.; et al. IgA2 Antibodies against SARS-CoV-2 Correlate with NET Formation and Fatal Outcome in Severely Diseased COVID-19 Patients. Cells 2020, 9, 2676. [Google Scholar] [CrossRef]
- Kober, C.; Manni, S.; Wolff, S.; Barnes, T.; Mukherjee, S.; Vogel, T.; Hoenig, L.; Vogel, P.; Hahn, A.; Gerlach, M.; et al. IgG3 and IgM Identified as Key to SARS-CoV-2 Neutralization in Convalescent Plasma Pools. PLoS ONE 2022, 17, e0262162. [Google Scholar] [CrossRef]
- LaSalle, T.J.; Gonye, A.L.K.; Freeman, S.S.; Kaplonek, P.; Gushterova, I.; Kays, K.R.; Manakongtreecheep, K.; Tantivit, J.; Rojas-Lopez, M.; Russo, B.C.; et al. Longitudinal Characterization of Circulating Neutrophils Uncovers Phenotypes Associated with Severity in Hospitalized COVID-19 Patients. Cell Rep. Med. 2022, 3, 100779. [Google Scholar] [CrossRef]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claër, L.; Quentric, P.; Fadlallah, J.; Devilliers, H.; Ghillani, P.; et al. IgA Dominates the Early Neutralizing Antibody Response to SARS-CoV-2. Sci. Transl. Med. 2021, 13, eabd2223. [Google Scholar] [CrossRef]
- Shan, M.; Yang, D.; Dou, H.; Zhang, L. Fucosylation in cancer biology and its clinical applications. Prog. Mol. Biol. Transl. Sci. 2019, 162, 93–119. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in Health and Disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Hoepel, W.; Allahverdiyeva, S.; Harbiye, H.; de Taeye, S.W.; van der Ham, A.J.; de Boer, L.; Zaat, S.A.J.; van Weeghel, M.; Baeten, D.L.P.; Houtkooper, R.H.; et al. IgG Subclasses Shape Cytokine Responses by Human Myeloid Immune Cells through Differential Metabolic Reprogramming. J. Immunol. 2020, 205, 3400–3407. [Google Scholar] [CrossRef]
- Iles, J.K.; Zmuidinaite, R.; Sadee, C.; Gardiner, A.; Lacey, J.; Harding, S.; Wallis, G.; Patel, R.; Roblett, D.; Heeney, J.; et al. Determination of IgG1 and IgG3 SARS-CoV-2 Spike Protein and Nucleocapsid Binding—Who Is Binding Who and Why? Int. J. Mol. Sci. 2022, 23, 6050. [Google Scholar] [CrossRef]
- Moura, A.D.; da Costa, H.H.M.; Correa, V.A.; de S. Lima, A.K.; Lindoso, J.A.L.; de Gaspari, E.; Hong, M.A.; Cunha-Junior, J.P.; Prudencio, C.R. Assessment of Avidity Related to IgG Subclasses in SARS-CoV-2 Brazilian Infected Patients. Sci. Rep. 2021, 11, 17642. [Google Scholar] [CrossRef]
- Szymczak, A.; Jedruchniewicz, N.; Torelli, A.; Kaczmarzyk-Radka, A.; Coluccio, R.; Kłak, M.; Konieczny, A.; Ferenc, S.; Witkiewicz, W.; Montomoli, E.; et al. Antibodies Specific to SARS-CoV-2 Proteins N, S and E in COVID-19 Patients in the Normal Population and in Historical Samples. J. Gen. Virol. 2021, 102, 001692. [Google Scholar] [CrossRef]
- Gunn, B.M.; Roy, V.; Karim, M.M.; Hartnett, J.N.; Suscovich, T.J.; Goba, A.; Momoh, M.; Sandi, J.D.; Kanneh, L.; Andersen, K.G.; et al. Survivors of Ebola Virus Disease Develop Polyfunctional Antibody Responses. J. Infect. Dis. 2020, 221, 156–161. [Google Scholar] [CrossRef]
- Larsen, M.D.; de Graaf, E.L.; Sonneveld, M.E.; Plomp, H.R.; Nouta, J.; Hoepel, W.; Chen, H.J.; Linty, F.; Visser, R.; Brinkhaus, M.; et al. Afucosylated IgG Characterizes Enveloped Viral Responses and Correlates with COVID-19 Severity. Science 2021, 371, eabc8378. [Google Scholar] [CrossRef] [PubMed]
- Bolton, M.; Arevalo, C.P.; Griesman, T.; Li, S.H.; Bates, P.; Wilson, P.C.; Hensley, S.E. IgG3 subclass antibodies recognize antigenically drifted influenza viruses and SARS-CoV-2 variants through efficient bivalent binding. bioRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Gasser, R.; Cloutier, M.; Prévost, J.; Fink, C.; Ducas, É.; Ding, S.; Dussault, N.; Landry, P.; Tremblay, T.; Laforce-Lavoie, A.; et al. Major role of IgM in the neutralizing activity of convalescent plasma against SARS-CoV-2. Cell Rep. 2021, 34, 108790. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Gonzalez, J.; Edwards, K.; Mallajosyula, V.; Buzzanco, A.S.; Sherwood, R.; Buffone, C.; Kathale, N.; Providenza, S.; Xie, M.M.; et al. Proinflammatory IgG Fc Structures in Patients with Severe COVID-19. Nat. Immunol. 2021, 22, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Gonzalez, J.C.; Sievers, B.L.; Mallajosyula, V.; Chakraborty, S.; Dubey, M.; Ashraf, U.; Cheng, B.Y.L.; Kathale, N.; Tran, K.Q.T.; et al. Early non-neutralizing, afucosylated antibody responses are associated with COVID-19 severity. Sci. Transl. Med. 2022, 14, abm7853. [Google Scholar] [CrossRef]
- Pattarabanjird, T.; Wilson, J.M.; Erickson, L.D.; Workman, L.J.; Qiao, H.; Ghosheh, Y.; Gulati, R.; Durant, C.; Vallejo, J.; Saigusa, R.; et al. Chemokine Receptor Activation Enhances Memory B Cell Class Switching Linked to IgE Sensitization to Alpha Gal and Cardiovascular Disease. Front. Cardiovasc. Med. 2022, 8, 2139. [Google Scholar] [CrossRef]
- Wasilko, D.J.; Johnson, Z.L.; Ammirati, M.; Che, Y.; Griffor, M.C.; Han, S.; Wu, H. Structural Basis for Chemokine Receptor CCR6 Activation by the Endogenous Protein Ligand CCL20. Nat. Commun. 2020, 11, 3031. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Ip, P.-P.; Liao, F. CCR6 Deficiency Impairs IgA Production and Dysregulates Antimicrobial Peptide Production, Altering the Intestinal Flora. Front. Immunol. 2017, 8, 805. [Google Scholar] [CrossRef]
- Kornek, B.; Leutmezer, F.; Rommer, P.S.; Koblischke, M.; Schneider, L.; Haslacher, H.; Thalhammer, R.; Zimprich, F.; Zulehner, G.; Bsteh, G.; et al. B Cell Depletion and SARS-CoV-2 Vaccine Responses in Neuroimmunologic Patients. Ann. Neurol. 2022, 91, 342–352. [Google Scholar] [CrossRef]
- Altevogt, P.; Sammar, M.; Hüser, L.; Kristiansen, G. Novel Insights into the Function of CD24: A Driving Force in Cancer. Int. J. Cancer 2021, 148, 546–559. [Google Scholar] [CrossRef]
- Parodis, I.; Gatto, M.; Sjöwall, C. B cells in systemic lupus erythematosus: Targets of new therapies and surveillance tools. Front. Med. 2022, 9, 952304. [Google Scholar] [CrossRef]
- Schickel, J.-N.; Glauzy, S.; Ng, Y.-S.; Chamberlain, N.; Massad, C.; Isnardi, I.; Katz, N.; Uzel, G.; Holland, S.M.; Picard, C.; et al. Self-Reactive VH4-34–Expressing IgG B Cells Recognize Commensal Bacteria. J. Exp. Med. 2017, 214, 1991–2003. [Google Scholar] [CrossRef]
- Santa Cruz, A.; Mendes-Frias, A.; Oliveira, A.I.; Dias, L.; Matos, A.R.; Carvalho, A.; Capela, C.; Pedrosa, J.; Castro, A.G.; Silvestre, R. Interleukin-6 Is a Biomarker for the Development of Fatal Severe Acute Respiratory Syndrome Coronavirus 2 Pneumonia. Front. Immunol. 2021, 12, 613422. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep Immune Profiling of COVID-19 Patients Reveals Distinct Immunotypes with Therapeutic Implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A Perspective on Potential Antibody-Dependent Enhancement of SARS-CoV-2. Nature 2020, 584, 353–363. [Google Scholar] [CrossRef]
- Woodruff, M.C.; Ramonell, R.P.; Nguyen, D.C.; Cashman, K.S.; Saini, A.S.; Haddad, N.S.; Ley, A.M.; Kyu, S.; Howell, J.C.; Ozturk, T.; et al. Extrafollicular B Cell Responses Correlate with Neutralizing Antibodies and Morbidity in COVID-19. Nat. Immunol. 2020, 21, 1506–1516. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C.M. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef]
- Busà, R.; Miele, M.; Sorrentino, M.C.; Amico, G.; Timoneri, F.; Miceli, V.; di Bella, M.; Russelli, G.; Gallo, A.; Zito, G.; et al. Long-Term Effectiveness of BNT162b2 Pfizer-BioNTech MRNA-Based Vaccine on B Cell Compartment: Efficient Recall of SARS-CoV-2-Specific Memory B Cells. Int. J. Mol. Sci. 2022, 23, 15046. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Ellebedy, A.H. The Germinal Centre B Cell Response to SARS-CoV-2. Nat. Rev. Immunol. 2022, 22, 7–18. [Google Scholar] [CrossRef]
- Golovkin, A.; Kalinina, O.; Bezrukikh, V.; Aquino, A.; Zaikova, E.; Karonova, T.; Melnik, O.; Vasilieva, E.; Kudryavtsev, I. Imbalanced Immune Response of T-Cell and B-Cell Subsets in Patients with Moderate and Severe COVID-19. Viruses 2021, 13, 1966. [Google Scholar] [CrossRef]
- Wen, C.; Dong, Z.; Wang, Y.; Ye, G.; Ma, Y.; Yi, X.; Zhou, Y.; Li, X.; Zheng, X.; Hou, J.; et al. CTLA4+CD4+CXCR5-FOXP3+ T Cells Associate with Unfavorable Outcome in Patients with Chronic HBV Infection. BMC Immunol. 2023, 24, 3. [Google Scholar] [CrossRef] [PubMed]
- Jo, N.; Hidaka, Y.; Kikuchi, O.; Fukahori, M.; Sawada, T.; Aoki, M.; Yamamoto, M.; Nagao, M.; Morita, S.; Nakajima, T.E.; et al. Impaired CD4+ T Cell Response in Older Adults Is Associated with Reduced Immunogenicity and Reactogenicity of MRNA COVID-19 Vaccination. Nat. Aging 2023, 3, 82–92. [Google Scholar] [CrossRef]
- Kallolimath, S.; Sun, L.; Palt, R.; Stiasny, K.; Mayrhofer, P.; Gruber, C.; Kogelmann, B.; Chen, Q.; Steinkellner, H. Highly Active Engineered IgG3 Antibodies against SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2107249118. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Kallolimath, S.; Palt, R.; Stiasny, K.; Mayrhofer, P.; Maresch, D.; Eidenberger, L.; Steinkellner, H. Increased in Vitro Neutralizing Activity of SARS-CoV-2 IgA1 Dimers Compared to Monomers and IgG. Proc. Natl. Acad. Sci. USA 2021, 118, e2107148118. [Google Scholar] [CrossRef]
- Zervou, F.N.; Louie, P.; Stachel, A.; Zacharioudakis, I.M.; Ortiz-Mendez, Y.; Thomas, K.; Aguero-Rosenfeld, M.E. SARS-CoV-2 Antibodies: IgA Correlates with Severity of Disease in Early COVID-19 Infection. J. Med. Virol. 2021, 93, 5409–5415. [Google Scholar] [CrossRef] [PubMed]
- Fine, N.; Tasevski, N.; McCulloch, C.A.; Tenenbaum, H.C.; Glogauer, M. The Neutrophil: Constant Defender and First Responder. Front. Immunol. 2020, 11, 571085. [Google Scholar] [CrossRef]
- Sachinidis, A.; Garyfallos, A. Double Negative (DN) B cells: A connecting bridge between rheumatic diseases and COVID-19? Mediterr. J. Rheumatol. 2021, 32, 192–199. [Google Scholar] [CrossRef]
