
Adult-Onset Familial Hemophagocytic Lymphohistiocytosis Presenting with Annular Erythema following COVID-19 Vaccination

Abstract
:1. Introduction
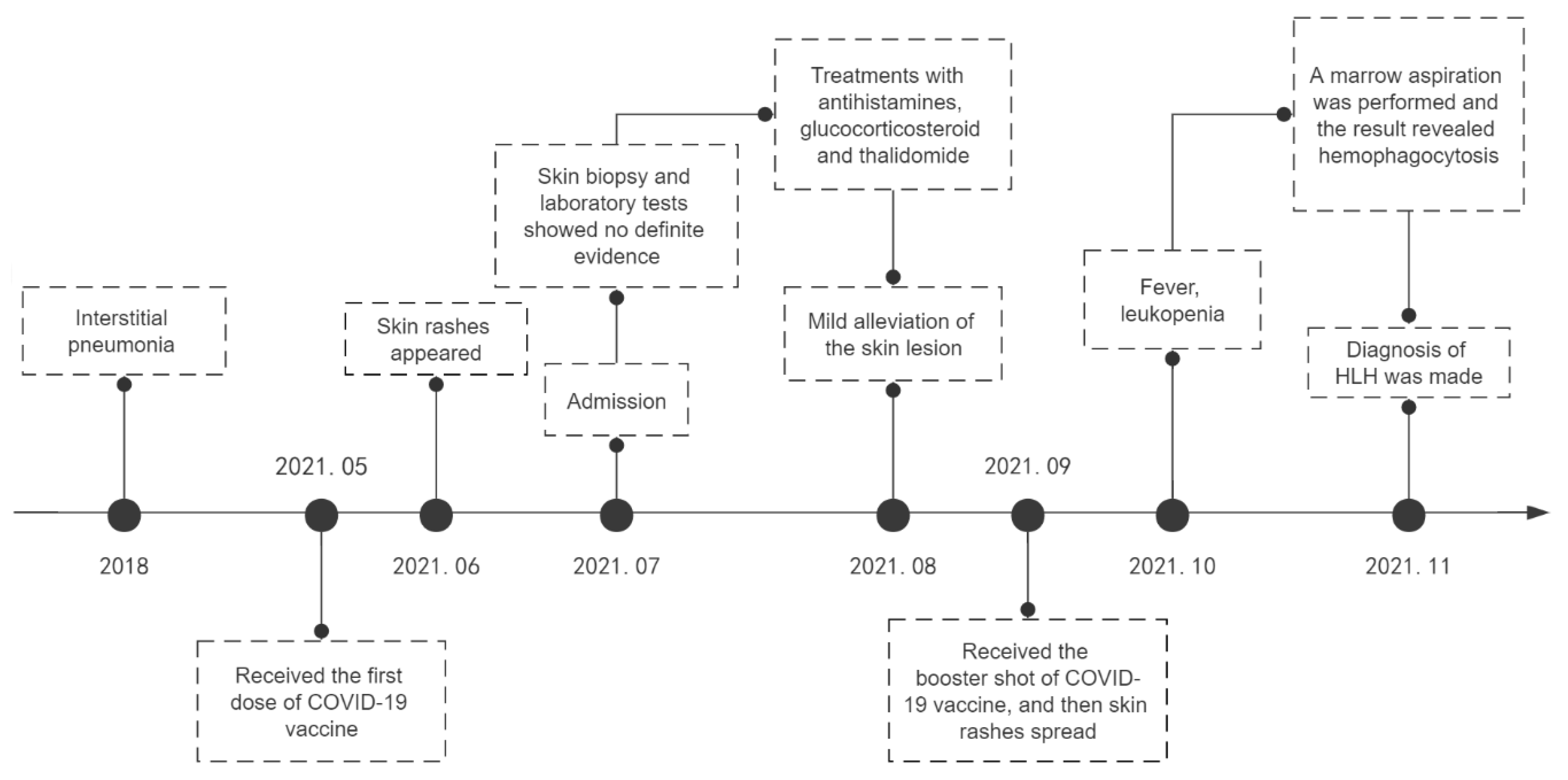
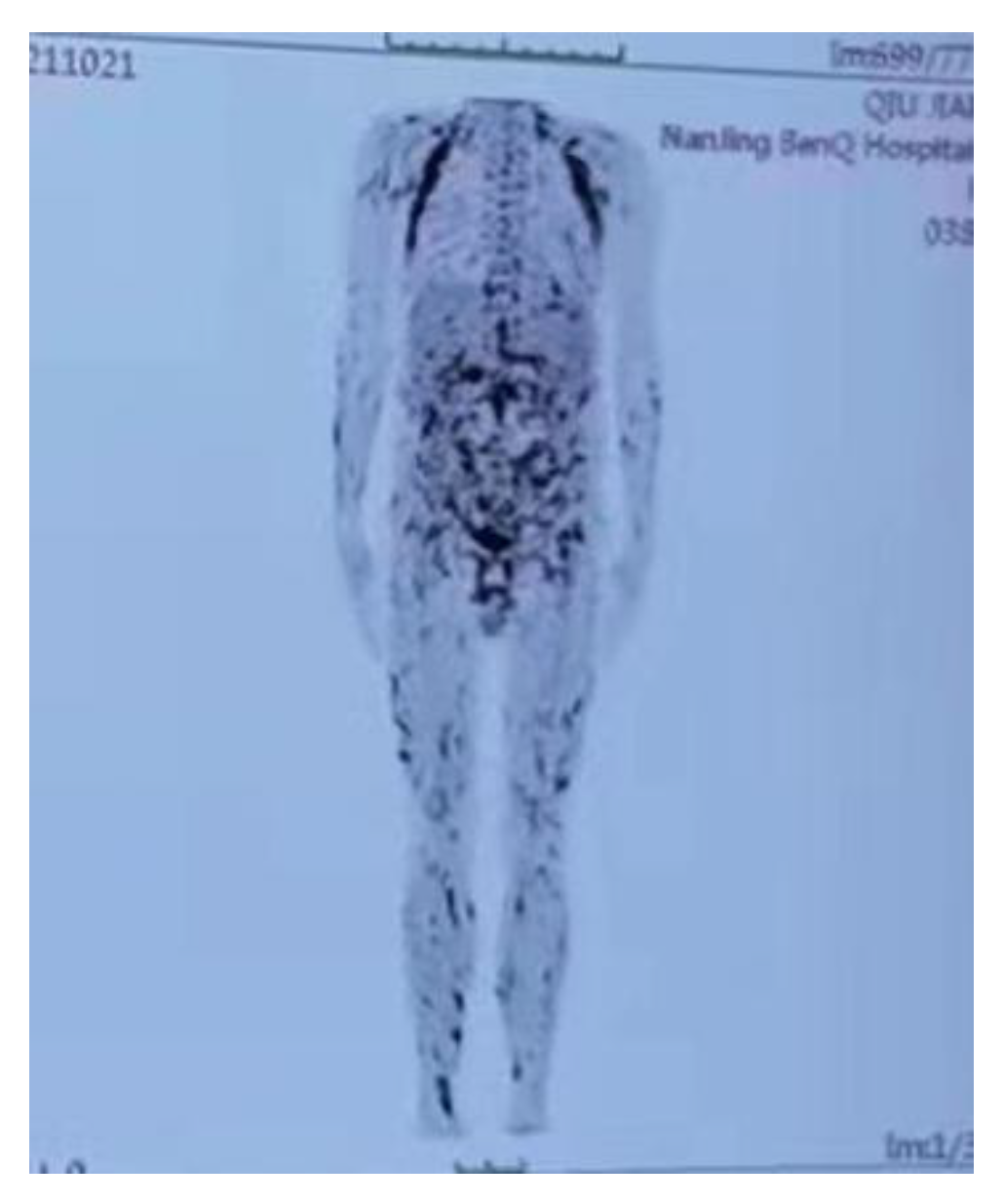
2. Case Report
3. Literature Review
3.1. HLH following COVID-19 Vaccination
3.2. Cutaneous Manifestations of HLH with Genetic Defects
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Samkari, H.; Berliner, N. Hemophagocytic Lymphohistiocytosis. Annu. Rev. Pathol. 2018, 13, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.; Berliner, N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 2015, 125, 2908–2914. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.A.; Hermiston, M.L.; Nichols, K.E.; Meyer, L.K. Digenic Inheritance: Evidence and Gaps in Hemophagocytic Lym-phohistiocytosis. Front. Immunol. 2021, 12, 777851. [Google Scholar] [CrossRef]
- Hayden, A.; Park, S.; Giustini, D.; Lee, A.Y.; Chen, L.Y. Hemophagocytic syndromes (HPSs) including hemophagocytic lym-phohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev. 2016, 30, 411–420. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; López-Guillermo, A.; Khamashta, M.A.; Bosch, X. Adult haemophagocytic syndrome. Lancet 2014, 383, 1503–1516. [Google Scholar] [CrossRef]
- Tang, L.V.; Hu, Y. Hemophagocytic lymphohistiocytosis after COVID-19 vaccination. J. Hematol. Oncol. 2021, 14, 1–5. [Google Scholar] [CrossRef]
- Hieber, M.L.; Sprute, R.; Eichenauer, D.A.; Hallek, M.; Jachimowicz, R.D. Hemophagocytic lymphohistiocytosis after SARS-CoV-2 vaccination. Infection 2022, 1–6. [Google Scholar] [CrossRef]
- Ai, S.; Awford, A.; Roncolato, F. Hemophagocytic lymphohistiocytosis following ChAdOx1 nCov-19 vaccination. J. Med. Virol. 2021, 94, 14–16. [Google Scholar] [CrossRef]
- Nasir, S.; Khan, S.R.; Iqbal, R.; Hashmi, A.P.; Moosajee, M.; Nasir, N. Inactivated COVID-19 vaccine triggering hemophagocytic lymphohistiocytosis in an immunocompetent adult—A case report. J. Clin. Transl. Res. 2022, 8, 152–155. [Google Scholar]
- Caocci, G.; Fanni, D.; Porru, M.; Greco, M.; Nemolato, S.; Firinu, D.; Faa, G.; Scuteri, A.; La Nasa, G. Kikuchi-Fujimoto disease associated with hemophagocytic lymphohistiocytosis following the BNT162b2 mRNA COVID-19 vaccination. Haematologica 2021, 107, 1222–1225. [Google Scholar] [CrossRef]
- Baek, D.W.; Hwang, S.; Kim, J.; Lee, J.M.; Cho, H.J.; Moon, J.-H.; Hwang, N.; Jeong, J.Y.; Lee, S.-W.; Sohn, S.K. Patients presenting high fever with lymphadenopathy after COVID-19 vaccination were diagnosed with hemophagocytic lymphohistiocytosis. Infect. Dis. 2021, 54, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Sassi, M.; Khefacha, L.; Merzigui, R.; Rakez, R.; Boukhriss, S.; Laatiri, M.A. Haemophagocytosis and atypical vacuolated lymphocytes in bone marrow and blood films after SARS-CoV-2 vaccination. Br. J. Haematol. 2021, 195, 649. [Google Scholar] [CrossRef] [PubMed]
