Oxidative Stress in Cardiovascular Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
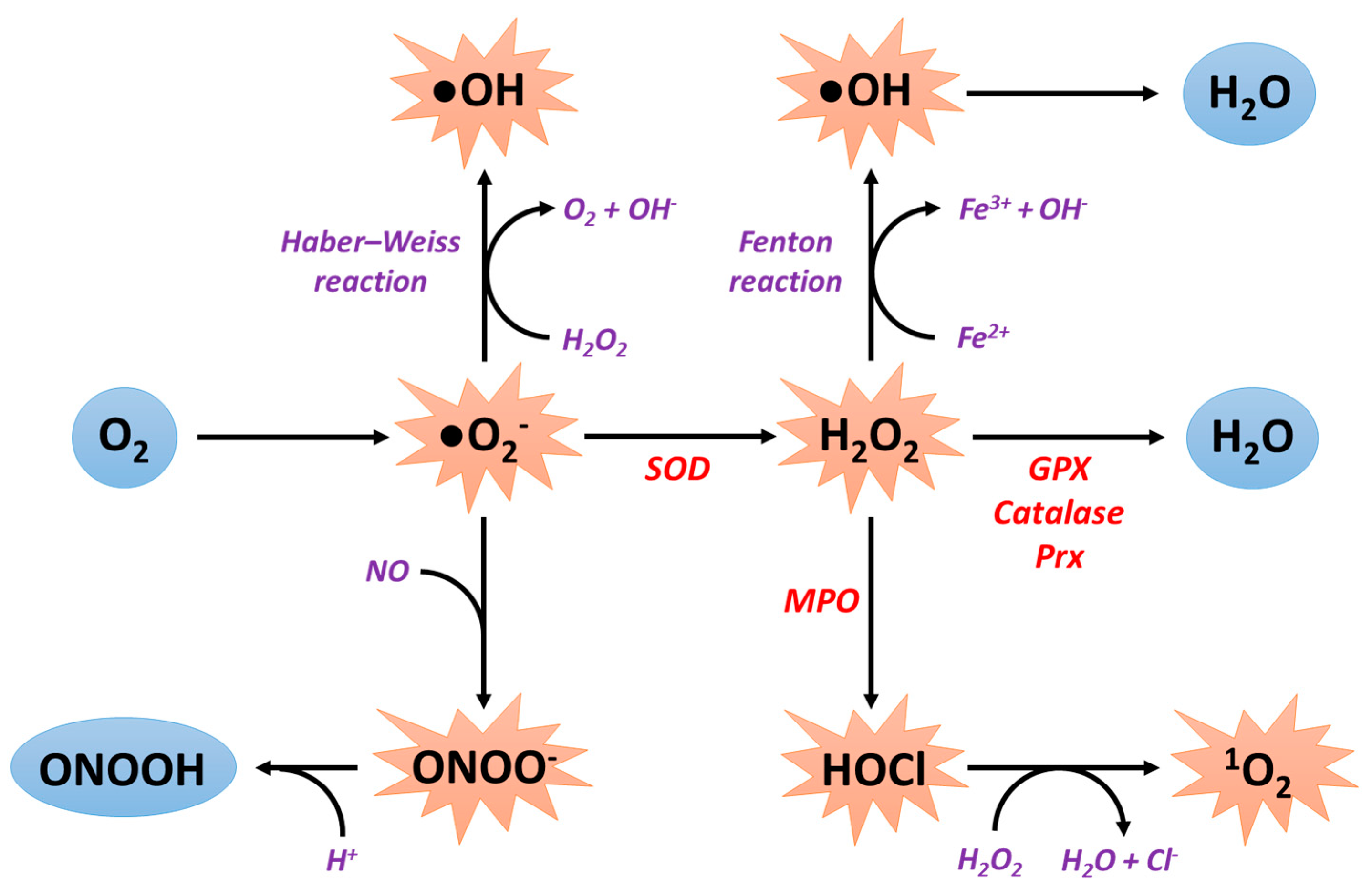
2. General Information about Reactive Oxygen Species Production
2.1. Sources of ROS Production