- Stewart, A.; Ng, J.C.-F.; Wallis, G.; Tsioligka, V.; Fraternali, F.; Dunn-Walters, D.K. Single-Cell Transcriptomic Analyses Define Distinct Peripheral B Cell Subsets and Discrete Development Pathways. Front. Immunol. 2021, 12, 602539. [Google Scholar] [CrossRef]
- Malone, R.W.; Tisdall, P.; Fremont-Smith, P.; Liu, Y.; Huang, X.-P.; White, K.M.; Miorin, L.; Moreno, E.; Alon, A.; Delaforge, E.; et al. COVID-19: Famotidine, Histamine, Mast Cells, and Mechanisms. Front. Pharmacol. 2021, 12, 633680. [Google Scholar] [CrossRef]
- Iwasaki, N.; Terawaki, S.; Shimizu, K.; Oikawa, D.; Sakamoto, H.; Sunami, K.; Tokunaga, F. Th2 Cells and Macrophages Cooperatively Induce Allergic Inflammation through Histamine Signaling. PLoS ONE 2021, 16, e0248158. [Google Scholar] [CrossRef]
- Kotani, Y.; Sumiyoshi, M.; Sasada, M.; Watanabe, T.; Matsuda, S. Arf1 Facilitates Mast Cell Proliferation via the MTORC1 Pathway. Sci. Rep. 2022, 12, 22297. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Reiding, K.R.; Lin, Y.-H.; van Alphen, F.P.J.; Meijer, A.B.; Heck, A.J.R. Neutrophil Azurophilic Granule Glycoproteins Are Distinctively Decorated by Atypical Pauci- and Phosphomannose Glycans. Commun. Biol. 2021, 4, 1012. [Google Scholar] [CrossRef]
- Xu, D.; Lu, W. Defensins: A Double-Edged Sword in Host Immunity. Front. Immunol. 2020, 11, 764. [Google Scholar] [CrossRef]
- Reusch, N.; De Domenico, E.; Bonaguro, L.; Schulte-Schrepping, J.; Baßler, K.; Schultze, J.L.; Aschenbrenner, A.C. Neutrophils in COVID-19. Front. Immunol. 2021, 12, 652470. [Google Scholar] [CrossRef]
- Duan, M.; Steinfort, D.P.; Smallwood, D.; Hew, M.; Chen, W.; Ernst, M.; Irving, L.B.; Anderson, G.P.; Hibbs, M.L. CD11b Immunophenotyping Identifies Inflammatory Profiles in the Mouse and Human Lungs. Mucosal. Immunol. 2016, 9, 550–563. [Google Scholar] [CrossRef]
- Rothman, J.E. The Gripping Story of Integrins. Cell 2022, 185, 3844–3848. [Google Scholar] [CrossRef]
- Dustin, M.L. Integrins and Their Role in Immune Cell Adhesion. Cell 2019, 177, 499–501. [Google Scholar] [CrossRef]
- De Filippo, K.; Rankin, S.M. CXCR4, the master regulator of neutrophil trafficking in homeostasis and disease. Eur. J. Clin. Investig. 2018, 48, e12949. [Google Scholar] [CrossRef] [Green Version]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 Antagonistically Regulate Neutrophil Trafficking from Murine Bone Marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef]
- Li, F.; Xu, X.; Geng, J.; Wan, X.; Dai, H. The Autocrine CXCR4/CXCL12 Axis Contributes to Lung Fibrosis through Modulation of Lung Fibroblast Activity. Exp. Ther. Med. 2020, 19, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.B.; Zhi, H.; Zhang, X.Y.; Liang, J.W.; He, J.; Su, C.; Xia, W.H.; Zhang, G.X.; Tao, J. Mitochondrial Dysfunction-Mediated Decline in Angiogenic Capacity of Endothelial Progenitor Cells Is Associated with Capillary Rarefaction in Patients with Hypertension via Downregulation of CXCR4/JAK2/SIRT5 Signaling. eBioMedicine 2019, 42, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Jaffar, J.; Griffiths, K.; Oveissi, S.; Duan, M.; Foley, M.; Glaspole, I.; Symons, K.; Organ, L.; Westall, G. CXCR4+ Cells Are Increased in Lung Tissue of Patients with Idiopathic Pulmonary Fibrosis. Respir. Res. 2020, 21, 221. [Google Scholar] [CrossRef] [PubMed]
- Azcutia, V.; Kelm, M.; Luissint, A.C.; Boerner, K.; Flemming, S.; Quiros, M.; Newton, G.; Nusrat, A.; Luscinskas, F.W.; Parkos, C.A. Neutrophil Expressed CD47 Regulates CD11b/CD18-Dependent Neutrophil Transepithelial Migration in the Intestine in Vivo. Mucosal. Immunol. 2021, 14, 331–341. [Google Scholar] [CrossRef]
- Alberca, R.W.; Andrade, M.M.D.S.; Branco, A.C.C.C.; Pietrobon, A.J.; Pereira, N.Z.; Fernandes, I.G.; Oliveira, L.D.M.; Teixeira, F.M.E.; Beserra, D.R.; de Oliveira, E.A.; et al. Frequencies of CD33+CD11b+HLA-DR–CD14–CD66b+ and CD33+CD11b+HLA-DR–CD14+CD66b– Cells in Peripheral Blood as Severity Immune Biomarkers in COVID-19. Front. Med. 2020, 7, 580677. [Google Scholar] [CrossRef]
- Kiaee, F.; Jamaati, H.; Shahi, H.; Roofchayee, N.D.; Varahram, M.; Folkerts, G.; Garssen, J.; Adcock, I.M.; Mortaz, E. Immunophenotype and Function of Circulating Myeloid Derived Suppressor Cells in COVID-19 Patients. Sci. Rep. 2022, 12, 22570. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, X.; Liu, X. NETosis and Neutrophil Extracellular Traps in COVID-19: Immunothrombosis and Beyond. Front. Immunol. 2022, 13, 838011. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.N.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef]
- McLeish, K.R.; Shrestha, R.; Vashishta, A.; Rane, M.J.; Barati, M.T.; Brier, M.E.; Lau, M.G.; Hu, X.; Chen, O.; Wessel, C.R.; et al. Differential Functional Responses of Neutrophil Subsets in Severe COVID-19 Patients. Front. Immunol. 2022, 13, 879686. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Beretta, A.; Cranage, M.; Zipeto, D. Is Cross-Reactive Immunity Triggering COVID-19 Immunopathogenesis? Front. Immunol. 2020, 11, 567710. [Google Scholar] [CrossRef]
- Cabrera, L.E.; Pekkarinen, P.T.; Alander, M.; Nowlan, K.H.A.; Nguyen, N.A.; Jokiranta, S.; Kuivanen, S.; Patjas, A.; Mero, S.; Pakkanen, S.H.; et al. Characterization of Low-Density Granulocytes in COVID-19. PLoS Pathog. 2021, 17, e1009721. [Google Scholar] [CrossRef]
- Guéant, J.; Guéant-Rodriguez, R.; Fromonot, J.; Oussalah, A.; Louis, H.; Chery, C.; Gette, M.; Gleye, S.; Callet, J.; Raso, J.; et al. Elastase and Exacerbation of Neutrophil Innate Immunity Are Involved in Multi-visceral Manifestations of COVID-19. Allergy 2021, 76, 1846–1858. [Google Scholar] [CrossRef]
- Tsou, E.P.S.; Sule, G.; Rubio, M.G.; Amin, M.A.; Zuo, Y.; Knight, J.; Kanthi, Y.; Fox, D. Identification of CD13 as a Potential Cause for SARS-CoV-2-Triggered Hyperinflammation and Thrombosis [Abstract 0069]. Arthritis Rheumatol. 2020, 72, 2. [Google Scholar]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wörsdörfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E.; et al. SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front. Cell Infect. Microbiol. 2021, 11, 701278. [Google Scholar] [CrossRef]
- Schimmel, L.; Chew, K.Y.; Stocks, C.J.; Yordanov, T.E.; Essebier, P.; Kulasinghe, A.; Monkman, J.; Santos Miggiolaro, A.F.R.; Cooper, C.; Noronha, L.; et al. Endothelial Cells Are Not Productively Infected by SARS-CoV-2. Clin. Transl. Immunol. 2021, 10, e1350. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, K.Y.; Huang, Y.; Lui, K.O. Endothelial Contribution to COVID-19: An Update on Mechanisms and Therapeutic Implications. J. Mol. Cell Cardiol. 2022, 164, 69–82. [Google Scholar] [CrossRef]
- Kuchroo, M.; Huang, J.; Wong, P.; Grenier, J.C.; Shung, D.; Tong, A.; Lucas, C.; Klein, J.; Burkhardt, D.B.; Gigante, S.; et al. Multiscale PHATE Identifies Multimodal Signatures of COVID-19. Nat. Biotechnol. 2022, 40, 681–691. [Google Scholar] [CrossRef]
- Frisoni, P.; Neri, M.; D’Errico, S.; Alfieri, L.; Bonuccelli, D.; Cingolani, M.; di Paolo, M.; Gaudio, R.M.; Lestani, M.; Marti, M.; et al. Cytokine Storm and Histopathological Findings in 60 Cases of COVID-19-Related Death: From Viral Load Research to Immunohistochemical Quantification of Major Players IL-1β, IL-6, IL-15 and TNF-α. Forensic. Sci. Med. Pathol. 2022, 18, 4–19. [Google Scholar] [CrossRef]
- Vanderbeke, L.; van Mol, P.; van Herck, Y.; de Smet, F.; Humblet-Baron, S.; Martinod, K.; Antoranz, A.; Arijs, I.; Boeckx, B.; Bosisio, F.M.; et al. Monocyte-Driven Atypical Cytokine Storm and Aberrant Neutrophil Activation as Key Mediators of COVID-19 Disease Severity. Nat. Commun. 2021, 12, 4117. [Google Scholar] [CrossRef]
- Osugi, Y.; Vuckovic, S.; Hart, D.N.J. Myeloid Blood CD11c+ Dendritic Cells and Monocyte-Derived Dendritic Cells Differ in Their Ability to Stimulate T Lymphocytes. Blood 2002, 100, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Ożańska, A.; Szymczak, D.; Rybka, J. Pattern of Human Monocyte Subpopulations in Health and Disease. Scand. J. Immunol. 2020, 92, e12883. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, P.B.; Marcovecchio, P.; Hamers, A.A.J.; Hedrick, C.C. Nonclassical Monocytes in Health and Disease. Annu. Rev. Immunol. 2019, 37, 439–456. [Google Scholar] [CrossRef] [PubMed]
- Burgener, S.S.; Schroder, K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb. Perspect. Biol. 2020, 12, a037028. [Google Scholar] [CrossRef]
- Knoll, R.; Schultze, J.L.; Schulte-Schrepping, J. Monocytes and Macrophages in COVID-19. Front. Immunol. 2021, 12, 720109. [Google Scholar] [CrossRef]
- Maher, A.K.; Burnham, K.L.; Jones, E.M.; Tan, M.M.H.; Saputil, R.C.; Baillon, L.; Selck, C.; Giang, N.; Argüello, R.; Pillay, C.; et al. Transcriptional Reprogramming from Innate Immune Functions to a Pro-Thrombotic Signature by Monocytes in COVID-19. Nat. Commun. 2022, 13, 7947. [Google Scholar] [CrossRef]
- Lee, J.; Tam, H.; Adler, L.; Ilstad-Minnihan, A.; Macaubas, C.; Mellins, E.D. The MHC Class II Antigen Presentation Pathway in Human Monocytes Differs by Subset and Is Regulated by Cytokines. PLoS ONE 2017, 12, e0183594. [Google Scholar] [CrossRef]
- Bardina, S.V.; Michlmayr, D.; Hoffman, K.W.; Obara, C.J.; Sum, J.; Charo, I.F.; Lu, W.; Pletnev, A.G.; Lim, J.K. Differential Roles of Chemokines CCL2 and CCL7 in Monocytosis and Leukocyte Migration during West Nile Virus Infection. J. Immunol. 2015, 195, 4306–4318. [Google Scholar] [CrossRef]
- Persaud, A.T.; Bennett, S.A.; Thaya, L.; Burnie, J.; Guzzo, C. Human Monocytes Store and Secrete Preformed CCL5, Independent of de Novo Protein Synthesis. J. Leukoc. Biol. 2022, 111, 573–583. [Google Scholar] [CrossRef]
- Yang, M.; Liu, Z.; Kang, K.; Yu, K.; Wang, C. Letter to the Editor: CD14 + HLA-DR + Cells in Patients May Be a Biomarker Reflecting the Progression of COVID-19. Viral. Immunol. 2021, 34, 579–581. [Google Scholar] [CrossRef]