- Rocco, J.M.; Mallarino-Haeger, C.; Randolph, A.H.; Ray, S.M.; Schechter, M.C.; Zerbe, C.S.; Holland, S.M.; Sereti, I. Hyperinflammatory Syndromes After Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Messenger RNA vaccination in Individuals With Underlying Immune Dysregulation. Clin. Infect. Dis. 2022, 75, e912–e915. [Google Scholar] [CrossRef]
- Attwell, L.; Zaw, T.; McCormick, J.; Marks, J.; McCarthy, H. Haemophagocytic lymphohistiocytosis after ChAdOx1 nCoV-19 vac-cination. J. Clin. Pathol. 2022, 75, 282–284. [Google Scholar] [CrossRef]
- Wu, V.; Lopez, C.A.; Hines, A.M.; Barrientos, J.C. Haemophagocytic lymphohistiocytosis following COVID-19 mRNA vaccina-tion. BMJ Case Rep. 2022, 15, e247022. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Yeh, Y.H.; Chen, L.W.; Cheng, C.-N.; Chang, C.; Roan, J.-N.; Shen, C.-F. Hemophagocytic Lymphohistiocytosis Following BNT162b2 mRNA COVID-19 Vaccina-tion. Vaccines 2022, 10, 573. [Google Scholar] [CrossRef]
- Cory, P.; Lawrence, H.; Abdulrahim, H.; Mahmood-Rao, H.; Hussein, A.; Gane, J. Lessons of the month 3: Haemophagocytic lym-phohistiocytosis following COVID-19 vaccination (ChAdOx1 nCoV-19). Clin. Med. 2021, 21, e677–e679. [Google Scholar] [CrossRef]
- Minocha, P.; Choudhary, R.; Agrawal, A.; Sitaraman, S. Griscelli syndrome subtype 2 with hemophagocytic lympho-histiocytosis: A case report and review of literature. Intractable Rare Dis. Res. 2017, 6, 76–79. [Google Scholar] [CrossRef]
- Mishra, K.; Singla, S.; Sharma, S.; Saxena, R.; Batra, V.V. Griscelli syndrome type 2: A novel mutation in RAB27A gene with different clinical features in 2 siblings: A diagnostic conundrum. Korean J. Pediatr. 2014, 57, 91–95. [Google Scholar] [CrossRef]
- Tewari, N.; Rajwar, A.; Mathur, V.; Chaudhari, P.K. Oral features of Griscelli syndrome type II: A rare case report. Spéc. Care Dent. 2018, 38, 421–425. [Google Scholar] [CrossRef]
- Panigrahi, I.; Suthar, R.; Rawat, A.; Behera, B. Seizure as the Presenting Manifestation in Griscelli Syndrome Type 2. Pediatr. Neurol. 2015, 52, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Meschede, I.; Santos, T.; Izidoro-Toledo, T.; Gurgel-Gianetti, J.; Espreafico, E. Griscelli syndrome-type 2 in twin siblings: Case report and update on RAB27A human mutations and gene structure. Braz. J. Med. Biol. Res. 2008, 41, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Gotesman, R.; Ramien, M.; Armour, C.M.; Pham-Huy, A.; Kirshen, C. Cutaneous granulomas as the presenting manifestation of Griscelli syndrome type 2. Pediatr. Dermatol. 2020, 38, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, Q.; Gao, J. Langerhans cell histiocytosis complicated with hemophagocytic lymphohistiocytosis in a boy with a novel XIAP mutation: A case report. Medicine 2018, 97, e13019. [Google Scholar] [CrossRef]
