
Genomic Tracking of SARS-CoV-2 Variants in Myanmar
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
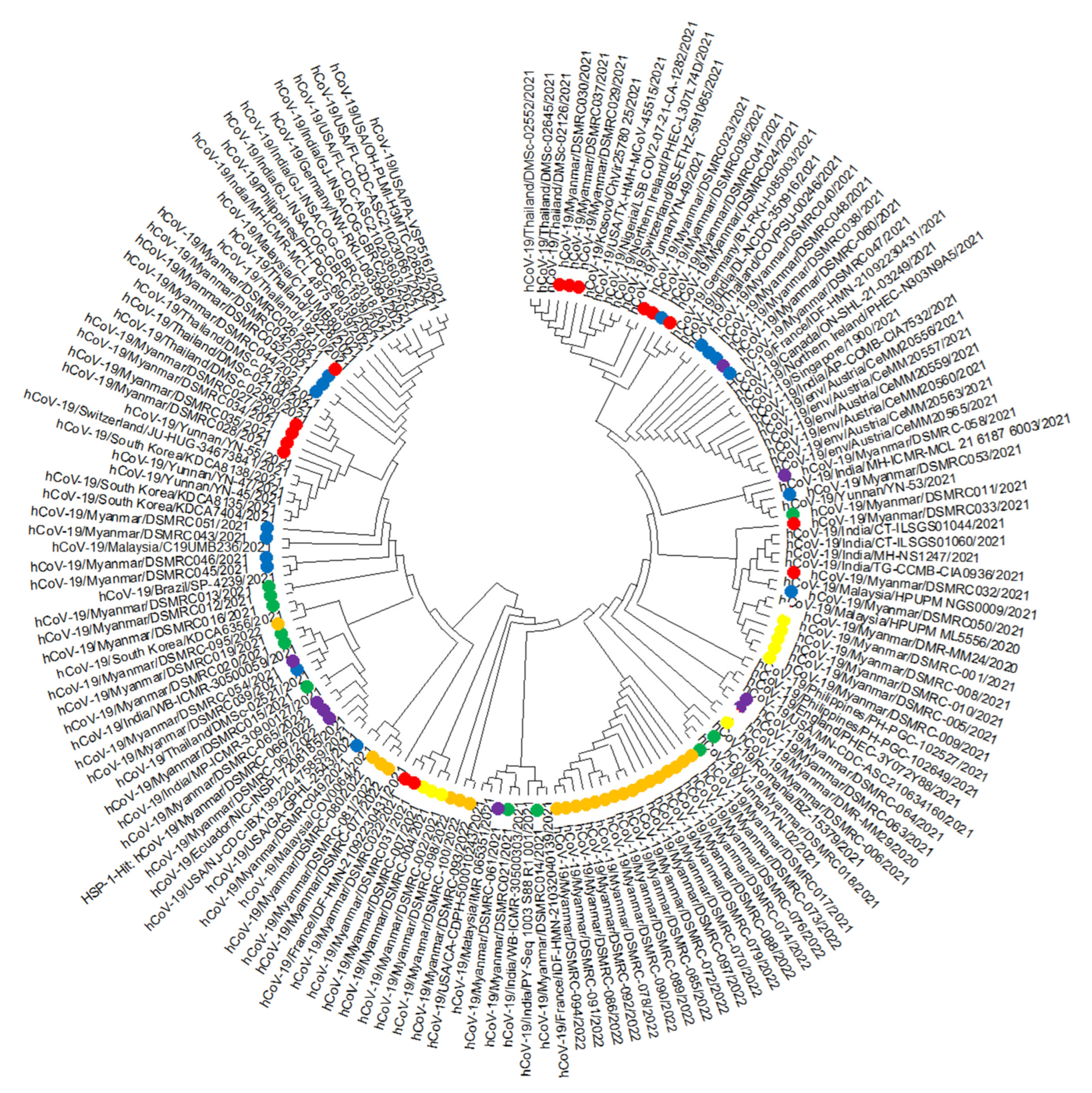
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, H.; Stratton, C.W.; Tang, Y.-W. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020, 92, 401–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- WHO. Coronavirus Disease 2019 (COVID-19) Situation Report–52. 2020. Available online: https://apps.who.int/iris/handle/10665/331476 (accessed on 20 June 2022).
- MOH. Ministry of Health Myanmar COVID-19 Surveillance Dashboard. 2022. Available online: https://mohs.gov.mm/ (accessed on 29 June 2022).
- WHO. Tracking SARS-CoV-2 Variants. 2022. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 22 June 2022).