- Wack, A. Monocyte and dendritic cell defects in COVID-19. Nat. Cell Biol. 2021, 23, 445–447. [Google Scholar] [CrossRef]
- Palacios, Y.; Ruiz, A.; Ramón-Luing, L.A.; Ocaña-Guzman, R.; Barreto-Rodriguez, O.; Sánchez-Monciváis, A.; Tecuatzi-Cadena, B.; Regalado-García, A.G.; Pineda-Gudiño, R.D.; García-Martínez, A.; et al. Severe COVID-19 Patients Show an Increase in Soluble TNFR1 and ADAM17, with a Relationship to Mortality. Int. J. Mol. Sci. 2021, 22, 8423. [Google Scholar] [CrossRef]
- Neurath, M.F. COVID-19 and Immunomodulation in IBD. Gut 2020, 69, 1335–1342. [Google Scholar] [CrossRef]
- Gómez-Rial, J.; Currás-Tuala, M.J.; Rivero-Calle, I.; Gómez-Carballa, A.; Cebey-López, M.; Rodríguez-Tenreiro, C.; Dacosta-Urbieta, A.; Rivero-Velasco, C.; Rodríguez-Núñez, N.; Trastoy-Pena, R.; et al. Increased Serum Levels of SCD14 and SCD163 Indicate a Preponderant Role for Monocytes in COVID-19 Immunopathology. Front. Immunol. 2020, 11, 560381. [Google Scholar] [CrossRef]
- Patterson, B.K.; Seethamraju, H.; Dhody, K.; Corley, M.J.; Kazempour, K.; Lalezari, J.; Pang, A.P.S.; Sugai, C.; Mahyari, E.; Francisco, E.B.; et al. CCR5 Inhibition in Critical COVID-19 Patients Decreases Inflammatory Cytokines, Increases CD8 T-Cells, and Decreases SARS-CoV2 RNA in Plasma by Day 14. Int. J. Infect. Dis. 2021, 103, 25–32. [Google Scholar] [CrossRef]
- Silva, C.M.S.; Wanderley, C.W.S.; Veras, F.P.; Gonçalves, A.V.; Lima, M.H.F.; Toller-Kawahisa, J.E.; Gomes, G.F.; Nascimento, D.C.; Monteiro, V.V.S.; Paiva, I.M.; et al. Gasdermin-D Activation by SARS-CoV-2 Triggers NET and Mediate COVID-19 Immunopathology. Crit. Care 2022, 26, 206. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Liu, X.; Ding, S.; Liu, P. The Roles of Gasdermin D in Coronavirus Infection and Evasion. Front. Microbiol. 2021, 12, 3482. [Google Scholar] [CrossRef]
- Junqueira, C.; Crespo, Â.; Ranjbar, S.; de Lacerda, L.B.; Lewandrowski, M.; Ingber, J.; Parry, B.; Ravid, S.; Clark, S.; Schrimpf, M.R.; et al. FcγR-Mediated SARS-CoV-2 Infection of Monocytes Activates Inflammation. Nature 2022, 606, 576–584. [Google Scholar] [CrossRef]
- Lage, S.L.; Amaral, E.P.; Hilligan, K.L.; Laidlaw, E.; Rupert, A.; Namasivayan, S.; Rocco, J.; Galindo, F.; Kellogg, A.; Kumar, P.; et al. Persistent Oxidative Stress and Inflammasome Activation in CD14highCD16- Monocytes From COVID-19 Patients. Front. Immunol. 2022, 12, 799558. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A Single-Cell Atlas of the Peripheral Immune Response in Patients with Severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Chilunda, V.; Martinez-Aguado, P.; Xia, L.C.; Cheney, L.; Murphy, A.; Veksler, V.; Ruiz, V.; Calderon, T.M.; Berman, J.W. Transcriptional Changes in CD16+ Monocytes May Contribute to the Pathogenesis of COVID-19. Front. Immunol. 2021, 12, 665773. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, F.R.; Govender, M.; Svanberg, C.; Nordgren, J.; Waller, H.; Nilsdotter-Augustinsson, Å.; Henningsson, A.J.; Hagbom, M.; Sjöwall, J.; Nyström, S.; et al. Major Alterations to Monocyte and Dendritic Cell Subsets Lasting More than 6 Months after Hospitalization for COVID-19. Front. Immunol. 2023, 13, 1082912. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic Reprogramming in Macrophages and Dendritic Cells in Innate Immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Kosyreva, A.; Dzhalilova, D.; Lokhonina, A.; Vishnyakova, P.; Fatkhudinov, T. The Role of Macrophages in the Pathogenesis of SARS-CoV-2-Associated Acute Respiratory Distress Syndrome. Front. Immunol. 2021, 12, 682871. [Google Scholar] [CrossRef]
- Lv, J.; Wang, Z.; Qu, Y.; Zhu, H.; Zhu, Q.; Tong, W.; Bao, L.; Lv, Q.; Cong, J.; Li, D.; et al. Distinct Uptake, Amplification, and Release of SARS-CoV-2 by M1 and M2 Alveolar Macrophages. Cell Discov. 2021, 7, 24. [Google Scholar] [CrossRef]
- Mitsi, E.; Kamng’ona, R.; Rylance, J.; Solórzano, C.; Reiné, J.J.; Mwandumba, H.C.; Ferreira, D.M.; Jambo, K.C. Human Alveolar Macrophages Predominately Express Combined Classical M1 and M2 Surface Markers in Steady State. Respir. Res. 2018, 19, 66. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Lurier, E.B.; Dalton, D.; Dampier, W.; Raman, P.; Nassiri, S.; Ferraro, N.M.; Rajagopalan, R.; Sarmady, M.; Spiller, K.L. Transcriptome Analysis of IL-10-Stimulated (M2c) Macrophages by next-Generation Sequencing. Immunobiology 2017, 222, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of Macrophage Development and Function in Peripheral Tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef]
- Lian, Q.; Zhang, K.; Zhang, Z.; Duan, F.; Guo, L.; Luo, W.; Mok, B.W.-Y.; Thakur, A.; Ke, X.; Motallebnejad, P.; et al. Differential Effects of Macrophage Subtypes on SARS-CoV-2 Infection in a Human Pluripotent Stem Cell-Derived Model. Nat. Commun. 2022, 13, 2028. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, X.; Han, G.; Shao, B.; Lin, L.; Jiang, S. Altered M1/M2 Polarization of Alveolar Macrophages Is Involved in the Pathological Responses of Acute Silicosis in Rats in Vivo. Toxicol. Ind. Health 2022, 38, 810–818. [Google Scholar] [CrossRef]
- Kolliniati, O.; Ieronymaki, E.; Vergadi, E.; Tsatsanis, C. Metabolic Regulation of Macrophage Activation. J. Innate Immun. 2022, 14, 51–68. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic Reprogramming in Macrophage Responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Koo, S.J.; Garg, N.J. Metabolic programming of macrophage functions and pathogens control. Redox Biol. 2019, 24, 101198. [Google Scholar] [CrossRef]
- Vyavahare, S.; Kumar, S.; Cantu, N.; Kolhe, R.; Bollag, W.B.; McGee-Lawrence, M.E.; Hill, W.D.; Hamrick, M.W.; Isales, C.M.; Fulzele, S. Tryptophan-Kynurenine Pathway in COVID-19-Dependent Musculoskeletal Pathology: A Minireview. Mediat. Inflamm. 2021, 2021, 2911578. [Google Scholar] [CrossRef]
- Guo, L.; Schurink, B.; Roos, E.; Nossent, E.J.; Duitman, J.W.; Vlaar, A.P.; Valk, P.; Vaz, F.M.; Yeh, S.; Geeraerts, Z.; et al. Indoleamine 2,3-dioxygenase (IDO)-1 and IDO-2 Activity and Severe Course of COVID-19. J. Pathol. 2022, 256, 256–261. [Google Scholar] [CrossRef]
- Wang, X.-F.; Wang, H.-S.; Wang, H.; Zhang, F.; Wang, K.-F.; Guo, Q.; Zhang, G.; Cai, S.-H.; Du, J. The Role of Indoleamine 2,3-Dioxygenase (IDO) in Immune Tolerance: Focus on Macrophage Polarization of THP-1 Cells. Cell Immunol. 2014, 289, 42–48. [Google Scholar] [CrossRef]
- Yu, S.; Ge, H.; Li, S.; Qiu, H.-J. Modulation of Macrophage Polarization by Viruses: Turning Off/On Host Antiviral Responses. Front. Microbiol. 2022, 13, 130. [Google Scholar] [CrossRef]
- Thiriot, J.D.; Martinez-Martinez, Y.B.; Endsley, J.J.; Torres, A.G. Hacking the Host: Exploitation of Macrophage Polarization by Intracellular Bacterial Pathogens. Pathog. Dis. 2020, 78, ftaa009. [Google Scholar] [CrossRef] [PubMed]
- Soroosh, P.; Doherty, T.A.; Duan, W.; Mehta, A.K.; Choi, H.; Adams, Y.F.; Mikulski, Z.; Khorram, N.; Rosenthal, P.; Broide, D.H.; et al. Lung-Resident Tissue Macrophages Generate Foxp3+ Regulatory T Cells and Promote Airway Tolerance. J. Exp. Med. 2013, 210, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Guan, T.; Zhou, X.; Zhou, W.; Lin, H. Regulatory T Cell and Macrophage Crosstalk in Acute Lung Injury: Future Perspectives. Cell Death Discov. 2023, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Shirai, M.; Asada, K.; Yasui, H.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; et al. Macrophage mannose receptor, CD206, predict prognosis in patients with pulmonary tuberculosis. Sci. Rep. 2018, 8, 13129. [Google Scholar] [CrossRef]
- Moghaddami, M.; Mayrhofer, G.; Cleland, L.G. MHC Class II Compartment, Endocytosis and Phagocytic Activity of Macrophages and Putative Dendritic Cells Isolated from Normal Tissues Rich in Synovium. Int. Immunol. 2005, 17, 1117–1130. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/Macrosialin: Not Just a Histochemical Marker. Lab. Investig. 2017, 97, 4–13. [Google Scholar] [CrossRef]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 Infection Triggers Profibrotic Macrophage Responses and Lung Fibrosis. Cell 2021, 184, 6243–6261.e27. [Google Scholar] [CrossRef]
- Sur, S.; Steele, R.; Isbell, T.S.; Ray, R.; Ray, R.B. Circulatory Exosomes from COVID-19 Patients Trigger NLRP3 Inflammasome in Endothelial Cells. mBio 2022, 13, e00951-22. [Google Scholar] [CrossRef]
- Shao, X.; Wu, B.; Cheng, L.; Li, F.; Zhan, Y.; Liu, C.; Ji, L.; Min, Z.; Ke, Y.; Sun, L.; et al. Distinct Alterations of CD68+CD163+ M2-like Macrophages and Myeloid-Derived Suppressor Cells in Newly Diagnosed Primary Immune Thrombocytopenia with or without CR after High-Dose Dexamethasone Treatment. J. Transl. Med. 2018, 16, 48. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Hijdra, D.; Vorselaars, A.D.M.; Grutters, J.C.; Claessen, A.M.E.; Rijkers, G.T. Phenotypic Characterization of Human Intermediate Monocytes. Front. Immunol. 2013, 4, 339. [Google Scholar] [CrossRef]
- Geurdes, H. Histamine Antagonists to Temper the Cytokine Overproduction in Gastrointestinal Cells Infected by SARS-CoV-2. Virol. Mycol. 2020, 9, 2. [Google Scholar] [CrossRef]
- Matic, S.; Popovic, S.; Djurdjevic, P.; Todorovic, D.; Djordjevic, N.; Mijailovic, Z.; Sazdanovic, P.; Milovanovic, D.; Ruzic Zecevic, D.; Petrovic, M.; et al. SARS-CoV-2 Infection Induces Mixed M1/M2 Phenotype in Circulating Monocytes and Alterations in Both Dendritic Cell and Monocyte Subsets. PLoS ONE 2020, 15, e0241097. [Google Scholar] [CrossRef]
- Blagov, A.V.; Markin, A.M.; Bogatyreva, A.I.; Tolstik, T.V.; Sukhorukov, V.N.; Orekhov, A.N. The Role of Macrophages in the Pathogenesis of Atherosclerosis. Cells 2023, 12, 522. [Google Scholar] [CrossRef]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting Edge: TREM-2 Attenuates Macrophage Activation. J. Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef]