- Kaya, Z.; Ehl, S.; Albayrak, M.; Maul-Pavicic, A.; Schwarz, K.; Kocak, U.; Ergun, M.A.; Gursel, T. A novel single point mutation of the LYST gene in two siblings with different phenotypic features of Chediak Higashi syndrome. Pediatr. Blood Cancer 2011, 56, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.; Agergaard, C.N.; Jakobsen, M.A.; Møller, M.B.; Fisker, N.; Barington, T. Infantile hemophagocytic lymphohistiocytosis in a case of chediak-higashi syndrome caused by a mutation in the LYST/CHS1 gene presenting with delayed umbilical cord de-tachment and diarrhea. J. Pediatr. Hematol. Oncol. 2015, 37, e73–e79. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, X.; Zhang, B.; Xuan, F.; Guo, H.; Ma, F. A novel frameshift mutation of Chediak-Higashi syndrome and treatment in the accelerated phase. Braz. J. Med. Biol. Res. 2017, 50, e5727. [Google Scholar] [CrossRef]
- Morrone, K.; Wang, Y.; Huizing, M.; Sutton, E.; White, J.G.; Gahl, W.A.; Moody, K. Two Novel Mutations Identified in an African-American Child with Chediak-Higashi Syndrome. Case Rep. Med. 2010, 2010, 967535. [Google Scholar] [CrossRef]
- Sheng, L.; Zhang, W.; Gu, J.; Shen, K.; Luo, H.; Yang, Y. Novel mutations of STXBP2 and LYST associated with adult haemophagocytic lymphohistiocytosis with Epstein-Barr virus infection: A case report. BMC Med. Genet. 2019, 20, 34. [Google Scholar] [CrossRef]
- Larson, K.N.; Gaitan, S.R.; Stahr, B.J.; Morrell, D.S. Hemophagocytic Lymphohistiocytosis in a Newborn Presenting as “Blueberry Muffin Baby”. Pediatr. Dermatol. 2017, 34, e150–e151. [Google Scholar] [CrossRef]
- Viñas-Giménez, L.; Rincón, R.; Colobran, R.; de la Cruz, X.; Celis, V.P.; Dapena, J.L.; Alsina, L.; Sayós, J.; Martínez-Gallo, M. Case Report: Characterizing the Role of the STXBP2-R190C Monoallelic Muta-tion Found in a Patient With Hemophagocytic Syndrome and Langerhans Cell Histiocytosis. Front. Immunol. 2021, 12, 723836. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Guo, X.; Li, Q.; Huang, Z. Familial hemophagocytic lymphohistiocytosis type 5 in a Chinese Tibetan patient caused by a novel compound heterozygous mutation in STXBP2. Medicine 2019, 98, e17674. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, C.; Jorini, M.; Carloni, I.; Giangiacomi, M.; Cetica, V.; Aricò, M.; de Benedictis, F.M. Cytophagic histiocytic panniculitis, hemophagocytic lymphohistiocytosis and unde-termined autoimmune disorder: Reconciling the puzzle. Ital. J. Pediatr. 2014, 40, 17. [Google Scholar] [CrossRef]
- Chen, R.-L.; Hsu, Y.-H.; Ueda, I.; Imashuku, S.; Takeuchi, K.; Tu, B.P.-H.; Chuang, S.-S. Cytophagic histiocytic panniculitis with fatal haemophagocytic lymphohistiocytosis in a paediatric patient with perforin gene mutation. J. Clin. Pathol. 2007, 60, 1168–1169. [Google Scholar] [CrossRef]