- Illumina COVIDSeq Test (RUO Version). Available online: https://www.illumina.com/products/by-type/clinical-research-products/covidseq.html (accessed on 29 September 2021).
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Cavalcante, I.J.M.; Vale, M.R. Epidemiological aspects of visceral leishmaniasis (kala-azar) in Ceará in the period 2007 to 2011. Rev. Bras. Epidemiol. 2014, 17, 911–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. bioRxiv 2020. [Google Scholar] [CrossRef]
- Singh, D.; Yi, S.V. On the origin and evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, W. Fast-spreading SARS-CoV-2 variants: Challenges to and new design strategies of COVID-19 vaccines. Signal Transduct. Target. Ther. 2021, 6, 226. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488.e4. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J. Med. Virol. 2021, 93, 5638–5643. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated im-munity. Cell 2021, 184, 1171–1187.e20. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Andreano, E.; Piccini, G.; Licastro, D.; Casalino, L.; Johnson, N.V.; Paciello, I.; Dal Monego, S.; Pantano, E.; Manganaro, N.; Manenti, A.; et al. SARS-CoV-2 escape in vitro from a highly neutralizing COVID-19 convalescent plasma. bioRxiv 2020. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Moghadasi, S.A.; Heilmann, E.; Moraes, S.N.; Kearns, F.L.; von Laer, D.; Amaro, R.E.; Harris, R.S. Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lee, J.T.; Yang, Q.; Gribenko, A.; Perrin, B.S., Jr.; Zhu, Y.; Cardin, R.; Liberator, P.A.; Anderson, A.S.; Hao, L. Genetic Surveillance of SARS-CoV-2 M(pro) Reveals High Sequence and Structural Conservation Prior to the Introduction of Protease Inhibitor Paxlovid. mBio 2022, 13, e0086922. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Shayestehpour, M.; Mirzaei, H. The impact of spike mutated variants of SARS-CoV2 [Alpha, Beta, Gamma, Delta, and Lambda] on the efficacy of subunit recombinant vaccines. Braz. J. Infect. Dis. 2021, 25, 101606. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. Understanding variants of SARS-CoV-2. Lancet 2021, 397, 462. [Google Scholar] [CrossRef]
- Khan, K.A.; Cheung, P. Presence of mismatches between diagnostic PCR assays and coronavirus SARS-CoV-2 genome. R. Soc. Open Sci. 2020, 7, 200636. [Google Scholar] [CrossRef]


{kind=link}
| Sequence Name | Age | Sex | Residence | Vaccination | Severity | Travel History | Collection Date (DD-MM-YY) | PANGO Lineage | GISAID Clade | GISAID Accession Number |
|---|---|---|---|---|---|---|---|---|---|---|
| 001/2021 | 40 | Male | NayPYiTaw | No | Mild | Local | 04-01-21 | B.1.36.16 | GH | EPI_ISL_833041 |
| 002/2021 | 37 | Male | NayPYiTaw | No | Mild | Local | 09-01-21 | B.1.36.16 | GH | EPI_ISL_849736 |
| 004/2021 | 38 | Male | NayPYiTaw | No | Mild | Local | 09-01-21 | B.1.36.16 | GH | EPI_ISL_849737 |
| 005/2021 | 41 | Male | NayPYiTaw | No | Mild | Local | 09-01-21 | B.1.36.16 | GH | EPI_ISL_849738 |
| 006/2021 | 42 | Male | NayPYiTaw | No | Mild | Local | 09-01-21 | B.1.36.16 | GH | EPI_ISL_849739 |