- Upadhyay, A.A.; Hoang, T.N.; Pino, M.; Boddapati, A.K.; Viox, E.G.; Lee, M.Y.H.; Corry, J.; Strongin, Z.; Cowan, D.A.; Beagle, E.N.; et al. TREM2+ and Interstitial Macrophages Orchestrate Airway Inflammation in SARS-CoV-2 Infection in Rhesus Macaques. bioRxiv, 2021; preprint. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, M.; Yin, H.; Ming, S.; Li, X.; Jiang, G.; Liu, Y.; Wang, P.; Zhou, G.; Liu, L.; et al. TREM-2 Is a Sensor and Activator of T Cell Response in SARS-CoV-2 Infection. Sci. Adv. 2021, 7, eabi6802. [Google Scholar] [CrossRef]
- Yalcinkaya, M.; Liu, W.; Islam, M.N.; Kotini, A.G.; Gusarova, G.A.; Fidler, T.P.; Papapetrou, E.P.; Bhattacharya, J.; Wang, N.; Tall, A.R. Modulation of the NLRP3 Inflammasome by Sars-CoV-2 Envelope Protein. Sci. Rep. 2021, 11, 24432. [Google Scholar] [CrossRef]
- Sefik, E.; Qu, R.; Junqueira, C.; Kaffe, E.; Mirza, H.; Zhao, J.; Brewer, J.R.; Han, A.; Steach, H.R.; Israelow, B.; et al. Inflammasome Activation in Infected Macrophages Drives COVID-19 Pathology. Nature 2022, 606, 585–593. [Google Scholar] [CrossRef]
- Garcia-Revilla, J.; Deierborg, T.; Venero, J.L.; Boza-Serrano, A. Hyperinflammation and Fibrosis in Severe COVID-19 Patients: Galectin-3, a Target Molecule to Consider. Front. Immunol. 2020, 11, 2069. [Google Scholar] [CrossRef]
- Grandvuillemin, A.; Rocher, F.; Valnet-Rabier, M.B.; Drici, M.-D.; Dautriche, A. Pharmacovigilance Follow-up of Patients in the Context of the COVID 19 Pandemic. Therapies, 2023; in Press, Journal Pre-proof. [Google Scholar] [CrossRef]
- Kushner, P.; McCarberg, B.H.; Grange, L.; Kolosov, A.; Haveric, A.L.; Zucal, V.; Petruschke, R.; Bissonnette, S. The Use of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) in COVID-19. NPJ Prim. Care Respir. Med. 2022, 32, 35. [Google Scholar] [CrossRef] [PubMed]
- Kragholm, K.; Torp-Pedersen, C.; Fosbol, E. Non-Steroidal Anti-Inflammatory Drug Use in COVID-19. Lancet Rheumatol. 2021, 3, e465–e466. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, W.; Ding, G.; Sun, L.; Chen, G.; Cao, X. DENDRITIC CELLS SUPPORT HEMATOPOIESIS OF BONE MARROW CELLS1. Transplantation 2001, 72, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Yokota, A.; Takeuchi, H.; Maeda, N.; Ohoka, Y.; Kato, C.; Song, S.-Y.; Iwata, M. GM-CSF and IL-4 Synergistically Trigger Dendritic Cells to Acquire Retinoic Acid-Producing Capacity. Int. Immunol. 2009, 21, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Usero, L.; Miralles, L.; Esteban, I.; Pastor-Quiñones, C.; Maleno, M.J.; Leal, L.; García, F.; Plana, M. Feasibility of Using Monocyte-Derived Dendritic Cells Obtained from Cryopreserved Cells for DC-Based Vaccines. J. Immunol. Methods 2021, 498, 113133. [Google Scholar] [CrossRef]
- Helft, J.; Manicassamy, B.; Guermonprez, P.; Hashimoto, D.; Silvin, A.; Agudo, J.; Brown, B.D.; Schmolke, M.; Miller, J.C.; Leboeuf, M.; et al. Cross-Presenting CD103+ Dendritic Cells Are Protected from Influenza Virus Infection. J. Clin. Investig. 2012, 122, 4037–4047. [Google Scholar] [CrossRef]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- Kim, M.K.; Kim, J. Properties of Immature and Mature Dendritic Cells: Phenotype, Morphology, Phagocytosis, and Migration. RSC Adv. 2019, 9, 11230–11238. [Google Scholar] [CrossRef]
- Lechmann, M.; Berchtold, S.; Steinkasserer, A.; Hauber, J. CD83 on Dendritic Cells: More than Just a Marker for Maturation. Trends Immunol. 2002, 23, 273–275. [Google Scholar] [CrossRef]
- Grosche, L.; Knippertz, I.; König, C.; Royzman, D.; Wild, A.B.; Zinser, E.; Sticht, H.; Muller, Y.A.; Steinkasserer, A.; Lechmann, M. The CD83 Molecule–An Important Immune Checkpoint. Front. Immunol. 2020, 11, 721. [Google Scholar] [CrossRef]
- Dudek, A.M.; Martin, S.; Garg, A.D.; Agostinis, P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a DC-Cancer Cells Interface That Augments Anticancer Immunity. Front. Immunol. 2013, 4, 438. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Burgueño-Bucio, E.; Xu, S.; Das, S.; Olguin-Alor, R.; Elmets, C.A.; Athar, M.; Raman, C.; Soldevila, G.; Xu, H. CD5 on Dendritic Cells Regulates CD4+ and CD8+ T Cell Activation and Induction of Immune Responses. PLoS ONE 2019, 14, e0222301. [Google Scholar] [CrossRef]
- Tran Janco, J.M.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-Infiltrating Dendritic Cells in Cancer Pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef]
- Cueto, F.J.; Sancho, D. The Flt3L/Flt3 Axis in Dendritic Cell Biology and Cancer Immunotherapy. Cancers 2021, 13, 1525. [Google Scholar] [CrossRef]
- Gujer, C.; Murer, A.; Müller, A.; Vanoaica, D.; Sutter, K.; Jacque, E.; Fournier, N.; Kalchschmidt, J.; Zbinden, A.; Capaul, R.; et al. Plasmacytoid Dendritic Cells Respond to Epstein-Barr Virus Infection with a Distinct Type I Interferon Subtype Profile. Blood Adv. 2019, 3, 1129–1144. [Google Scholar] [CrossRef]
- Perez-Zsolt, D.; Martinez-Picado, J.; Izquierdo-Useros, N. When Dendritic Cells Go Viral: The Role of Siglec-1 in Host Defense and Dissemination of Enveloped Viruses. Viruses 2019, 12, 8. [Google Scholar] [CrossRef]
- Turesson, C. Endothelial Expression of MHC Class II Molecules in Autoimmune Disease. Curr. Pharm. Des. 2004, 10, 129–143. [Google Scholar] [CrossRef]
- Amersfoort, J.; Eelen, G.; Carmeliet, P. Immunomodulation by Endothelial Cells—Partnering up with the Immune System? Nat. Rev. Immunol. 2022, 22, 576–588. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, W.; Zhao, M.; Fu, L.; Liu, L.; Wu, J.; Luo, S.; Wang, L.; Wang, Z.; Lin, L.; et al. T Cell Receptor β Repertoires as Novel Diagnostic Markers for Systemic Lupus Erythematosus and Rheumatoid Arthritis. Ann. Rheum. Dis. 2019, 78, 1070–1078. [Google Scholar] [CrossRef]
- van Lummel, M.; van Veelen, P.A.; Zaldumbide, A.; de Ru, A.; Janssen, G.M.C.; Moustakas, A.K.; Papadopoulos, G.K.; Drijfhout, J.W.; Roep, B.O.; Koning, F. Type 1 Diabetes-Associated HLA-DQ8 Transdimer Accommodates a Unique Peptide Repertoire. J. Biol. Chem. 2012, 287, 9514–9524. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, S.; Zhou, C.; Wu, J.; Jiao, Y.; Lin, L.; Lu, X.; Yang, B.; Zhang, W.; Xiao, X.; et al. Comprehensive TCR Repertoire Analysis of CD4+ T-Cell Subsets in Rheumatoid Arthritis. J. Autoimmun. 2020, 109, 102432. [Google Scholar] [CrossRef] [PubMed]
- Wordsworth, B.P.; Lanchbury, J.S.; Sakkas, L.I.; Welsh, K.I.; Panayi, G.S.; Bell, J.I. HLA-DR4 Subtype Frequencies in Rheumatoid Arthritis Indicate That DRB1 Is the Major Susceptibility Locus within the HLA Class II Region. Proc. Natl. Acad. Sci. USA 1989, 86, 10049–10053. [Google Scholar] [CrossRef] [PubMed]
- Cianciotti, B.C.; Ruggiero, E.; Campochiaro, C.; Oliveira, G.; Magnani, Z.I.; Baldini, M.; Doglio, M.; Tassara, M.; Manfredi, A.A.; Baldissera, E.; et al. CD4+ Memory Stem T Cells Recognizing Citrullinated Epitopes Are Expanded in Patients With Rheumatoid Arthritis and Sensitive to Tumor Necrosis Factor Blockade. Arthritis Rheumatol. 2020, 72, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Geginat, J.; Vasco, M.; Gerosa, M.; Tas, S.W.; Pagani, M.; Grassi, F.; Flavell, R.A.; Meroni, P.; Abrignani, S. IL-10 Producing Regulatory and Helper T-Cells in Systemic Lupus Erythematosus. Semin. Immunol. 2019, 44, 101330. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Li, J.-L.; Wang, D.-G.; Zhou, D. Targeting IL-10 in Auto-Immune Diseases. Cell Biochem. Biophys. 2014, 70, 37–49. [Google Scholar] [CrossRef]
- Kheiri, F.; Rostami-Nejad, M.; Amani, D.; Sadeghi, A.; Moradi, A.; Aghamohammadi, E.; Sahebkar, A.; Zali, M.R. Expression of Tolerogenic Dendritic Cells in the Small Intestinal Tissue of Patients with Celiac Disease. Heliyon 2022, 8, e12273. [Google Scholar] [CrossRef]
- Mbongue, J.C.; Nieves, H.A.; Torrez, T.W.; Langridge, W.H.R. The Role of Dendritic Cell Maturation in the Induction of Insulin-Dependent Diabetes Mellitus. Front. Immunol. 2017, 8, 327. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of CDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Chong, S.Z.; Wong, K.L.; Lin, G.; Yang, C.M.; Wong, S.-C.; Angeli, V.; MacAry, P.A.; Kemeny, D.M. Human CD8+ T Cells Drive Th1 Responses through the Differentiation of TNF/INOS-Producing Dendritic Cells. Eur. J. Immunol. 2011, 41, 1639–1651. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 Axis for Immune Activation–A Target for Novel Cancer Therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Croxford, A.L.; Kulig, P.; Becher, B. IL-12-and IL-23 in Health and Disease. Cytokine Growth Factor Rev. 2014, 25, 415–421. [Google Scholar] [CrossRef]
- Yin, X.; Yu, H.; Jin, X.; Li, J.; Guo, H.; Shi, Q.; Yin, Z.; Xu, Y.; Wang, X.; Liu, R.; et al. Human Blood CD1c+ Dendritic Cells Encompass CD5high and CD5low Subsets That Differ Significantly in Phenotype, Gene Expression, and Functions. J. Immunol. 2017, 198, 1553–1564. [Google Scholar] [CrossRef]
- Megjugorac, N.J.; Young, H.A.; Amrute, S.B.; Olshalsky, S.L.; Fitzgerald-Bocarsly, P. Virally Stimulated Plasmacytoid Dendritic Cells Produce Chemokines and Induce Migration of T and NK Cells. J. Leukoc. Biol. 2004, 75, 504–514. [Google Scholar] [CrossRef]
- Tzelepis, F.; Verway, M.; Daoud, J.; Gillard, J.; Hassani-Ardakani, K.; Dunn, J.; Downey, J.; Gentile, M.E.; Jaworska, J.; Sanchez, A.M.J.; et al. Annexin1 Regulates DC Efferocytosis and Cross-Presentation during Mycobacterium Tuberculosis Infection. J. Clin. Investig. 2015, 125, 752–768. [Google Scholar] [CrossRef]
- Cytlak, U.; Resteu, A.; Pagan, S.; Green, K.; Milne, P.; Maisuria, S.; McDonald, D.; Hulme, G.; Filby, A.; Carpenter, B.; et al. Differential IRF8 Transcription Factor Requirement Defines Two Pathways of Dendritic Cell Development in Humans. Immunity 2020, 53, 353–370.e8, PMCID:PMC7798577. [Google Scholar] [CrossRef] [PubMed]