- Akyol, S.; Ozcan, A.; Sekine, T.; Chiang, S.C.C.; Yilmaz, E.; Karakurkcu, M.; Patiroglu, T.; Bryceson, Y.; Unal, E. Different Clinical Presentation of 3 Children With Familial Hemophagocytic Lymphohisti-ocytosis With 2 Novel Mutations. J. Pediatr. Hematol. Oncol. 2020, 42, e627–e629. [Google Scholar] [CrossRef] [PubMed]
- Zengin, H.B.; Reyes-Barron, C.; Cusick, E.; Cordisco, M.; Katzman, P.J.; Burack, W.R.; Scott, G.A. Young Boy With Hemophagocytic Lymphohistiocytosis Presenting With Vac-cine-Related Granulomatous Dermatitis: A Case Report and Literature Review. Am. J. Dermatopathol. 2021, 43, e267–e272. [Google Scholar] [CrossRef]
- Wysocki, C.A. Comparing hemophagocytic lymphohistiocytosis in pediatric and adult patients. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 405–413. [Google Scholar] [CrossRef]
- Mascellino, M.T.; Di Timoteo, F.; De Angelis, M.; Oliva, A. Overview of the Main Anti-SARS-CoV-2 Vaccines: Mechanism of Action, Efficacy and Safety. Infect. Drug Resist. 2021, 14, 3459–3476. [Google Scholar] [CrossRef]
- Sprute, R.; Hieber, M.L.; Jachimowicz, R.D. Correspondence to: Hemophagocytic lymphohistiocytosis after SARS-CoV-2 vaccination. Infection 2022, 1–2. [Google Scholar] [CrossRef]
- Miao, Y.; Zhu, H.-Y.; Qiao, C.; Xia, Y.; Kong, Y.; Zou, Y.-X.; Miao, Y.-Q.; Chen, X.; Cao, L.; Wu, W.; et al. Pathogenic Gene Mutations or Variants Identified by Targeted Gene Sequencing in Adults With Hemophagocytic Lymphohistiocytosis. Front. Immunol. 2019, 10, 395. [Google Scholar] [CrossRef]
- Usmani, G.N.; Woda, B.A.; Newburger, P.E. Advances in understanding the pathogenesis of HLH. Br. J. Haematol. 2013, 161, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Risma, K.A.; Marsh, R.A. Hemophagocytic Lymphohistiocytosis: Clinical Presentations and Diagnosis. J. Allergy Clin. Immunol. Pract. 2018, 7, 824–832. [Google Scholar] [CrossRef]
- Zerah, M.L.; DeWitt, C.A. Cutaneous Findings in Hemophagocytic Lymphohistiocytosis. Dermatology 2015, 230, 234–243. [Google Scholar] [CrossRef]
- Fardet, L.; Galicier, L.; Vignon-Pennamen, M.D.; Regnier, S.; Noguera, M.E.; de Labarthe, A.; Raffoux, E.; Martinez, V.; Buyse, S.; Viguier, M.; et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with re-active haemophagocytic syndrome. Br. J. Dermatol. 2010, 162, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Morrell, D.S.; Pepping, M.A.; Scott, J.P.; Esterly, N.B.; Drolet, B.A. Cutaneous manifestations of hemophagocytic lymphohistio-cytosis. Arch. Dermatol. 2002, 138, 1208–1212. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tests on Admission | Results | Normal Ranges | Tests on Admission | Results | Normal Ranges |
|---|---|---|---|---|---|
| Complete blood count | Hepatic and renal function | ||||
| White blood cell (109/L) | 2.09 | 3.5–9.5 | ALT (U/L) | 27.8 | 5–46 |
| Neutrophil count (109/L) | 1.50 | 1.8–6.3 | AST (U/L) | 66.7 | 8–40 |
| Lymphocyte count (109/L) | 0.43 | 1.1–3.2 | Lactate dehydrogenase (U/L) | 668 | 100–245 |