| 007/2021 | 37 | Male | NayPYiTaw | No | Mild | Local | 09-01-21 | B.1.36.16 | GH | EPI_ISL_849740 |
| 008/2021 | 38 | Male | NayPYiTaw | No | Mild | Local | 09-01-21 | B.1.36.16 | GH | EPI_ISL_849741 |
| 009/2021 | 41 | Male | NayPYiTaw | No | Mild | Local | 09-01-21 | B.1.36.16 | GH | EPI_ISL_849742 |
| 010/2021 | 42 | Male | NayPYiTaw | No | Mild | Local | 09-01-21 | B.1.36.16 | GH | EPI_ISL_849743 |
| 011/2021 | 35 | Female | Kalay | Not known | Mild | Local | 28-05-21 | B.1.617.2 | GK | EPI_ISL_2612300 |
| 012/2021 | 55 | Male | Myeik | Not known | Mild | Local | 28-05-21 | B.1.1.7 | GR | EPI_ISL_2592630 |
| 013/2021 | 22 | Female | Myeik | Not known | Mild | Local | 28-05-21 | B.1.617.1 | G | EPI_ISL_2593764 |
| 014/2021 | 51 | Female | Myeik | Not known | Mild | Local | 28-05-21 | B.1.1.7 | GR | EPI_ISL_2595725 |
| 015/2021 | 28 | Female | Mandalay | Not known | Mild | Local | 01-06-21 | B.1.617.2 | GK | EPI_ISL_2595726 |
| 016/2021 | 7 | Female | Mandalay | Not known | Mild | Local | 01-06-21 | B.1.617.2 | GK | EPI_ISL_2595860 |
| 017/2021 | 25 | Female | Mandalay | Not known | Mild | Local | 01-06-21 | B.1.617.2 | GK | EPI_ISL_2596341 |
| 018/2021 | 18 | Female | Mandalay | Not known | Mild | Local | 01-06-21 | B.1.617.2 | GK | EPI_ISL_2596803 |
| 019/2021 | 23 | Male | Tamu | Not known | Mild | Local | 02-06-21 | B.1.617.1 | G | EPI_ISL_2597228 |
| 020/2021 | 42 | Male | Tamu | Not known | Mild | Local | 02-06-21 | B.1.617.1 | G | EPI_ISL_2597229 |
| 021/2021 | 42 | Male | Yangon | Not known | Mild | Local | 26-05-21 | B.1.617.1 | G | EPI_ISL_2597312 |
| 022/2021 | 43 | Male | Nay Pyi Taw | No | Mild | Local | 07-07-21 | AY.30 | GK | EPI_ISL_5424999 |
| 023/2021 | 55 | Female | Mandalay | No | Dead | Local | 25-06-21 | B.1.617.2 | GK | EPI_ISL_5427023 |
| 024/2021 | 76 | Female | Monywa | No | Severe/ICU | Local | 12-08-21 | B.1.617.2 | GK | EPI_ISL_5427644 |
| 026/2021 | 52 | Female | Kalay | Covidshield | Mild | Local | 11-08-21 | B.1.617.2 | GK | EPI_ISL_5427934 |
| 027/2021 | 30 | Male | Mawlamyine | Covidshield | Asymptomatic | Local | 09-08-21 | AY.30 | GK | EPI_ISL_5428660 |
| 028/2021 | 28 | Male | Lashio | Sinopharm | Mild | Local | 11-08-21 | B.1.617.2 | GK | EPI_ISL_5428815 |
| 029/2021 | 4 | Female | Nay Pyi Taw | No | Mild | Local | 16-08-21 | AY.30 | GK | EPI_ISL_5428817 |
| 030/2021 | 31 | Male | Nay Pyi Taw | No | Critical/ICU | Local | 16-08-21 | AY.30 | GK | EPI_ISL_5428988 |
| 031/2021 | 46 | Female | Sittwe | Sinopharm | Mild | Local | 25-08-21 | AY.30 | GK | EPI_ISL_5428993 |
| 032/2021 | 26 | Male | Yangon | Covaxin | Mild | Local | 07-06-21 | B.1.617.2 | GK | EPI_ISL_5428995 |
| 033/2021 | 31 | Male | Yangon | Covaxin | Severe/ICU | Local | 25-08-21 | B.1.617.2 | GK | EPI_ISL_5429000 |
| 034/2021 | 45 | Female | Nay Pyi Taw | Covaxin | Mild | Local | 06-07-21 | AY.30 | GK | EPI_ISL_5429003 |
| 035/2021 | 37 | Female | Taungoo | Covidshield | Mild | Local | 05-07-21 | B.1.617.2 | GK | EPI_ISL_5429005 |
| 036/2021 | 3 | Female | Nay Pyi Taw | No | Mild | Local | 16-08-21 | B.1.617.2 | GK | EPI_ISL_5429009 |