- Psarras, A.; Antanaviciute, A.; Alase, A.; Carr, I.; Wittmann, M.; Emery, P.; Tsokos, G.C.; Vital, E.M. TNF-α Regulates Human Plasmacytoid Dendritic Cells by Suppressing IFN-α Production and Enhancing T Cell Activation. J. Immunol. 2021, 206, 785–796. [Google Scholar] [CrossRef]
- Stephenson, E.; Reynolds, G.; Botting, R.A.; Calero-Nieto, F.J.; Morgan, M.D.; Tuong, Z.K.; Bach, K.; Sungnak, W.; Worlock, K.B.; Yoshida, M.; et al. Single-Cell Multi-Omics Analysis of the Immune Response in COVID-19. Nat. Med. 2021, 27, 904–916. [Google Scholar] [CrossRef]
- Pérez-Gómez, A.; Vitallé, J.; Gasca-Capote, C.; Gutierrez-Valencia, A.; Trujillo-Rodriguez, M.; Serna-Gallego, A.; Muñoz-Muela, E.; Jimenez-Leon, M.D.L.R.; Rafii-El-Idrissi Benhnia, M.; Rivas-Jeremias, I.; et al. Dendritic Cell Deficiencies Persist Seven Months after SARS-CoV-2 Infection. Cell Mol. Immunol. 2021, 18, 2128–2139. [Google Scholar] [CrossRef]
- Huang, J.J.; Gaines, S.B.; Amezcua, M.L.; Lubell, T.R.; Dayan, P.S.; Dale, M.; Boneparth, A.D.; Hicar, M.D.; Winchester, R.; Gorelik, M. Upregulation of Type 1 Conventional Dendritic Cells Implicates Antigen Cross-Presentation in Multisystem Inflammatory Syndrome. J. Allergy Clin. Immunol. 2022, 149, 912–922. [Google Scholar] [CrossRef]
- Borcherding, L.; Teksen, A.S.; Grosser, B.; Schaller, T.; Hirschbühl, K.; Claus, R.; Spring, O.; Wittmann, M.; Römmele, C.; Sipos, É.; et al. Impaired Dendritic Cell Homing in COVID-19. Front. Med. 2021, 8, 761372. [Google Scholar] [CrossRef]
- Cai, G.; Du, M.; Bossé, Y.; Albrecht, H.; Qin, F.; Luo, X.; Androulakis, X.M.; Cheng, C.; Nagarkatti, M.; Nagarkatti, P.; et al. SARS-CoV-2 Impairs Dendritic Cells and Regulates DC-SIGN Gene Expression in Tissues. Int. J. Mol. Sci. 2021, 22, 9228. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, C.-A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; van den Hoogen, L.L.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 2019, 51, 573–589.e8. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, L.; Protti, G.; Facchini, F.A.; Valache, M.; Mingozzi, F.; Ranzani, V.; Putignano, A.R.; Salviati, L.; Bevilacqua, V.; Curti, S.; et al. Maturation Signatures of Conventional Dendritic Cell Subtypes in COVID-19 Suggest Direct Viral Sensing. Eur. J. Immunol. 2022, 52, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Tjan, L.H.; Furukawa, K.; Nagano, T.; Kiriu, T.; Nishimura, M.; Arii, J.; Hino, Y.; Iwata, S.; Nishimura, Y.; Mori, Y. Early Differences in Cytokine Production by Severity of Coronavirus Disease 2019. J. Infect. Dis. 2021, 223, 1145–1149. [Google Scholar] [CrossRef]
- Moll-Bernardes, R.; de Sousa, A.S.; Macedo, A.V.S.; Lopes, R.D.; Vera, N.; Maia, L.C.R.; Feldman, A.; Arruda, G.D.A.S.; Castro, M.J.C.; Pimentel-Coelho, P.M.; et al. IL-10 and IL-12 (P70) Levels Predict the Risk of Covid-19 Progression in Hypertensive Patients: Insights From the BRACE-CORONA Trial. Front. Cardiovasc. Med. 2021, 8, 702507. [Google Scholar] [CrossRef]
- Niles, M.A.; Gogesch, P.; Kronhart, S.; Ortega Iannazzo, S.; Kochs, G.; Waibler, Z.; Anzaghe, M. Macrophages and Dendritic Cells Are Not the Major Source of Pro-Inflammatory Cytokines Upon SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 647824. [Google Scholar] [CrossRef]
- Lévy, Y.; Wiedemann, A.; Hejblum, B.P.; Durand, M.; Lefebvre, C.; Surénaud, M.; Lacabaratz, C.; Perreau, M.; Foucat, E.; Déchenaud, M.; et al. CD177, a Specific Marker of Neutrophil Activation, Is Associated with Coronavirus Disease 2019 Severity and Death. iScience 2021, 24, 102711. [Google Scholar] [CrossRef]
- Langton, D.J.; Bourke, S.C.; Lie, B.A.; Reiff, G.; Natu, S.; Darlay, R.; Burn, J.; Echevarria, C. The Influence of HLA Genotype on the Severity of COVID-19 Infection. HLA 2021, 98, 14–22. [Google Scholar] [CrossRef]
- Nagler, A.; Lanier, L.L.; Cwirla, S.; Phillips, J.H. Comparative studies of human FcRIII-positive and negative natural killer cells. J. Immunol. 1989, 143, 3183–3191. [Google Scholar] [CrossRef]
- Poli, A.; Michel, T.; Thérésine, M.; Andrès, E.; Hentges, F.; Zimmer, J. CD56 Bright Natural Killer (NK) Cells: An Important NK Cell Subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef]
- de Andrade, L.F.; Lu, Y.; Luoma, A.; Ito, Y.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Yuan, G.-C.; Wucherpfennig, K.W. Discovery of Specialized NK Cell Populations Infiltrating Human Melanoma Metastases. JCI Insight 2019, 4, e133103. [Google Scholar] [CrossRef]
- Lord, S.J.; Rajotte, R.V.; Korbutt, G.S.; Bleackley, R.C. Granzyme B: A Natural Born Killer. Immunol. Rev. 2003, 193, 31–38. [Google Scholar] [CrossRef]
- Prager, I.; Liesche, C.; van Ooijen, H.; Urlaub, D.; Verron, Q.; Sandström, N.; Fasbender, F.; Claus, M.; Eils, R.; Beaudouin, J.; et al. NK Cells Switch from Granzyme B to Death Receptor–Mediated Cytotoxicity during Serial Killing. J. Exp. Med. 2019, 216, 2113–2127. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Dunstone, M.A.; Baran, K.; Whisstock, J.C.; Trapani, J.A. Perforin: Structure, Function, and Role in Human Immunopathology. Immunol. Rev. 2010, 235, 35–54. [Google Scholar] [CrossRef]
- Feng, Y.; Daley-Bauer, L.P.; Mocarski, E.S. Caspase-8-Dependent Control of NK- and T Cell Responses during Cytomegalovirus Infection. Med. Microbiol. Immunol. 2019, 208, 555–571. [Google Scholar] [CrossRef]
- Erokhina, S.A.; Streltsova, M.A.; Kanevskiy, L.M.; Telford, W.G.; Sapozhnikov, A.M.; Kovalenko, E.I. HLA-DR + NK Cells Are Mostly Characterized by Less Mature Phenotype and High Functional Activity. Immunol. Cell Biol. 2018, 96, 212–228. [Google Scholar] [CrossRef]
- Erokhina, S.A.; Streltsova, M.A.; Kanevskiy, L.M.; Grechikhina, M.V.; Sapozhnikov, A.M.; Kovalenko, E.I. HLA-DR-expressing NK Cells: Effective Killers Suspected for Antigen Presentation. J. Leukoc. Biol. 2021, 109, 327–337. [Google Scholar] [CrossRef]
- Pahl, J.H.W.; Koch, J.; Götz, J.-J.; Arnold, A.; Reusch, U.; Gantke, T.; Rajkovic, E.; Treder, M.; Cerwenka, A. CD16A Activation of NK Cells Promotes NK Cell Proliferation and Memory-Like Cytotoxicity against Cancer Cells. Cancer Immunol. Res. 2018, 6, 517–527. [Google Scholar] [CrossRef]
- Maucourant, C.; Filipovic, I.; Ponzetta, A.; Aleman, S.; Cornillet, M.; Hertwig, L.; Strunz, B.; Lentini, A.; Reinius, B.; Brownlie, D.; et al. Natural Killer Cell Immunotypes Related to COVID-19 Disease Severity. Sci. Immunol. 2020, 5, eabd6832. [Google Scholar] [CrossRef]
- Bulygin, A.S.; Khantakova, J.N.; Shkaruba, N.S.; Shiku, H.; Sennikov, S.S. The Role of Metabolism on Regulatory T Cell Development and Its Impact in Tumor and Transplantation Immunity. Front. Immunol. 2022, 13, 1016670. [Google Scholar] [CrossRef]
- Srivastava, R.; Dhanushkodi, N.; Prakash, S.; Coulon, P.G.; Vahed, H.; Zayou, L.; Quadiri, A.; BenMohamed, L. High Frequencies of Phenotypically and Functionally Senescent and Exhausted CD56sup/SupCD57sup/SupPD-1sup/Sup Natural Killer Cells, SARS-CoV-2-Specific Memory CD4sup/Sup and CD8sup/Sup T Cells Associated with Severe Disease in Unvaccinated COVID-19 Patients. bioXriv, 2022; pre-print. [Google Scholar] [CrossRef]
- Xie, M.; Yunis, J.; Yao, Y.; Shi, J.; Yang, Y.; Zhou, P.; Liang, K.; Wan, Y.; Mehdi, A.; Chen, Z.; et al. High Levels of Soluble CD25 in COVID-19 Severity Suggest a Divergence between Anti-viral and Pro-inflammatory T-cell Responses. Clin. Transl. Immunol. 2021, 10, e1251. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Wood, J.; Jaycox, J.; Lu, P.; Dhodapkar, R.M.; Gehlhausen, J.R.; Tabachnikova, A.; Tabacof, L.; Malik, A.A.; Kamath, K.; et al. Distinguishing Features of Long COVID Identified through Immune Profiling. medRxiv, 2022; pre-print. [Google Scholar] [CrossRef]
- Bi, J. NK Cell Dysfunction in Patients with COVID-19. Cell. Mol. Immunol. 2022, 19, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Kokuina, E.; Breff-Fonseca, M.C.; Villegas-Valverde, C.A.; Mora-Díaz, I. Normal Values of T, B and NK Lymphocyte Subpopulations in Peripheral Blood of Healthy Cuban Adults. MEDICC Rev. 2019, 21, 16–21. [Google Scholar] [CrossRef]
- Song, M.; Graubard, B.I.; Rabkin, C.S.; Engels, E.A. Neutrophil-to-Lymphocyte Ratio and Mortality in the United States General Population. Sci. Rep. 2021, 11, 464. [Google Scholar] [CrossRef]
- Bernard, A.; Boumsell, L.; Hill, C. Joint Report of the First International Workshop on Human Leucocyte Differentiation Antigens by the Investigators of the Participating Laboratories. In Leucocyte Typing; Springer: Berlin/Heidelberg, Germany, 1984; pp. 9–142. [Google Scholar]
- Jarjour, N.N.; Masopust, D.; Jameson, S.C. T Cell Memory: Understanding COVID-19. Immunity 2021, 54, 14–18. [Google Scholar] [CrossRef]
- Wu, Z.; Zheng, Y.; Sheng, J.; Han, Y.; Yang, Y.; Pan, H.; Yao, J. CD3+CD4-CD8- (Double-Negative) T Cells in Inflammation, Immune Disorders and Cancer. Front. Immunol. 2022, 13, 816005. [Google Scholar] [CrossRef]
- Frumento, G.; Verma, K.; Croft, W.; White, A.; Zuo, J.; Nagy, Z.; Kissane, S.; Anderson, G.; Moss, P.; Chen, F.E. Homeostatic Cytokines Drive Epigenetic Reprogramming of Activated T Cells into a “Naive-Memory” Phenotype. iScience 2020, 23, 100989. [Google Scholar] [CrossRef]
- He, Q.; Jiang, X.; Zhou, X.; Weng, J. Targeting Cancers through TCR-Peptide/MHC Interactions. J. Hematol. Oncol. 2019, 12, 139. [Google Scholar] [CrossRef]
- Quinti, I.; Lougaris, V.; Milito, C.; Cinetto, F.; Pecoraro, A.; Mezzaroma, I.; Mastroianni, C.M.; Turriziani, O.; Bondioni, M.P.; Filippini, M.; et al. A Possible Role for B Cells in COVID-19? Lesson from Patients with Agammaglobulinemia. J. Allergy Clin. Immunol. 2020, 146, 211–213.e4. [Google Scholar] [CrossRef]
- Soresina, A.; Moratto, D.; Chiarini, M.; Paolillo, C.; Baresi, G.; Focà, E.; Bezzi, M.; Baronio, B.; Giacomelli, M.; Badolato, R. Two X-linked Agammaglobulinemia Patients Develop Pneumonia as COVID-19 Manifestation but Recover. Pediatr. Allergy Immunol. 2020, 31, 565–569. [Google Scholar] [CrossRef]