| Hemoglobin (g/L) | 147.0 | 130–175 | Serum creatinine (umol/L) | 69.3 | 35–115 |
| Platelet (109/L) | 111 | 125–350 | Blood urea nitrogen (mmol/L) | 4.97 | 2.9–8.2 |
| Coagulation | Fasting triglycerides (mg/dL) | 1.96 | 0.46–2.25 | ||
| APTT (s) | 34.00 | 25–37 | Ferritin (ng/mL) | 18,669.00 | 24.00–336.2 |
| PT (s) | 11.80 | 9–13.5 | Soluble IL-2 receptor (U/mL) | 103,915 | <6400 |
| Thrombin time (s) | 18.80 | 10.3–16.6 | NK cell (%) | 11.54 | ≥15.11 |
| Fibrinogen (g/L) | 1.83 | 2–4 | |||
| D-Dimer (mg/mL) | 322.00 | 0–550 |
| Case | Sex, Age | Symptoms Onset after Vaccination | Clinical Manifestations | Medical History | Other Cause of HLH | Gene Mutations | Treatment | Outcome |
|---|---|---|---|---|---|---|---|---|
| Our case | M, 38 y | 4 weeks | Annular erythema, fever, fatigue | Interstitial lung disease | None | UNC13D | Methylprednisolone, etoposide, doxorubicin, HSCT | Improved and under follow-up |
| Tang et al., J Hematol Oncol, 2021 [6] | M, 43 y | Shortly after vaccination | Fever, vomiting, malaise | None | EBV infection | Absent | Dexamethasone | Discharged |
| Hieber et al., Infection, 2022 [7] | F, 24 y | 10 days | Fatigue, fever, chills, nausea | None | None | Not tested | IVIG, dexamethasone, Anakinra | Discharged |
| Ai et al., J Med Virol, 2022 [8] | M, 68 y | 10 days | Fevers, rigors, lethargy, night sweats | Hypertension, gout, Bowen’s disease | None | Not tested | No therapy | Spontaneous improvement |
| Nasir et al., J Clin Transl Res, 2022 [9] | M, 46 y | 3 weeks | Fever, fatigue, disturbed sleep, reduced appetite, skin rashes, oral ulcers | None | None | Not tested | Dexamethasone | Improved |
| Caocci et al., Haematologica, 2021 [10] | M, 38 y | 21 days | Fever, chills, fatigue, erythematous papules | None | None | Not tested | Methylprednisolone | Fully recovered |
| Baek et al., Infect Dis (Lond), 2021 [11] | M, 20 y | 2 days | Fever, myalgia, nausea, skin rashes | None | Not provided | Not tested | Dexamethasone | Immediate improvement |
| F, 71 y | 7 days | Fever, neurologic symptoms | Hypertension | Not provided | Not tested | Dexamethasone, etoposide | Discharged | |
| Sassi et al., Br J Haematol, 2021 [12] | M, 85 y | Shortly after vaccination | Anorexia, asthenia, pruritus | None | Not provided | Not tested | Not provided | Not provided |
| Rocco et al., Clin Infect Dis, 2021 [13] | M, 52 y | 1 day | Fever, abdominal pain | A viral syndrome | T-cell lymphoma, EBV viremia | Not tested | Dexamethasone, etoposide | Death |
| M, 53 y | 4 days | Fever, worsening hypoxia | Interstitial lung disease | EBV viremia | Not tested | Dexamethasone, Anakinra, IVIG, rituximab | Discharged to rehab facility | |
| M, 57 y | 12 days | Malaise, nausea | Heart failure, HIV infection, Mycobacterium avium, KSHV viremia | Kaposi sarcoma herpesvirus viremia | Not tested | Methylprednisolone | Death | |