| 037/2021 | 33 | Male | Mandalay | Covidshield | Mild | Local | 25-06-21 | AY.30 | GK | EPI_ISL_5429011 |
| 038/2021 | 34 | Female | Taunggyi | Covidshield | Mild | Local | 23-11-21 | B.1.617.2 | GK | EPI_ISL_7337304 |
| 039/2021 | 38 | Male | ThinganNyinaung | Covidshield | Mild | Thailand | 12-11-21 | AY.85 | GK | EPI_ISL_7337866 |
| 040/2021 | 29 | Male | Mawlamyine | Covidshield | Mild | Thailand | 03-11-21 | AY.85 | GK | EPI_ISL_7337871 |
| 041/2021 | 20 | Male | Kengtung | Covidshield | Mild | Laos | 08-11-21 | B.1.617.2 | GK | EPI_ISL_7337873 |
| 042/2021 | 21 | Female | Kengtung | Covidshield | Mild | Laos | 08-11-21 | B.1.617.2 | GK | EPI_ISL_7338123 |
| 043/2021 | 39 | Male | Shan | Covidshield | Mild | Local | 24-11-21 | AY.79 | GK | EPI_ISL_7338109 |
| 044/2021 | 34 | Male | Naypyitaw | Covidshield | Mild | Local | 01-12-21 | B.1.617.2 | GK | EPI_ISL_7338111 |
| 045/2021 | 36 | Male | Naypyidaw | Covidshield | Mild | Local | 03-12-21 | AY.129 | GK | EPI_ISL_7338113 |
| 046/2021 | 47 | Female | Naypyitaw | Covidshield | Mild | Local | 03-12-21 | AY.98 | GK | EPI_ISL_7338114 |
| 047/2021 | 52 | Male | Yangon | Covidshield | Mild | Local | 30-11-21 | AY.129 | GK | EPI_ISL_7338119 |
| 048/2021 | 25 | Male | Yangon | Covidshield | Mild | Malaysia | 01-12-21 | B.1.617.2 | G | EPI_ISL_7338201 |
| 049/2021 | 36 | Male | Yangon | Covidshield | Mild | Malaysia | 01-12-21 | AY.114 | G | EPI_ISL_7338374 |
| 050/2021 | 54 | Male | Mawlamyine | Covidshield | Mild | Thailand | 04-12-21 | AY.59 | GK | EPI_ISL_7338375 |
| 051/2021 | 17 | Female | Mawlamyine | Covidshield | Mild | Thailand | 04-12-21 | AY.59 | GK | EPI_ISL_7338376 |
| 052/2021 | 18 | Male | Mandalay | Covidshield | Mild | Local | 03-12-21 | B.1.617.2 | GK | EPI_ISL_7338402 |
| 053/2021 | 54 | Male | Mandalay | Covidshield | Mild | Local | 03-12-21 | B.1.617.2 | GK | EPI_ISL_7338766 |
| 054/2021 | 52 | Male | Tachileik | Not known | Mild | Thailand | 06-12-21 | AY.129 | GK | EPI_ISL_8920487 |
| 058/2021 | 65 | Male | Kalay | Not known | Mild | Local | 27-12-21 | B.1.617.2 | GK | EPI_ISL_8920488 |
| 060/2021 | 38 | Male | Yangon | Not known | Mild | Local | 27-12-21 | AY.79 | GK | EPI_ISL_8920490 |
| 061/2021 | 43 | Male | Yangon | Not known | Mild | Local | 27-12-21 | AY.79 | GK | EPI_ISL_8920491 |
| 063/2021 | 29 | Male | Yangon | Not known | Mild | UAE | 30-11-21 | BA.1 | GRA | EPI_ISL_8920492 |
| 064/2021 | 29 | Male | Yangon | Not known | Mild | UAE | 30-11-21 | BA.1 | GRA | EPI_ISL_8920493 |
| 065/2022 | 4 | Female | Yangon | Not known | Mild | Philippines | 04-01-22 | BA.2.3 | GRA | EPI_ISL_8920494 |
| 066/2022 | 44 | Male | Yangon | Not known | Mild | Philippines | 04-01-22 | BA.2.3 | GRA | EPI_ISL_8920501 |
| 067/2022 | 33 | Male | Yangon | Not known | Mild | Malaysia | 04-01-22 | BA.1.1 | GRA | EPI_ISL_8920500 |
| 070/2022 | 42 | Male | Nay Pyi Taw | Covaxin | Mild | Local | 12-03-22 | BA.2 | GRA | EPI_ISL_11149371 |
| 072/2022 | 34 | Male | Nay Pyi Taw | Covaxin | Mild | Local | 12-03-22 | BA.2 | GRA | EPI_ISL_11149374 |