- Devassikutty, F.M.; Jain, A.; Edavazhippurath, A.; Joseph, M.C.; Peedikayil, M.M.T.; Scaria, V.; Sandhya, P.; Govindaraj, G.M. X-Linked Agammaglobulinemia and COVID-19: Two Case Reports and Review of Literature. Pediatr. Allergy Immunol. Pulmonol. 2021, 34, 115–118. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and Cross-Reactive SARS-CoV-2 T Cell Epitopes in Unexposed Humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. SARS-CoV-2-Reactive T Cells in Healthy Donors and Patients with COVID-19. Nature 2020, 587, 270–274. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Meckiff, B.J.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4+ T Cells in COVID-19. Cell 2020, 183, 1340–1353.e16. [Google Scholar] [CrossRef]
- Xu, Q.; Milanez-Almeida, P.; Martins, A.J.; Radtke, A.J.; Hoehn, K.B.; Oguz, C.; Chen, J.; Liu, C.; Tang, J.; Grubbs, G.; et al. Adaptive immune responses to SARS-CoV-2 persist in the pharyngeal lymphoid tissue of children. Nat. Immunol. 2023, 24, 186–199. [Google Scholar] [CrossRef]
- Choi, S.J.; Kim, D.U.; Noh, J.Y.; Kim, S.; Park, S.H.; Jeong, H.W.; Shin, E.C. T cell epitopes in SARS-CoV-2 proteins are substantially conserved in the Omicron variant. Cell. Mol. Immunol. 2022, 19, 447–448. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef]
- Nelde, A.; Bilich, T.; Heitmann, J.S.; Maringer, Y.; Salih, H.R.; Roerden, M.; Lübke, M.; Bauer, J.; Rieth, J.; Wacker, M.; et al. SARS-CoV-2-Derived Peptides Define Heterologous and COVID-19-Induced T Cell Recognition. Nat. Immunol. 2021, 22, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Bacher, P.; Rosati, E.; Esser, D.; Martini, G.R.; Saggau, C.; Schiminsky, E.; Dargvainiene, J.; Schröder, I.; Wieters, I.; Khodamoradi, Y.; et al. Low-Avidity CD4+ T Cell Responses to SARS-CoV-2 in Unexposed Individuals and Humans with Severe COVID-19. Immunity 2020, 53, 1258–1271.e5. [Google Scholar] [CrossRef] [PubMed]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-Specific T Cell Immunity in Cases of COVID-19 and SARS, and Uninfected Controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Niessl, J.; Sekine, T.; Lange, J.; Konya, V.; Forkel, M.; Maric, J.; Rao, A.; Mazzurana, L.; Kokkinou, E.; Weigel, W.; et al. Identification of Resident Memory CD8+ T Cells with Functional Specificity for SARS-CoV-2 in Unexposed Oropharyngeal Lymphoid Tissue. Sci. Immunol. 2021, 6, eabk0894. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Loyal, L.; Braun, J.; Henze, L.; Kruse, B.; Dingeldey, M.; Reimer, U.; Kern, F.; Schwarz, T.; Mangold, M.; Unger, C.; et al. Cross-Reactive CD4+ T Cells Enhance SARS-CoV-2 Immune Responses upon Infection and Vaccination. Science 2021, 374, eabh1823. [Google Scholar] [CrossRef]
- Becerra-Artiles, A.; Calvo-Calle, J.M.; Co, M.D.; Nanaware, P.P.; Cruz, J.; Weaver, G.C.; Lu, L.; Forconi, C.; Finberg, R.W.; Moormann, A.M.; et al. Broadly Recognized, Cross-Reactive SARS-CoV-2 CD4 T Cell Epitopes Are Highly Conserved across Human Coronaviruses and Presented by Common HLA Alleles. Cell Rep. 2022, 39, 110952. [Google Scholar] [CrossRef]
- Low, J.S.; Vaqueirinho, D.; Mele, F.; Foglierini, M.; Jerak, J.; Perotti, M.; Jarrossay, D.; Jovic, S.; Perez, L.; Cacciatore, R.; et al. Clonal Analysis of Immunodominance and Crossreactivity of the CD4 T Cell Response to SARS-CoV-2. Science 2021, 372, 1336–1341. [Google Scholar] [CrossRef]
- Lu, X.; Hosono, Y.; Nagae, M.; Ishizuka, S.; Ishikawa, E.; Motooka, D.; Ozaki, Y.; Sax, N.; Maeda, Y.; Kato, Y.; et al. Identification of Conserved Sars-Cov-2 Spike Epitopes That Expand Public Ctfh Clonotypes in Mild Covid-19 Patients. J. Exp. Med. 2021, 218, e20211327. [Google Scholar] [CrossRef]
- Bartolo, L.; Afroz, S.; Pan, Y.G.; Xu, R.; Williams, L.; Lin, C.F.; Tanes, C.; Bittinger, K.; Friedman, E.S.; Gimotty, P.A.; et al. SARS-CoV-2-Specific T Cells in Unexposed Adults Display Broad Trafficking Potential and Cross-React with Commensal Antigens. Sci. Immunol. 2022, 7, eabn3127. [Google Scholar] [CrossRef]
- Eggenhuizen, P.J.; Ng, B.H.; Chang, J.; Fell, A.L.; Cheong, R.M.Y.; Wong, W.Y.; Gan, P.Y.; Holdsworth, S.R.; Ooi, J.D. BCG Vaccine Derived Peptides Induce SARS-CoV-2 T Cell Cross-Reactivity. Front. Immunol. 2021, 12, 3034. [Google Scholar] [CrossRef]
- Bukhari, S.; Henick, B.S.; Winchester, R.J.; Lerrer, S.; Adam, K.; Gartshteyn, Y.; Maniar, R.; Lin, Z.; Khodadadi-Jamayran, A.; Tsirigos, A.; et al. Single-Cell RNA Sequencing Reveals Distinct T Cell Populations in Immune-Related Adverse Events of Checkpoint Inhibitors. Cell Rep. Med. 2022, 12, 100868. [Google Scholar] [CrossRef]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef]
- Ahn, E.; Araki, K.; Hashimoto, M.; Li, W.; Riley, J.L.; Cheung, J.; Sharpe, A.H.; Freeman, G.J.; Irving, B.A.; Ahmed, R. Role of PD-1 during Effector CD8 T Cell Differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, 4749–4754. [Google Scholar] [CrossRef]
- Aghbash, P.S.; Eslami, N.; Shamekh, A.; Entezari-Maleki, T.; Baghi, H.B. SARS-CoV-2 Infection: The Role of PD-1/PD-L1 and CTLA-4 Axis. Life Sci. 2021, 270, 119124. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.; Okazaki, T. LAG-3: From Molecular Functions to Clinical Applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef]
- Chocarro, L.; Blanco, E.; Zuazo, M.; Arasanz, H.; Bocanegra, A.; Fernández-Rubio, L.; Morente, P.; Fernández-Hinojal, G.; Echaide, M.; Garnica, M.; et al. Understanding LAG-3 Signaling. Int. J. Mol. Sci. 2021, 22, 5282. [Google Scholar] [CrossRef]
- Krishnaswamy, J.K.; Gowthaman, U.; Zhang, B.; Mattsson, J.; Szeponik, L.; Liu, D.; Wu, R.; White, T.; Calabro, S.; Xu, L.; et al. Migratory CD11b + Conventional Dendritic Cells Induce T Follicular Helper Cell–Dependent Antibody Responses. Sci. Immunol. 2017, 2, eaam9169. [Google Scholar] [CrossRef]
- André, S.; Picard, M.; Cezar, R.; Roux-Dalvai, F.; Alleaume-Butaux, A.; Soundaramourty, C.; Cruz, A.S.; Mendes-Frias, A.; Gotti, C.; Leclercq, M.; et al. T Cell Apoptosis Characterizes Severe Covid-19 Disease. Cell Death Differ. 2022, 29, 1486–1499. [Google Scholar] [CrossRef]
- Bohan, D.; van Ert, H.; Ruggio, N.; Rogers, K.J.; Badreddine, M.; Aguilar Briseño, J.A.; Elliff, J.M.; Rojas Chavez, R.A.; Gao, B.; Stokowy, T.; et al. Phosphatidylserine Receptors Enhance SARS-CoV-2 Infection. PLoS Pathog. 2021, 17, e1009743. [Google Scholar] [CrossRef] [PubMed]
- Sage, P.T.; Alvarez, D.; Godec, J.; von Andrian, U.H.; Sharpe, A.H. Circulating T Follicular Regulatory and Helper Cells Have Memory-like Properties. J. Clin. Investig. 2014, 124, 5191–5204. [Google Scholar] [CrossRef] [PubMed]
- Hanson, A.; Cohen, H.; Wang, H.; Shekhar, N.; Shah, C.; Harvey, B.W.; Murray, R.; Harvey, C.J. Impaired ICOS Signaling between Tfh and B Cells Distinguishes Hospitalized from Ambulatory CoViD-19 patients. medRxiv, 2020; pre-print. [Google Scholar] [CrossRef]
- Kramer, K.J.; Wilfong, E.M.; Voss, K.; Barone, S.M.; Shiakolas, A.R.; Raju, N.; Roe, C.E.; Suryadevara, N.; Walker, L.M.; Wall, S.C.; et al. Single-Cell Profiling of the Antigen-Specific Response to BNT162b2 SARS-CoV-2 RNA Vaccine. Nat. Commun. 2022, 13, 3466. [Google Scholar] [CrossRef] [PubMed]
- Gazzinelli-Guimaraes, P.H.; Sanku, G.; Sette, A.; Weiskopf, D.; Schaughency, P.; Lack, J.; Nutman, T.B. Antigenic Determinants of SARS-CoV-2-Specific CD4+ T Cell Lines Reveals M Protein-Driven Dysregulation of Interferon Signaling. Front. Immunol. 2022, 13, 883159. [Google Scholar] [CrossRef]
- Asashima, H.; Mohanty, S.; Comi, M.; Ruff, W.E.; Hoehn, K.B.; Wong, P.; Klein, J.; Lucas, C.; Cohen, I.; Coffey, S.; et al. PD-1highCXCR5–CD4+ Peripheral Helper T Cells Promote CXCR3+ Plasmablasts in Human Acute Viral Infection. Cell Rep. 2023, 42, 111895. [Google Scholar] [CrossRef]
- Hanna, S.J.; Codd, A.S.; Gea-Mallorqui, E.; Scourfield, D.O.; Richter, F.C.; Ladell, K.; Borsa, M.; Compeer, E.B.; Moon, O.R.; Galloway, S.A.; et al. T cell phenotypes in COVID-19—A living review. Oxf. Open Immunol. 2020, 2, iqaa007, PMCID:PMC7798577. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ephraim, Y.E.; Koning, J.J.; Burniol Ruiz, E.; Konijn, T.; Mourits, V.P.; Lakeman, K.A.; Boon, L.; Bögels, M.; van Maanen, J.P.; Den Haan, J.M.M.; et al. CD62L Is a Functional and Phenotypic Marker for Circulating Innate Lymphoid Cell Precursors. J. Immunol. 2019, 202, 171–182. [Google Scholar] [CrossRef]
- Paulin, D.; Lilienbaum, A.; Kardjian, S.; Agbulut, O.; Li, Z. Vimentin: Regulation and Pathogenesis. Biochimie 2022, 197, 96–112. [Google Scholar] [CrossRef]
- Adam, L.; Rosenbaum, P.; Quentric, P.; Parizot, C.; Bonduelle, O.; Guillou, N.; Corneau, A.; Dorgham, K.; Miyara, M.; Luyt, C.E.; et al. CD8+PD-L1+CXCR3+ polyfunctional T cell abundances are associated with survival in critical SARS-CoV-2-infected patients. JCI Insight 2021, 6, e151571. [Google Scholar] [CrossRef]
- Saris, A.; Reijnders, T.D.Y.; Reijm, M.; Hollander, J.C.; de Buck, K.; Schuurman, A.R.; Duitman, J.; Heunks, L.; Aman, J.; Bogaard, H.J.; et al. Enrichment of CCR6 CD8 T Cells and CCL20 in the Lungs of Mechanically Ventilated Patients with COVID-19. Eur. J. Immunol. 2021, 51, 1535–1538. [Google Scholar] [CrossRef]
- Elsner, R.A.; Shlomchik, M.J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150. [Google Scholar] [CrossRef]
- Bobcakova, A.; Barnova, M.; Vysehradsky, R.; Petriskova, J.; Kocan, I.; Diamant, Z.; Jesenak, M. Activated CD8+CD38+ Cells Are Associated With Worse Clinical Outcome in Hospitalized COVID-19 Patients. Front. Immunol. 2022, 13, 961666. [Google Scholar] [CrossRef]
- Du, J.; Wei, L.; Li, G.; Hua, M.; Sun, Y.; Wang, D.; Han, K.; Yan, Y.; Song, C.; Song, R.; et al. Persistent High Percentage of HLA-DR+CD38high CD8+ T Cells Associated With Immune Disorder and Disease Severity of COVID-19. Front. Immunol. 2021, 12, 735125. [Google Scholar] [CrossRef]