| F, 55 y | 3 days | Fever | Adult-onset Still’s disease, pulmonary aspergillosis, GATA2 deficiency | Not found | Not tested | Anakinra | Slowly recovered | |
| F, 48 y | 4 days | Fever, cough, pleuritic chest pain | HIV | Not found | Not tested | Prednisone, infliximab | Improvement within 72 h | |
| Attwell et al., J Clin Pathol, 2022 [14] | M, 65 y | 5 days | Breathlessness, fever, myalgia | Type 2 diabetes mellitus | Not found | Not tested | Methylprednisolone, IVIG, Anakinra | Deteriorated |
| F, 75 y | 7 days | Night sweats, breathlessness, myalgia, fever, cough | JAK2-mutation positive essential thrombocythaemia, breast cancer | Not found | Not tested | Methylprednisolone, IVIG, Anakinra | Death | |
| M, 35 y | 8 days | Fever, diarrhea, sore throat, pruritic rash, breathlessness | Ankylosing spondylitis | Not found | Not tested | Methylprednisolone | Responded well | |
| Wu et al., BMJ Case Rep,2022 [15] | M, 60 y | 6 days | Fevers, night sweats, loss of appetite, delirium, non-ambulatory | Barrett’s esophagus | Not found | Not tested | Dexamethasone, etoposide | Discharged, but relapsed and deteriorated |
| F, 32 y | 4 weeks | Fever | None | Not found | Not provided | Dexamethasone, etoposide, emapalumab-lzsg | Discharged, but deteriorated | |
| Lin et al., Vaccines, 2022 [16] | F, 14 y | 15 days | Fever, headache, nausea, tachypnea, drowsy consciousness, mottling skin, jaundice | None | EBV viremia | Not tested | Methylprednisolone, IVIG | Discharged |
| Cory et al., Clin Med (Lond), 2021 [17] | F, 36 y | 9 days | Fever, myalgia, sore throat, mild facial swelling | None | Not found | Not tested | Methylprednisolone, IVIG | Improvement |
| Case | Sex, Age | Gene Defects | HLH Type | Initial Symptoms | Cutaneous Manifestations | Skin Histologic Findings | Treatments | Outcome |
|---|---|---|---|---|---|---|---|---|
| Minocha et al., Intractable Rare Dis Res, 2017 [18] | M, 20 m | RAB27A | Griscelli syndrome type 2 | Fever, jaundice, pallor, weight loss | Icterus, silvery-gray hair, and hypopigmented skin | Not provided | Mycophenolate mofetil, HSCT | Improved and under follow-up |
| Mishra et al., Korean J Pediatr, 2014 [19] | M, 5 y | RAB27A | Griscelli syndrome type 2 | Fever, skin lesion | A generalized hypopigmented skin and multiple erythematous to hyperpigmented, nodular lesions, extending from midthighs to feet | Widening of septae in the subcutaneous tissue, infiltration of the periphery of the fat lobule by chronic inflammatory cells | Prednisolone, HSCT | Not provided |
| Tewari et al., Spec Care Dentist, 2018 [20] | M, 4 y | RAB27A | Griscelli syndrome type 2 | Pain in the oral cavity and tooth decay | Silvery white hair and white skin | Not provided | Not provided | Not provided |
| Panigrahi et al., Pediatr Neurol, 2015 [21] | F, 1 y | RAB27A | Griscelli syndrome type 2 | Fever | Silvery white hairs, silvery eyelashes | Not provided | Methylprednisolone | Died |