| 073/2022 | 28 | Male | Myanaung | Not known | Mild | Local | 14-03-22 | BA.2 | GRA | EPI_ISL_11149375 |
| 074/2022 | 43 | Female | Nay Pyi Taw | Covaxin | Mild | Local | 14-03-22 | BA.2 | GRA | EPI_ISL_11149531 |
| 076/2022 | 36 | Female | Nay Pyi Taw | Sinopharm | Mild | Local | 14-03-22 | BA.2 | GRA | EPI_ISL_11149533 |
| 077/2022 | 30 | Male | Taungdwingyi | Covidshield | Mild | Local | 14-03-22 | BA.2 | GRA | EPI_ISL_11149535 |
| 078/2022 | 55 | Female | Taungoo | Sinopharm | Mild | Local | 14-03-22 | BA.2.10.1 | GRA | EPI_ISL_11149537 |
| 079/2022 | 49 | Male | Nay Pyi Taw | Covaxin | Mild | Local | 14-03-22 | BA.2 | GRA | EPI_ISL_11149539 |
| 080/2022 | 30 | Female | Nay Pyi Taw | Covidshield | Mild | Local | 14-03-22 | BA.2 | GRA | EPI_ISL_11149543 |
| 081/2022 | 23 | Female | Nay Pyi Taw | Covidshield | Mild | Local | 14-03-22 | BA.2 | GRA | EPI_ISL_11149547 |
| 085/2022 | 27 | Female | Nay Pyi Taw | Covaxin | Mild | Local | 11-03-22 | BA.2 | GRA | EPI_ISL_11149552 |
| 086/2022 | 57 | Female | Thayet | Covaxin | Mild | Local | 11-03-22 | BA.2 | GRA | EPI_ISL_11149554 |
| 088/2022 | 32 | Female | Nay Pyi Taw | Sinopharm | Mild | Local | 11-03-22 | BA.2.10 | GRA | EPI_ISL_11149559 |
| 089/2022 | 29 | Female | Nay Pyi Taw | Sinopharm | Mild | Local | 11-03-22 | BA.2.10.1 | GRA | EPI_ISL_11149562 |
| 090/2022 | 32 | Female | Homalin | Covidshield | Mild | Local | 19-02-22 | BA.2.10.1 | GRA | EPI_ISL_11149564 |
| 091/2022 | 26 | Male | Homalin | Covidshield | Mild | Local | 19-02-22 | BA.2 | GRA | EPI_ISL_11149614 |
| 092/2022 | 17 | Female | Homalin | Covidshield | Mild | Local | 19-02-22 | BA.2.10 | GRA | EPI_ISL_11149615 |
| 093/2022 | 60 | Female | Homalin | Covidshield | Mild | Local | 19-02-22 | BA.2.10.1 | GRA | EPI_ISL_11149617 |
| 094/2022 | 56 | Male | Homalin | Covidshield | Mild | Local | 19-02-22 | BA.2 | GRA | EPI_ISL_11149639 |
| 095/2022 | 17 | Male | Homalin | Covidshield | Mild | Local | 19-02-22 | BA.2 | GRA | EPI_ISL_11149640 |
| 097/2022 | 42 | Male | Nay Pyi Taw | Covidshield | Mild | Local | 11-03-22 | BA.2 | GRA | EPI_ISL_11149643 |
| 098/2022 | 42 | Male | Nay Pyi Taw | Covaxin | Mild | Local | 11-03-22 | BA.2.38 | GRA | EPI_ISL_11149644 |
| 100/2022 | 34 | Female | Nay Pyi Taw | Covaxin | Mild | Local | 11-03-22 | BA.2.10 | GRA | EPI_ISL_11149739 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oo, K.Z.; Htun, Z.W.; Aung, N.M.; Win, K.K.; Linn, K.Z.; Htoo, S.P.; Aung, P.K.; Oo, T.W.; Zaw, M.T.; Ko, L.Y.; et al. Genomic Tracking of SARS-CoV-2 Variants in Myanmar. Vaccines 2023, 11, 6. https://doi.org/10.3390/vaccines11010006
Oo KZ, Htun ZW, Aung NM, Win KK, Linn KZ, Htoo SP, Aung PK, Oo TW, Zaw MT, Ko LY, et al. Genomic Tracking of SARS-CoV-2 Variants in Myanmar. Vaccines. 2023; 11(1):6. https://doi.org/10.3390/vaccines11010006
Chicago/Turabian StyleOo, Khine Zaw, Zaw Win Htun, Nay Myo Aung, Ko Ko Win, Kyaw Zawl Linn, Sett Paing Htoo, Phyo Kyaw Aung, Thet Wai Oo, Myo Thiha Zaw, Linn Yuzana Ko, and et al. 2023. "Genomic Tracking of SARS-CoV-2 Variants in Myanmar" Vaccines 11, no. 1: 6. https://doi.org/10.3390/vaccines11010006