- Takiguchi, S.; Tomita, Y.; Uehara, S.; Tateishi, K.; Yamamoto, N.; Nakamura, M. Immunological Imprint on Peripheral Blood in Kidney Transplant Recipients after Two Doses of SARS-CoV-2 MRNA Vaccination in Japan. Front. Med. 2022, 9, 2939. [Google Scholar] [CrossRef]
- Santopaolo, M.; Gregorova, M.; Hamilton, F.; Arnold, D.; Long, A.; Lacey, A.; Oliver, E.; Halliday, A.; Baum, H.; Hamilton, K.; et al. Prolonged T-Cell Activation and Long COVID Symptoms Independently Associate with Severe Disease at 3 Months in a UK Cohort of Hospitalized COVID-19 Patients. MedRxiv, 2022; pre-print. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Ding, X.; Zhang, M.; He, M.; Zhao, Y.; Hu, S.; Zhao, F.; Wang, J.; Xie, B.; et al. The Proportion of Peripheral Blood Tregs among the CD4+ T Cells of Autoimmune Thyroid Disease Patients: A Meta-Analysis. Endocr. J. 2020, 67, 317–326. [Google Scholar] [CrossRef]
- Pearce, E.N.; Farwell, A.P.; Braverman, L.E. Thyroiditis. N. Engl. J. Med. 2003, 348, 2646–2655. [Google Scholar] [CrossRef]
- Huang, S.-C.; Gau, S.-Y.; Huang, J.-Y.; Wu, W.-J.; Wei, J.C.-C. Increased Risk of Hypothyroidism in People with Asthma: Evidence from a Real-World Population-Based Study. J. Clin. Med. 2022, 11, 2776. [Google Scholar] [CrossRef]
- Patterson, S.J.; Pesenacker, A.M.; Wang, A.Y.; Gillies, J.; Mojibian, M.; Morishita, K.; Tan, R.; Kieffer, T.J.; Verchere, C.B.; Panagiotopoulos, C.; et al. T Regulatory Cell Chemokine Production Mediates Pathogenic T Cell Attraction and Suppression. J. Clin. Investig. 2016, 126, 1039–1051. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, J.; Islam, M.S.; Yang, Y.; Hu, Y.; Chen, X. The Role of CD4 FoxP3+ Regulatory T Cells in the Immunopathogenesis of COVID-19: Implications for Treatment. Int. J. Biol. Sci. 2021, 17, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Seepathomnarong, P.; Ongarj, J.; Sophonmanee, R.; Seeyankem, B.; Chusri, S.; Surasombatpattana, S.; Pinpathomrat, N. Regulatory T Cells Decreased during Recovery from Mild COVID-19. Viruses 2022, 14, 1688. [Google Scholar] [CrossRef] [PubMed]
- Falck-Jones, S.; Österberg, B.; Smed-Sörensen, A. Respiratory and systemic monocytes, dendritic cells, and myeloid-derived suppressor cells in COVID-19: Implications for disease severity. J. Intern. Med. 2023, 293, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ma, W.; Zhang, T.; Wu, L.; Qi, H. Phenotypic Tfh Development Promoted by CXCR5-Controlled Re-Localization and IL-6 from Radiation-Resistant Cells. Protein. Cell 2015, 6, 825–832. [Google Scholar] [CrossRef]
- Lee, S.H.; eun Kwon, J.; Cho, M.-L. Immunological Pathogenesis of Inflammatory Bowel Disease. Intestig. Res. 2018, 16, 26. [Google Scholar] [CrossRef]
- Zúñiga, L.A.; Jain, R.; Haines, C.; Cua, D.J. Th17 Cell Development: From the Cradle to the Grave. Immunol. Rev. 2013, 252, 78–88. [Google Scholar] [CrossRef]
- Karin, N. CXCR3 Ligands in Cancer and Autoimmunity, Chemoattraction of Effector T Cells, and Beyond. Front. Immunol. 2020, 11, 976. [Google Scholar] [CrossRef]
- Yamada, A.; Arakaki, R.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Role of Regulatory T Cell in the Pathogenesis of Inflammatory Bowel Disease. World J. Gastroenterol. 2016, 22, 2195–2205. [Google Scholar] [CrossRef]
- Oparaugo, N.C.; Ouyang, K.; Nguyen, N.P.N.; Nelson, A.M.; Agak, G.W. Human Regulatory T Cells: Understanding the Role of Tregs in Select Autoimmune Skin Diseases and Post-Transplant Nonmelanoma Skin Cancers. Int. J. Mol. Sci. 2023, 24, 1527. [Google Scholar] [CrossRef]
- Sałkowska, A.; Karaś, K.; Karwaciak, I.; Walczak-Drzewiecka, A.; Krawczyk, M.; Sobalska-Kwapis, M.; Dastych, J.; Ratajewski, M. Identification of Novel Molecular Markers of Human Th17 Cells. Cells 2020, 9, 1611. [Google Scholar] [CrossRef]
- Agalioti, T.; Villablanca, E.J.; Huber, S.; Gagliani, N. T H 17 cell Plasticity: The Role of Dendritic Cells and Molecular Mechanisms. J. Autoimmun. 2018, 87, 50–60. [Google Scholar] [CrossRef]
- Honey, K. CCL3 and CCL4 actively recruit CD8+ T cells. Nat. Rev. Immunol. 2006, 6, 427. [Google Scholar] [CrossRef]
- Baggiolini, M. CXCL8-the First Chemokine. Front. Immunol. 2015, 6, 285. [Google Scholar] [CrossRef]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef]
- Mishra, A.; Suman, K.H.; Nair, N.; Majeed, J.; Tripathi, V. An Updated Review on the Role of the CXCL8-CXCR1/2 Axis in the Progression and Metastasis of Breast Cancer. Mol. Biol. Rep. 2021, 48, 6551–6561. [Google Scholar] [CrossRef]
- Reschke, R.; Gajewski, T.F. CXCL9 and CXCL10 bring the heat to tumors. Sci. Immunol. 2022, 7, eabq6509. [Google Scholar] [CrossRef] [PubMed]
- Gudowska-Sawczuk, M.; Mroczko, B. What Is Currently Known about the Role of CXCL10 in SARS-CoV-2 Infection? Int. J. Mol. Sci. 2022, 23, 3673, PMCID:PMC8998241. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, A.; Tahmasebi, S.; Mahmood, A.; Kuznetsova, M.; Valizadeh, H.; Taghizadieh, A.; Nazemiyeh, M.; Aghebati-Maleki, L.; Jadidi-Niaragh, F.; Abbaspour-Aghdam, S.; et al. Th17 and Treg Cells Function in SARS-CoV2 Patients Compared with Healthy Controls. J. Cell Physiol. 2021, 236, 2829–2839. [Google Scholar] [CrossRef]
- Gurlevik, S.L.; Ozsurekci, Y.; Sağ, E.; Derin Oygar, P.; Kesici, S.; Akca, Ü.K.; Cuceoglu, M.K.; Basaran, O.; Göncü, S.; Karakaya, J.; et al. The Difference of the Inflammatory Milieu in MIS-C and Severe COVID-19. Pediatr. Res. 2022, 92, 1805–1814. [Google Scholar] [CrossRef]
- Mahmoud Salehi Khesht, A.; Karpisheh, V.; Qubais Saeed, B.; Olegovna Zekiy, A.; Yapanto, L.M.; Nabi Afjadi, M.; Aksoun, M.; Nasr Esfahani, M.; Aghakhani, F.; Movahed, M.; et al. Different T Cell Related Immunological Profiles in COVID-19 Patients Compared to Healthy Controls. Int. Immunopharmacol. 2021, 97, 107828. [Google Scholar] [CrossRef]
- Pérez-Martínez, A.; Martín-Quirós, A.; Ferreras, C.; Al-Akioui, K.; Mora-Rillo, M.; Goterris, R.; Briones, M.L.; Gasior, M.; Amat, P.; de Paz, R.; et al. A Phase I/II Dose-Escalation Multi-Center Study to Evaluate the Safety of Infusion of Natural Killer Cells or Memory T Cells As Adoptive Therapy in Coronavirus Pneumonia and/or Lymphopenia: (RELEASE NCT04578210). Blood 2021, 138, 1765. [Google Scholar] [CrossRef]
- Carding, S.R.; Egan, P.J. Γδ T Cells: Functional Plasticity and Heterogeneity. Nat. Rev. Immunol. 2002, 2, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.-H.; Wu, F.-T.; Pang, L.-T.; Zhang, T.-B.; Chen, Z. Role of ΓδT Cells in Liver Diseases and Its Relationship with Intestinal Microbiota. World J. Gastroenterol. 2020, 26, 2559–2569. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.; Mezouar, S.; Cano, C.; Frohna, P.; Madakamutil, L.; Mège, J.-L.; Olive, D. Role of Vγ9vδ2 T Lymphocytes in Infectious Diseases. Front. Immunol. 2022, 13, 928441. [Google Scholar] [CrossRef] [PubMed]
- Harly, C.; Guillaume, Y.; Nedellec, S.; Peigné, C.-M.; Mönkkönen, H.; Mönkkönen, J.; Li, J.; Kuball, J.; Adams, E.J.; Netzer, S.; et al. Key Implication of CD277/Butyrophilin-3 (BTN3A) in Cellular Stress Sensing by a Major Human Γδ T-Cell Subset. Blood 2012, 120, 2269–2279. [Google Scholar] [CrossRef]
- Gu, S.; Sachleben, J.R.; Boughter, C.T.; Nawrocka, W.I.; Borowska, M.T.; Tarrasch, J.T.; Skiniotis, G.; Roux, B.; Adams, E.J. Phosphoantigen-Induced Conformational Change of Butyrophilin 3A1 (BTN3A1) and Its Implication on Vγ9Vδ2 T Cell Activation. Proc. Natl. Acad. Sci. USA 2017, 114, E7311–E7320. [Google Scholar] [CrossRef]
- Uldrich, A.P.; Rigau, M.; Godfrey, D.I. Immune Recognition of Phosphoantigen-butyrophilin Molecular Complexes by Γδ T Cells. Immunol. Rev. 2020, 298, 74–83. [Google Scholar] [CrossRef]
- Sant, S.; Jenkins, M.R.; Dash, P.; Watson, K.A.; Wang, Z.; Pizzolla, A.; Koutsakos, M.; Nguyen, T.H.; Lappas, M.; Crowe, J.; et al. Human Γδ T-cell Receptor Repertoire Is Shaped by Influenza Viruses, Age and Tissue Compartmentalisation. Clin. Transl. Immunol. 2019, 8, e1079. [Google Scholar] [CrossRef]
- Poccia, F.; Agrati, C.; Castilletti, C.; Bordi, L.; Gioia, C.; Horejsh, D.; Ippolito, G.; Chan, P.K.S.; Hui, D.S.C.; Sung, J.J.Y.; et al. Anti–Severe Acute Respiratory Syndrome Coronavirus Immune Responses: The Role Played by Vγ9Vδ2 T Cells. J. Infect. Dis. 2006, 193, 1244–1249. [Google Scholar] [CrossRef]
- Gay, L.; Rouviere, S.M.; Mezouar, S.; Richaud, M.; Gorvel, G.; Foucher, E.; Madakamutil, L.; la Scola, B.; Menard, A.; Allardet-Servent, J.; et al. Vγ9Vδ2 T Cells Are Potent Inhibitors of SARS-Co V-2 Replication and Exert Effector Phenotypes in COVID-19 Patients. bioRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Atmeh, P.A.; Gay, L.; Levasseur, A.; la Scola, B.; Olive, D.; Mezouar, S.; Gorvel, J.-P.; Mege, J.-L. Macrophages and Γδ T Cells Interplay during SARS-CoV-2 Variants Infection. Front. Immunol. 2022, 13, 1078741. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against Type I IFNs in Patients with Life-Threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Karbuz, A.; Gervais, A.; Tayoun, A.A.; Aiuti, A.; Belot, A.; Bolze, A.; Gaudet, A.; Bondarenko, A.; et al. Human Genetic and Immunological Determinants of Critical COVID-19 Pneumonia. Nature 2022, 603, 587–598. [Google Scholar] [CrossRef]
- Von Stemann, J.H.; Rigas, A.S.; Thørner, L.W.; Rasmussen, D.G.K.; Pedersen, O.B.; Rostgaard, K.; Erikstrup, C.; Ullum, H.; Hansen, M.B. Prevalence and Correlation of Cytokine-Specific Autoantibodies with Epidemiological Factors and C-Reactive Protein in 8972 Healthy Individuals: Results from the Danish Blood Donor Study. PLoS ONE 2017, 12, e0179981. [Google Scholar] [CrossRef]
- Pfeifer, J.; Thurner, B.; Kessel, C.; Fadle, N.; Kheiroddin, P.; Regitz, E.; Hoffmann, M.-C.; Kos, I.A.; Preuss, K.-D.; Fischer, Y.; et al. Autoantibodies against Interleukin-1 Receptor Antagonist in Multisystem Inflammatory Syndrome in Children: A Multicentre, Retrospective, Cohort Study. Lancet Rheumatol. 2022, 4, e329–e337. [Google Scholar] [CrossRef]