| Meschede et al., Braz J Med Biol Res, 2008 [22] | M, 3 y | RAB27A | Griscelli syndrome type 2 | Fever | Light silvery-gray colored scalp hair and eyebrows | Not provided | Corticoid, cyclosporine | Died |
| Gotesman et al., Pediatr Dermatol, 2020 [23] | F, 18 m | RAB27A | Griscelli syndrome type 2 | Skin lesion | Non-pruritic, erythematous-violaceous papules, and dry, coarse, silvery-gray hair | A granulomatous inflammatory process | HSCT | Improvement |
| Guo et al., Medicine, 2018 [24] | M, 13 m | XIAP | X-linked lymphoproliferative syndrome | Fever, skin lesion, recurrent ear discharge | Widespread hemorrhagic skin eruptions | Langerhans cell histiocytosis (LCH) | HLH-2004- directed chemotherapy | Died |
| Kaya et al., Pediatr Blood Cancer, 2011 [25] | F, 4 y | LYST | Chediak–Higashi syndrome | Fever, pallor, lethargy, poor appetite | Speckled hypopigmented areas | Not provided | HLH-2004 therapy | Complete remission |
| Nielsen et al., J Pediatr Hematol Oncol, 2015 [26] | F, 2 m | LYST | Chediak–Higashi syndrome | Coryza, coughing, skin eruption | A pustule skin eruption, fair hair, pale and wax-like skin | Numerous large inclusion bodies in mast cells, and an epidermis virtually absent of melanin in both melanocytes and keratinocytes | Etoposide, dexamethasone, cyclosporine, IVIG | Died |
| Wu et al., Braz J Med Biol Res, 2017 [27] | M, 9 m | LYST | Chediak–Higashi syndrome | Fever | Mild pallor, gray hair, patchy hypopigmentation of the skin, red rashes on the trunk | Not provided | Cyclosporine A, dexamethasone, etoposide; HSCT has been planned | Temporary remission |
| Morrone et al., Case Rep Med, 2010 [28] | F, 16 m | CHD1 | Chediak–Higashi syndrome | Fever, decreased activity, increased sleepiness, irritability | Silvery hair, pale skin, edematous eyelids | Not provided | Etoposide, dexamethasone, cyclosporine | Died |
| Sheng et al., BMC Med Genet, 2019 [29] | F, 30 y | STXBP2, LYST | Not provided | Fever, fatigue | Oedematose swelling of the face and coexistent skin lesions | Not provided | Etoposide, dexamethasone, currently waiting for HSCT | Well-controlled for a month |
| Larson et al., Pediatr Dermatol, 2017 [30] | M, 6 d | PRF1 | F-HLH type 2 | Skin lesion | Multiple blue–purple violaceous nodules | An intact epidermis and an underlying dermal infiltrate of mononuclear cells | Not provided | Not provided |
| Viñas et al., Front Immunol, 2021 [31] | M, 16 m | STXBP2 | F-HLH type 5 | Skin lesion, fever, vomit, diarrhea, edema | An exacerbation of cutaneous Langerhans cell histiocytosis, edema | Not provided | Not provided | Not provided |
| Tang et al., Medicine, 2019 [32] | F, 9 y | STXBP2 | F-HLH type 5 | Fever | Ecchymosis and edema of the lower extremities | Not provided | HLH-2004-directed chemotherapy, HSCT | Died |
| Pasqualin et al., Ital J Pediatr, 2014 [33] | M, 11 y | STX11 | F-HLH type 4 | Fever, dyspnea | A warm, painful, indurated plaque with a brownish, hyperpigmented over the right thigh | Mixed septal and lobular inflammatory infiltrate of benign-appearing histiocytes, plasma cells and lymphocytes, and diffuse fat necrosis | Methylprednisolone, cyclosporine | Remission was sustained at 6-month follow-up |
| Chen et al., J Clin Pathol, 2007 [34] | F, 11 y | PRF1 | F-HLH type 2 | Fever, skin lesion | Indurated skin nodules over the left thigh | Lobular panniculitis with lymphocytic infiltration with occasional benign histiocytes showing hemophagocytosis | 13-cis retinoic acid, prednisolone | Died |
| Akyol et al., J Pediatr Hematol Oncol, 2020 [35] | F, 21 m | UNC13D | F-HLH type 3 | Fever, skin lesion | Widespread maculopapular rash | Not provided | Not provided | Not provided |
| Zengin et al., Am J Dermatopathol, 2021 [36] | M, 4 y | UNC13D | F-HLH type 3 | Skin lesion | A widespread popular–pustular rash | A cup-shaped depression of the epidermis, which exhibited perforation with necrobiotic collagen. Necrobiosis with palisading histiocytes and lymphoplasmacytic inflammatory cell infiltration in the dermis | Dexamethasone, HSCT | Not provided |
| our case | M, 36 y | UNC13D | F-HLH type 3 | Skin lesion | Widespread annular erythema, facial edema | Lymphohistiocytic infiltration in the dermis, as well as reactive hyperplasias of lymphoid tissue dominated by cytotoxic T-cells | Methylprednisolone, etoposide, doxorubicin, HSCT | Improved and under follow-up |
| Type of HLH | Defective Gene | |
|---|---|---|
| Familial HLH (F-HLH) | F-HLH type 2 | PRF1 |
| F-HLH type 3 | UNC13D | |
| F-HLH type 4 | STX11 | |
| F-HLH type 5 | STXBP2 | |
| Immuno-deficiency syndromes | Griscelli syndrome type 2 | RAB27A |
| Chediak–Higashi syndrome | LYST | |
| Hermansky–Pudlak syndrome type 2 | AP3B1 | |
| EBV-driven | X-linked lymphoproliferative disorder type 1 (XLP-1) | SH2D1A |
| X-linked lymphoproliferative disorder type 2 (XLP-2) | BIRC4 | |
| IL2-inducible T-cell kinase deficiency | ITK | |
| CD27 deficiency | CD27 | |
| X-linked immunodeficiency with magnesium defect (XMEN) | MAGT1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Hui, Y.; Liu, H.; Wu, Y.; Sang, H.; Liu, F. Adult-Onset Familial Hemophagocytic Lymphohistiocytosis Presenting with Annular Erythema following COVID-19 Vaccination. Vaccines 2022, 10, 1436. https://doi.org/10.3390/vaccines10091436
He Y, Hui Y, Liu H, Wu Y, Sang H, Liu F. Adult-Onset Familial Hemophagocytic Lymphohistiocytosis Presenting with Annular Erythema following COVID-19 Vaccination. Vaccines. 2022; 10(9):1436. https://doi.org/10.3390/vaccines10091436
Chicago/Turabian StyleHe, Yifan, Yun Hui, Haibo Liu, Yifan Wu, Hong Sang, and Fang Liu. 2022. "Adult-Onset Familial Hemophagocytic Lymphohistiocytosis Presenting with Annular Erythema following COVID-19 Vaccination" Vaccines 10, no. 9: 1436. https://doi.org/10.3390/vaccines10091436