- Ward, K.E.; Steadman, L.; Karim, A.R.; Reynolds, G.M.; Pugh, M.; Chua, W.; Faustini, S.E.; Veenith, T.; Thwaites, R.S.; Openshaw, P.J.; et al. SARS-CoV-2 Infection Is Associated with Anti-Desmoglein 2 Autoantibody Detection. medrXiv, 2022; preprint. [Google Scholar] [CrossRef]
- Taeschler, P.; Cervia, C.; Zurbuchen, Y.; Hasler, S.; Pou, C.; Tan, Z.; Adamo, S.; Raeber, M.E.; Bächli, E.; Rudiger, A.; et al. Autoantibodies in COVID-19 Correlate with Antiviral Humoral Responses and Distinct Immune Signatures. Allergy 2022, 77, 2415–2430. [Google Scholar] [CrossRef]
- De Vries, R.; Duprex, W.; de Swart, R. Morbillivirus Infections: An Introduction. Viruses 2015, 7, 699–706. [Google Scholar] [CrossRef]
- Amrute, J.M.; Perry, A.M.; Anand, G.; Cruchaga, C.; Hock, K.G.; Farnsworth, C.W.; Randolph, G.J.; Lavine, K.J.; Steed, A.L. Cell Specific Peripheral Immune Responses Predict Survival in Critical COVID-19 Patients. Nat. Commun. 2022, 13, 882. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody Evasion by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 Humoral Immunity Induces Convergent Omicron RBD Evolution. Nature 2022, 424, 1–3. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Zhou, H.; Zhu, H.; Jiang, S.; Wang, P. Broadly neutralizing antibodies to SARS-CoV-2 and other human coronaviruses. Nat. Rev. Immunol. 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chandele, A.; Sharma, A. Current Status of Therapeutic Monoclonal Antibodies against SARS-CoV-2. PLoS Pathog. 2021, 17, e1009885. [Google Scholar] [CrossRef] [PubMed]
- Doshi, P. COVID-19: Do Many People Have Pre-Existing Immunity? BMJ 2020, 370, m3563. [Google Scholar] [CrossRef]
- Agrati, C.; Sacchi, A.; Tartaglia, E.; Vergori, A.; Gagliardini, R.; Scarabello, A.; Bibas, M. The Role of P-Selectin in COVID-19 Coagulopathy: An Updated Review. Int. J. Mol. Sci. 2021, 22, 7942. [Google Scholar] [CrossRef] [PubMed]
- Fenyves, B.G.; Mehta, A.; MGH COVID-19 Collection & Processing Team; Kays, K.R.; Beakes, C.; Margolin, J.; Goldberg, M.B.; Hacohen, N.; Filbin, M.R. Plasma P-selectin is an early marker of thromboembolism in COVID-19. Am. J. Hematol. 2021, 96, E468–E471. [Google Scholar] [CrossRef]
- Watany, M.M.; Abdou, S.; Elkolaly, R.; Elgharbawy, N.; Hodeib, H. Evaluation of Admission Levels of P, E and L Selectins as Predictors for Thrombosis in Hospitalized COVID-19 Patients. Clin. Exp. Med. 2022, 22, 567–575. [Google Scholar] [CrossRef]
- Osman, I.O.; Garrec, C.; de Souza, G.A.P.; Zarubica, A.; Belhaouari, D.B.; Baudoin, J.-P.; Lepidi, H.; Mege, J.-L.; Malissen, B.; la Scola, B.; et al. Control of CDH1/E-Cadherin Gene Expression and Release of a Soluble Form of E-Cadherin in SARS-CoV-2 Infected Caco-2 Intestinal Cells: Physiopathological Consequences for the Intestinal Forms of COVID-19. Front. Cell Infect. Microbiol. 2022, 12, 438. [Google Scholar] [CrossRef]
- Mezu-Ndubuisi, O.J.; Maheshwari, A. The role of integrins in inflammation and angiogenesis. Pediatr. Res. 2021, 89, 1619–1626. [Google Scholar] [CrossRef]
- Yao, S.; Luo, N.; Liu, J.; Zha, H.; Ai, Y.; Luo, J.; Shi, S.; Wu, K. Elevated Serum Levels of Progranulin and Soluble Vascular Cell Adhesion Molecule-1 in Patients with COVID-19. J. Inflamm. Res. 2021, 14, 4785–4794. [Google Scholar] [CrossRef]
- Spadaro, S.; Fogagnolo, A.; Campo, G.; Zucchetti, O.; Verri, M.; Ottaviani, I.; Tunstall, T.; Grasso, S.; Scaramuzzo, V.; Murgolo, F.; et al. Markers of Endothelial and Epithelial Pulmonary Injury in Mechanically Ventilated COVID-19 ICU Patients. Crit. Care 2021, 25, 74. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A Master Regulator of Cellular Responses in Inflammation, Injury Resolution, and Tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Ramkumar, K.; Cargill, K.R.; Cardnell, R.J.; Nilsson, M.B.; Heeke, S.; Park, E.M.; Kundu, S.T.; Diao, L.; et al. Lung Cancer Models Reveal Severe Acute Respiratory Syndrome Coronavirus 2–Induced Epithelial-to-Mesenchymal Transition Contributes to Coronavirus Disease 2019 Pathophysiology. J. Thorac. Oncol. 2021, 16, 1821–1839. [Google Scholar] [CrossRef]
- Davila, M.L.; Xu, M.; Huang, C.; Gaddes, E.R.; Winter, L.; Cantorna, M.T.; Wang, Y.; Xiong, N. CCL27 is a crucial regulator of immune homeostasis of the skin and mucosal tissues. iScience 2022, 25, 104426, PMCID:PMC9157018. [Google Scholar] [CrossRef] [PubMed]
- Mildner, M.; Prior, M.; Gschwandtner, M.; Schuster, C.; Tschachler, E.; Elbe-Bürger, A. Epidermal CCL27 Expression Is Regulated during Skin Development and Keratinocyte Differentiation. J. Investig. Dermatol. 2014, 134, 855–858. [Google Scholar] [CrossRef]
- Mai, C.-L.; Tan, Z.; Xu, Y.-N.; Zhang, J.-J.; Huang, Z.-H.; Wang, D.; Zhang, H.; Gui, W.-S.; Zhang, J.; Lin, Z.-J.; et al. CXCL12-Mediated Monocyte Transmigration into Brain Perivascular Space Leads to Neuroinflammation and Memory Deficit in Neuropathic Pain. Theranostics 2021, 11, 1059–1078. [Google Scholar] [CrossRef]
- Peled, A.; Klein, S.; Beider, K.; Burger, J.A.; Abraham, M. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine 2018, 109, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Godinez, D.R.; Brust-Mascher, I.; Miller, E.N.; Gareau, M.G.; Reardon, C. Neuroanatomy of the Spleen: Mapping the Relationship between Sympathetic Neurons and Lymphocytes. PLoS ONE 2017, 12, e0182416. [Google Scholar] [CrossRef]
- Li, C.; Kim, H.K.; Prakhar, P.; Luo, S.; Crossman, A.; Ligons, D.L.; Luckey, M.A.; Awasthi, P.; Gress, R.E.; Park, J.-H. Chemokine Receptor CCR9 Suppresses the Differentiation of CD4+CD8αα+ Intraepithelial T Cells in the Gut. Mucosal. Immunol. 2022, 15, 882–895. [Google Scholar] [CrossRef]
- Chu, F.; Li, H.S.; Liu, X.; Cao, J.; Ma, W.; Ma, Y.; Weng, J.; Zhu, Z.; Cheng, X.; Wang, Z.; et al. CXCR5+CD8+ T Cells Are a Distinct Functional Subset with an Antitumor Activity. Leukemia 2019, 33, 2640–2653. [Google Scholar] [CrossRef]
- Singh, P.; Mohammad, K.S.; Pelus, L.M. CXCR4 Expression in the Bone Marrow Microenvironment Is Required for Hematopoietic Stem and Progenitor Cell Maintenance and Early Hematopoietic Regeneration after Myeloablation. Stem. Cells 2020, 38, 849–859. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]













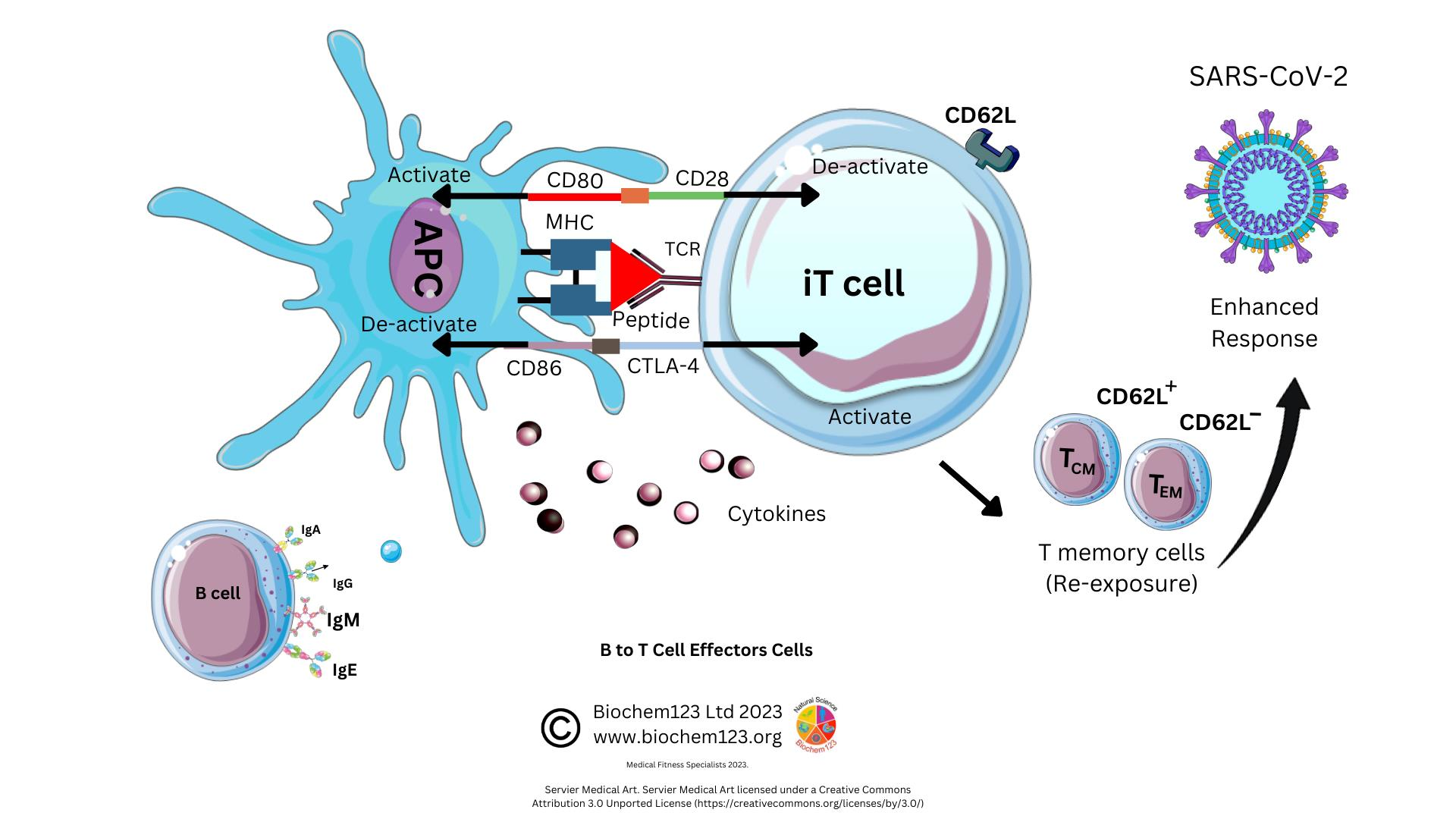{kind=link}
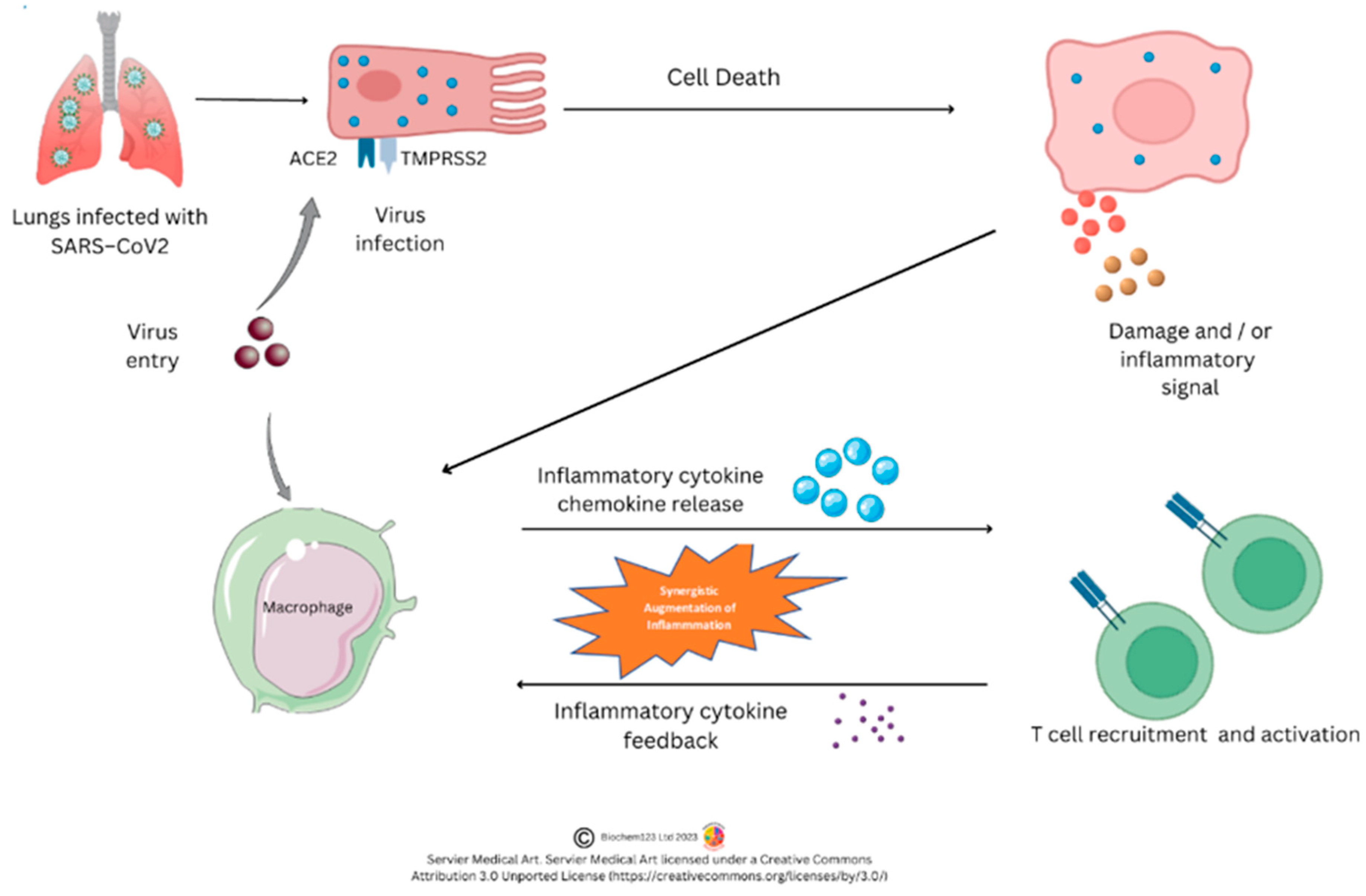{kind=link}
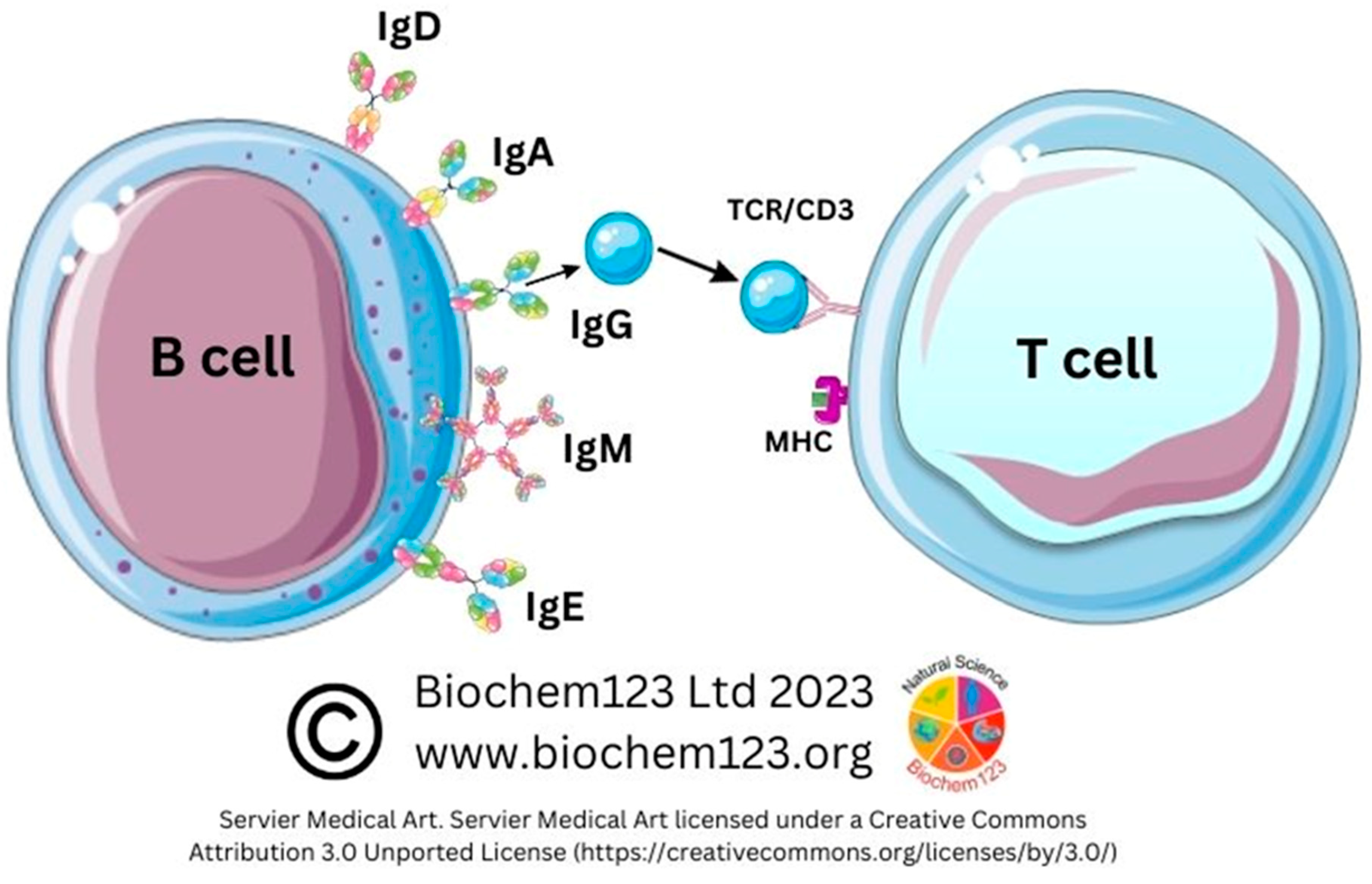{kind=link}
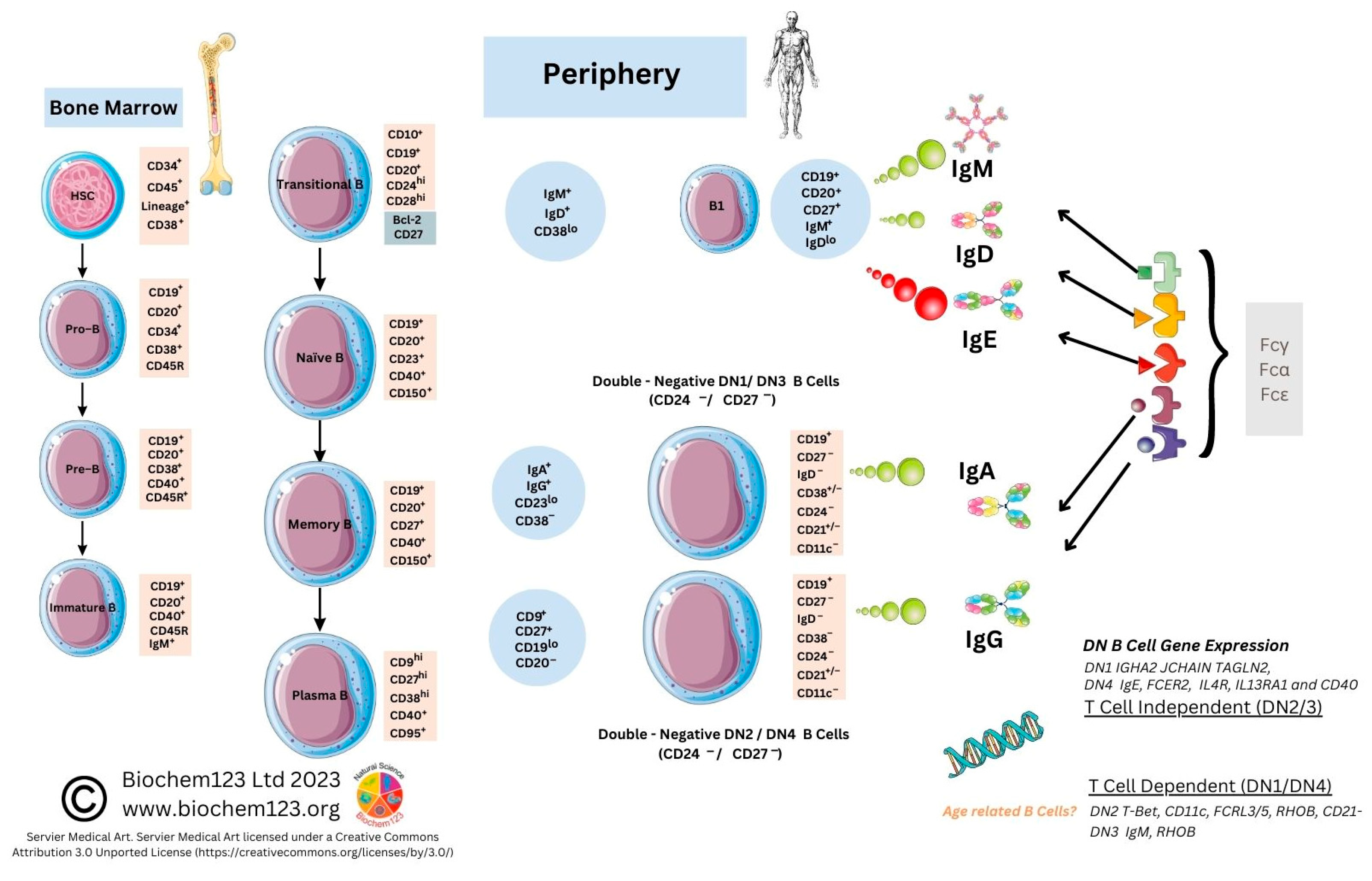{kind=link}
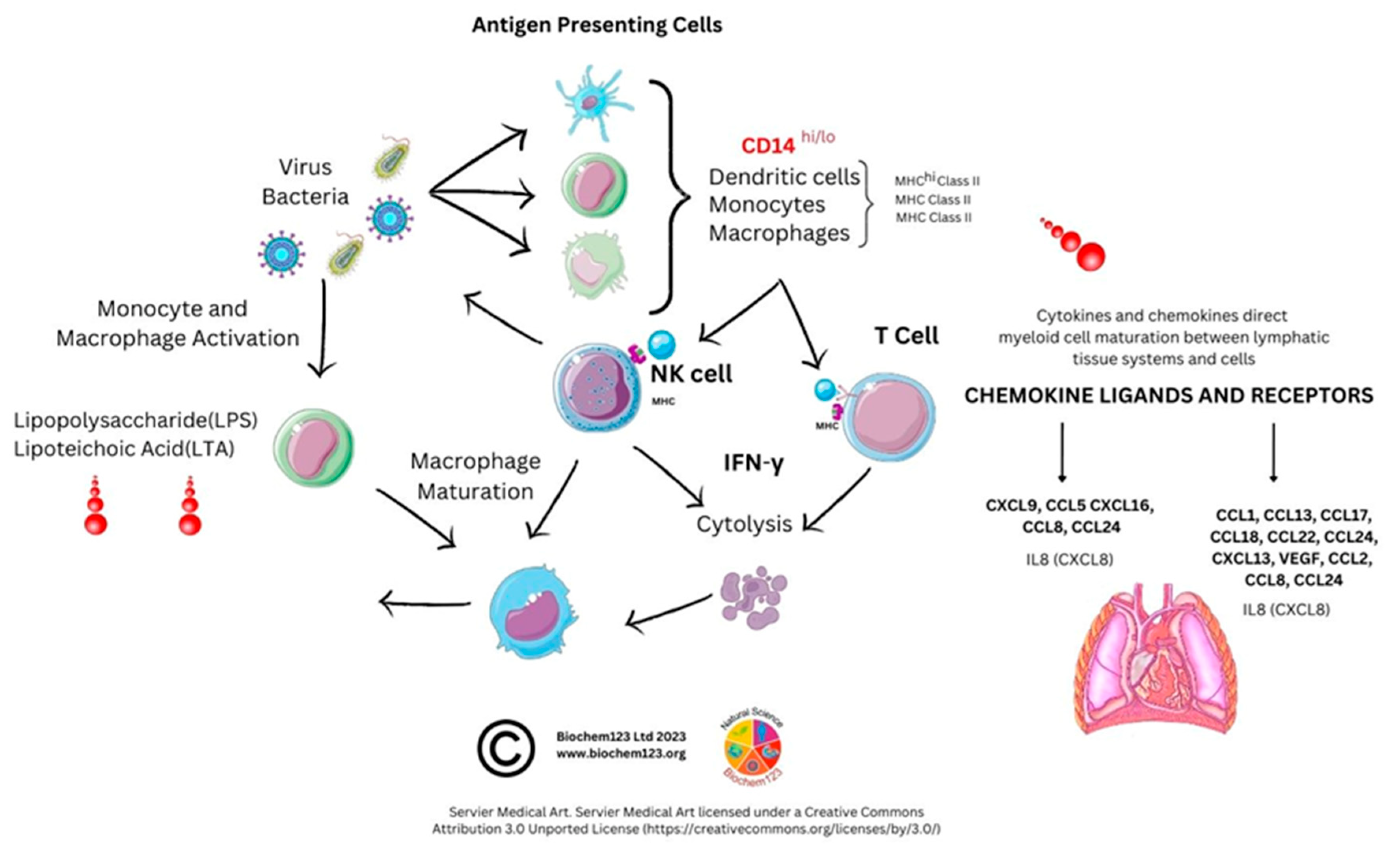{kind=link}
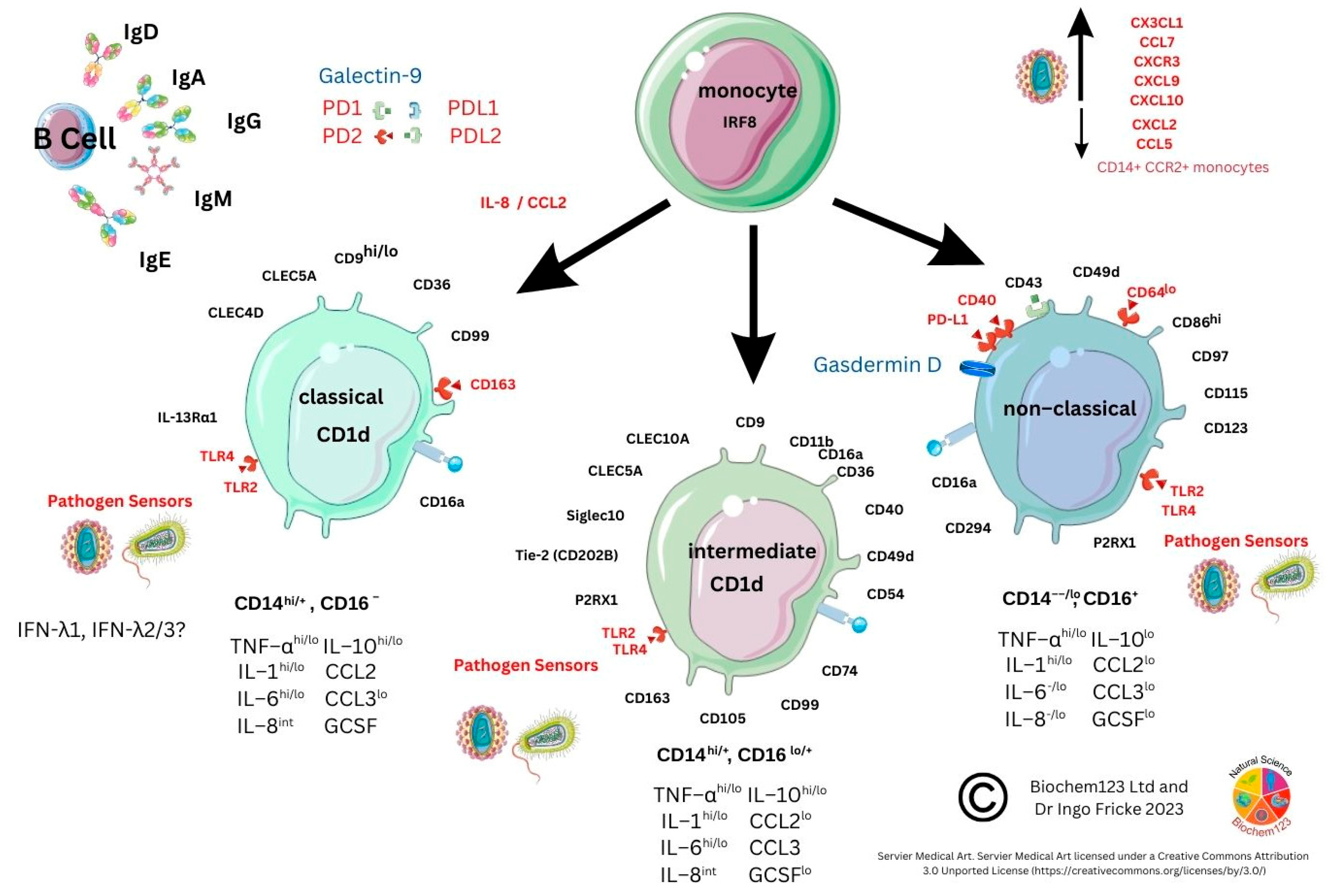{kind=link}
{kind=link}
{kind=link}
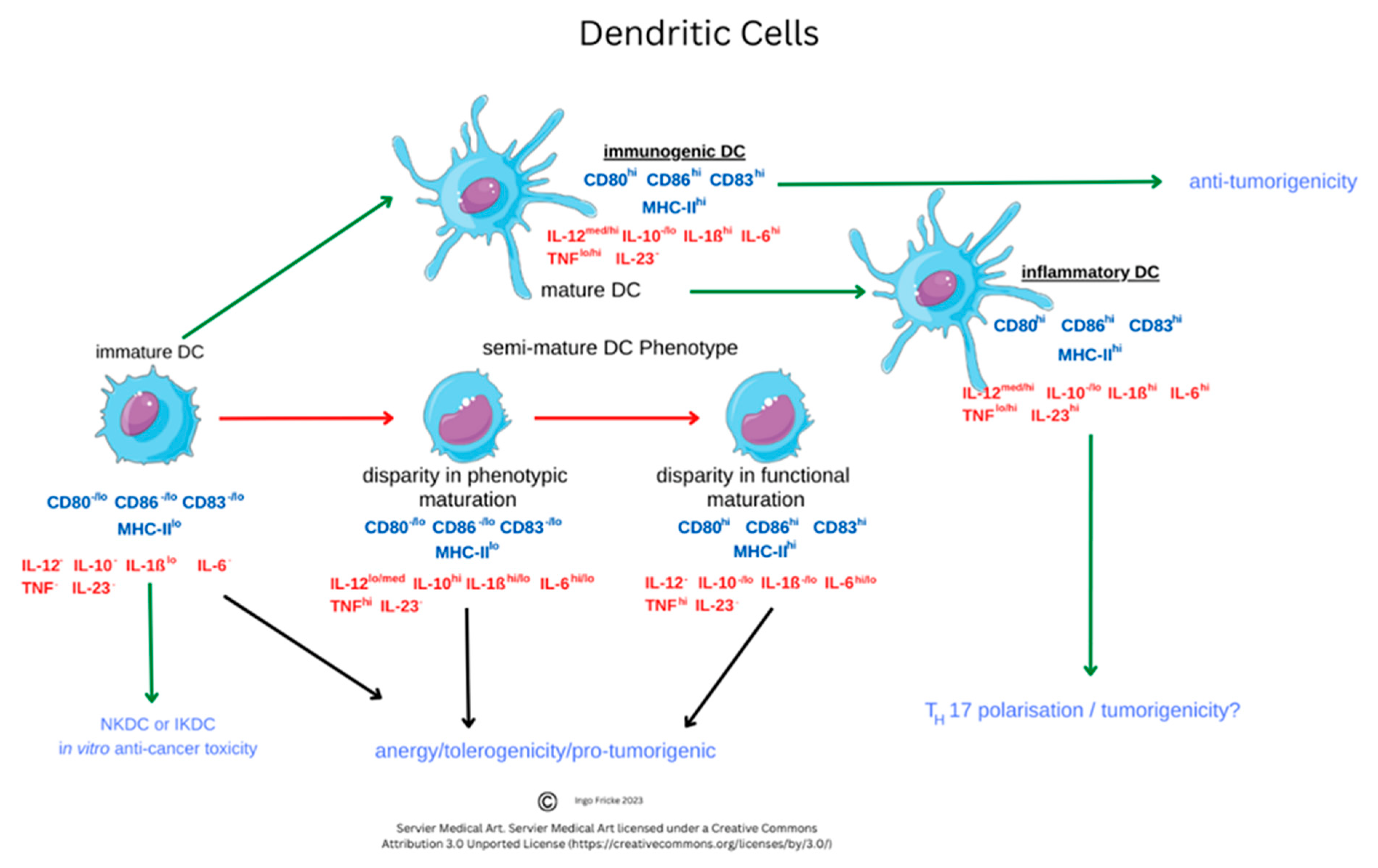{kind=link}
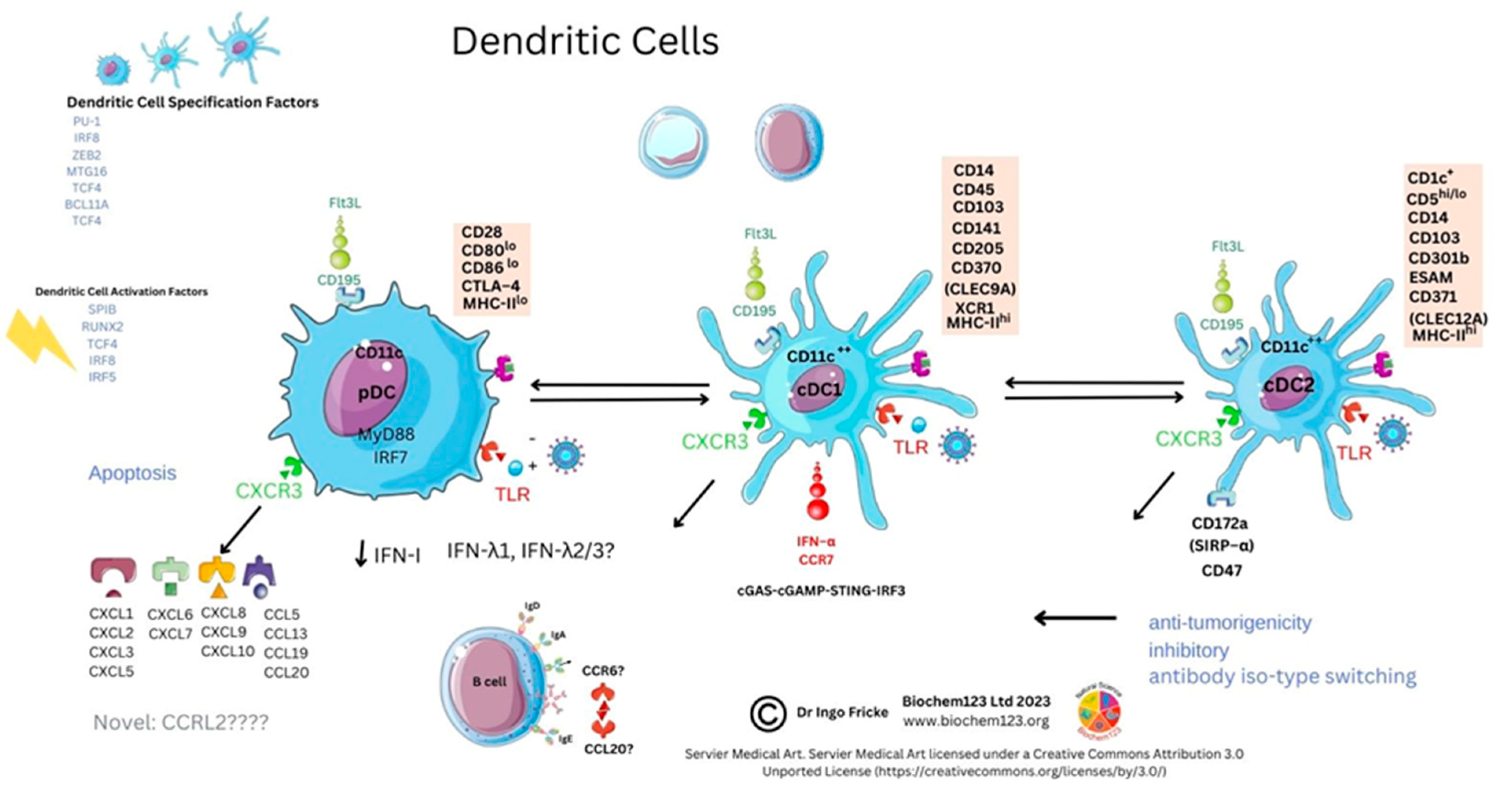{kind=link}
{kind=link}
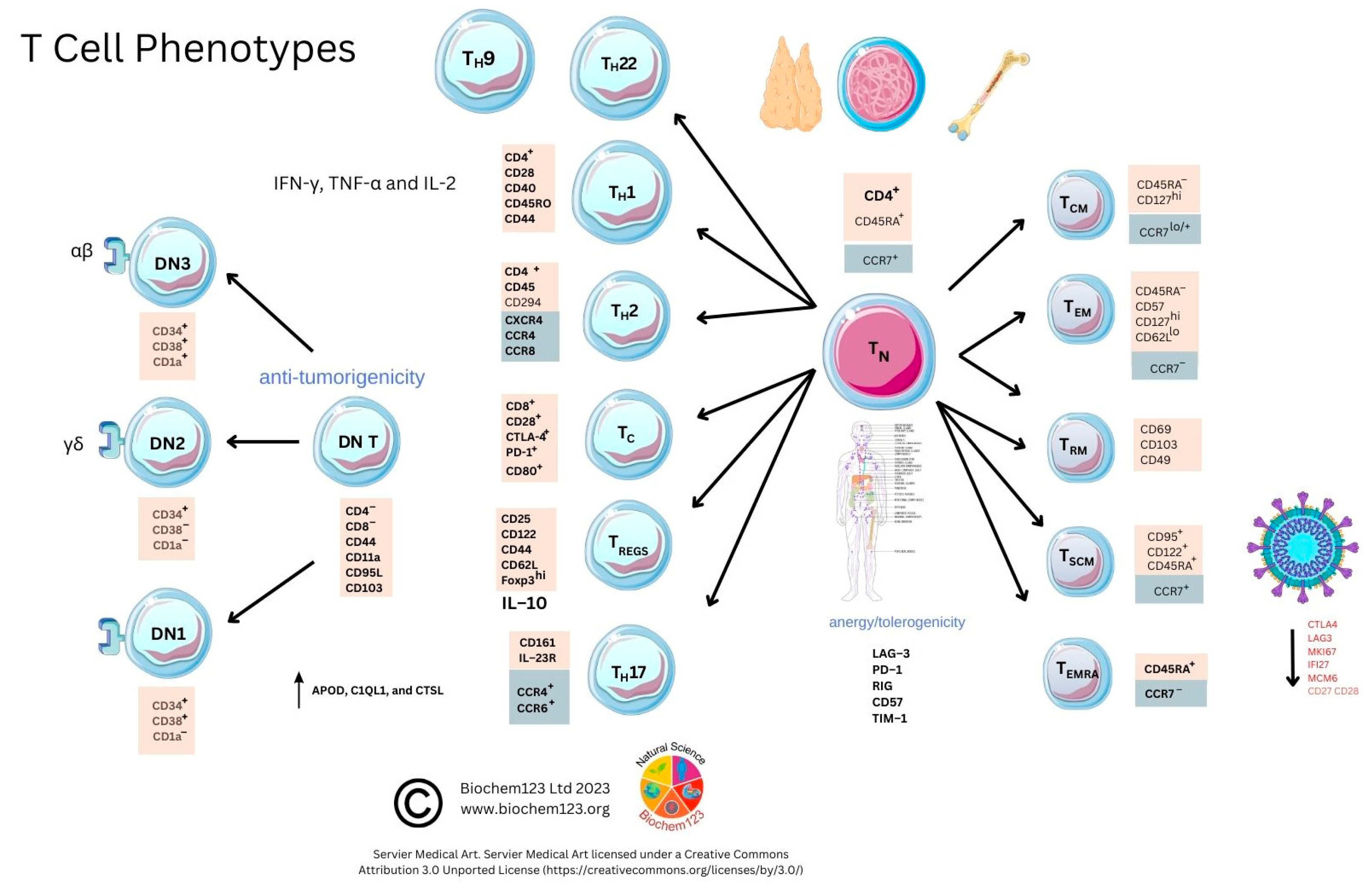{kind=link}
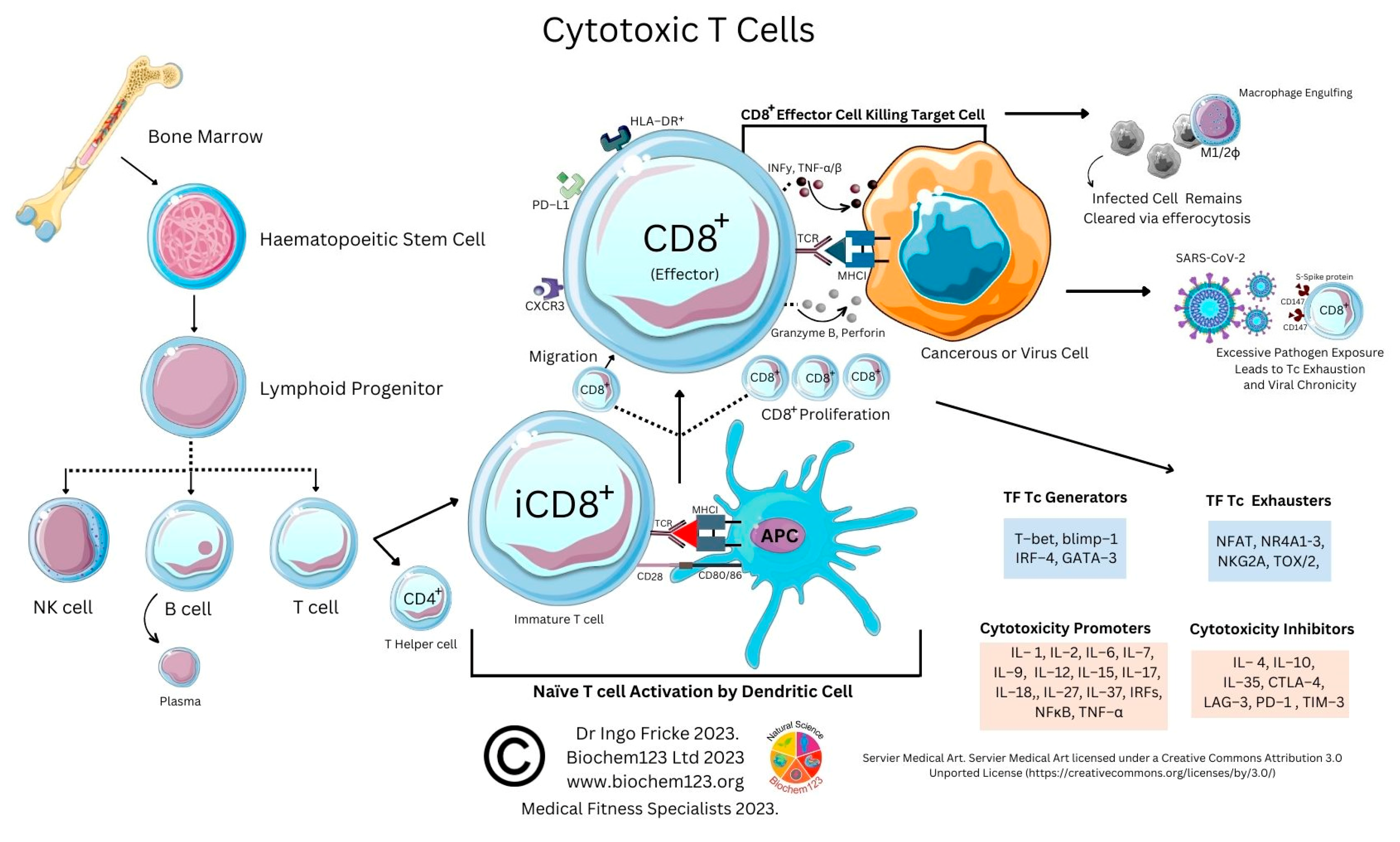{kind=link}
| Isotype | Serum Level (mg/mL) | Molecular Weight (Monomer) | Complement Activation | Half Life |
|---|---|---|---|---|
| IgA1 | 0.6–3 | 160 (monomer) | – | 5.5 |
| IgA2 | 0.06–0.6 | 160 | – | 5.5 |
| IgM | 1.5 | 970 | +++ | 5–10 |
| IgE | 5 × 10−5 | 188 | – | 2 |
| IgG1 | 3.8–11.4 | 146 | ++ | 23 |
| IgG2 | 1.5–6.9 | 146 | + | 23 |
| IgG3 | 0.2–1.7 | 165 | +++ | 7 |
| IgG4 | 0.08–1.4 | 146 | – | 23 |
| Antigen | Mild | Moderate | Severe | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IgG | IgA | IgE | IgG | IgA | IgE | IgG | IgA | IgE | IgG | IgA | IgE | |
| Spike (FP) | 94.7 | 16.7 | 66.7 | 100 | 38.5 | 92.3 | 100 | 54.5 | 90 | 97.3 | 30.1 | 79.7 |
| Stable Spike Trimer | 97.4 | 25 | 12.1 | 100 | 42.3 | 50 | 100 | 54.5 | 60 | 98.7 | 35.6 | 33.3 |
| RBD | 92.1 | 22.2 | 0 | 100 | 42.3 | 3.85 | 100 | 63.6 | 10 | 96 | 35.6 | 2.9 |
| S1 | 89.5 | 22.2 | 0 | 100 | 42.3 | 34.6 | 100 | 63.6 | 20 | 94.7 | 35.6 | 15.9 |
| B Cells | Markers | Chemokines | Ig | Amount | |||
|---|---|---|---|---|---|---|---|
| Naïve B Cells | CD27+ | ND | ND | ND | IgD+ | 29–69 | [106,113,114,115,116,117,118] |
| Unswitched B Cells | CD27+ | ND | ND | ND | IgD+ | 5–22 | [114] |
| Switched B Cells | CD27+ | ND | ND | ND | IgD− | 8–43 | [114] |
| B Cell DN1 | CD27− | CD21+ | CD11c− | CXCR5+ | IgD− | 4–6 | [107,115] |
| B Cell DN2 | CD27+ | CD21− | CD11c+ | CXCR5− | IgD− | ND | [116,117] |
| B Cell DN3 | CD27− | CD21− | CD11c− | ND | IgD− | ND | [118] |
| B Cell DN4 | CD27− | ND | ND | ND | IgD− | ND | [118] |
| pDC | cDC1 | cDC2 | moDC | Tissue DC | |
|---|---|---|---|---|---|
| Cellular Differentiation Marker | CD11c− | CD11c++ | CD11c++ | CD11b/c+ | CD11clo/hi |
| CD303 | CD141+ | CD1c+ | CD1c+ | CD1c+ | |
| CD123 | CD370 (CLEC9) | CD172a+ (SIRPα) | CD1a+ | CD1a+/− | |
| CD45RA | CD14 | CD14 | CD206/CD209 | CD14+/− | |
| CD45+ CD103+ | CD5hi/lo CD103+ CD123− | CD172a+ (SIRPα) | |||
| Cytokine/ Chemokine | IFN–α, CCR7 | XCR1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, B.; Ojha, V.; Fricke, I.; Al-Sheboul, S.A.; Imarogbe, C.; Gravier, T.; Green, M.; Peterson, L.; Koutsaroff, I.P.; Demir, A.; et al. Innate and Adaptive Immunity during SARS-CoV-2 Infection: Biomolecular Cellular Markers and Mechanisms. Vaccines 2023, 11, 408. https://doi.org/10.3390/vaccines11020408
Brown B, Ojha V, Fricke I, Al-Sheboul SA, Imarogbe C, Gravier T, Green M, Peterson L, Koutsaroff IP, Demir A, et al. Innate and Adaptive Immunity during SARS-CoV-2 Infection: Biomolecular Cellular Markers and Mechanisms. Vaccines. 2023; 11(2):408. https://doi.org/10.3390/vaccines11020408
Chicago/Turabian StyleBrown, Brent, Vanshika Ojha, Ingo Fricke, Suhaila A Al-Sheboul, Chinua Imarogbe, Tanya Gravier, Michael Green, Lori Peterson, Ivoyl P. Koutsaroff, Ayça Demir, and et al. 2023. "Innate and Adaptive Immunity during SARS-CoV-2 Infection: Biomolecular Cellular Markers and Mechanisms" Vaccines 11, no. 2: 408. https://doi.org/10.3390/vaccines11020408