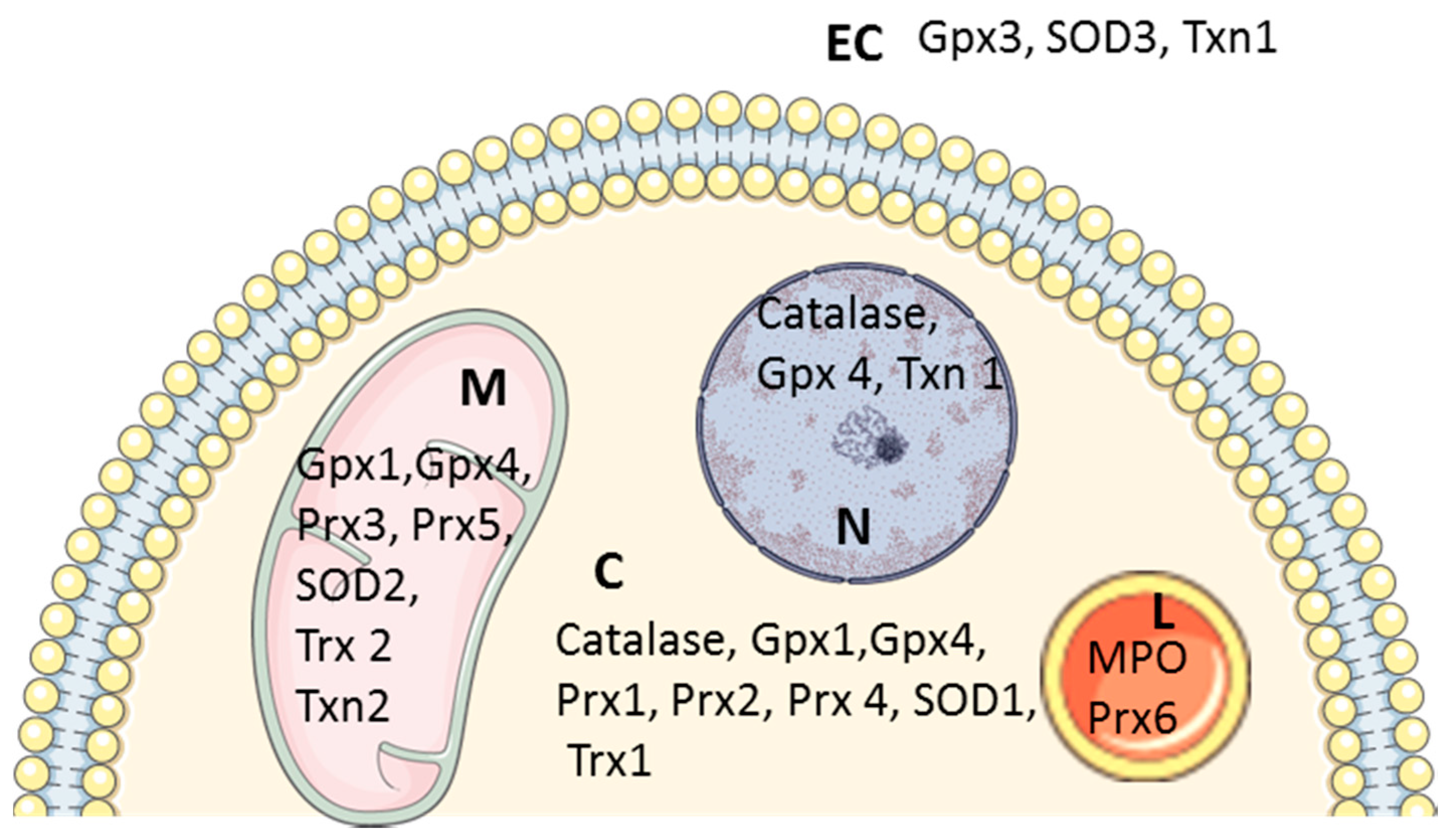
2.2. Antioxidant Systems
2.2.1. Superoxide Dismutases
2.2.2. Catalase
2.2.3. Peroxiredoxins
2.2.4. Glutathione Peroxidases
2.2.5. Non-Enzymatic Antioxidant Defense
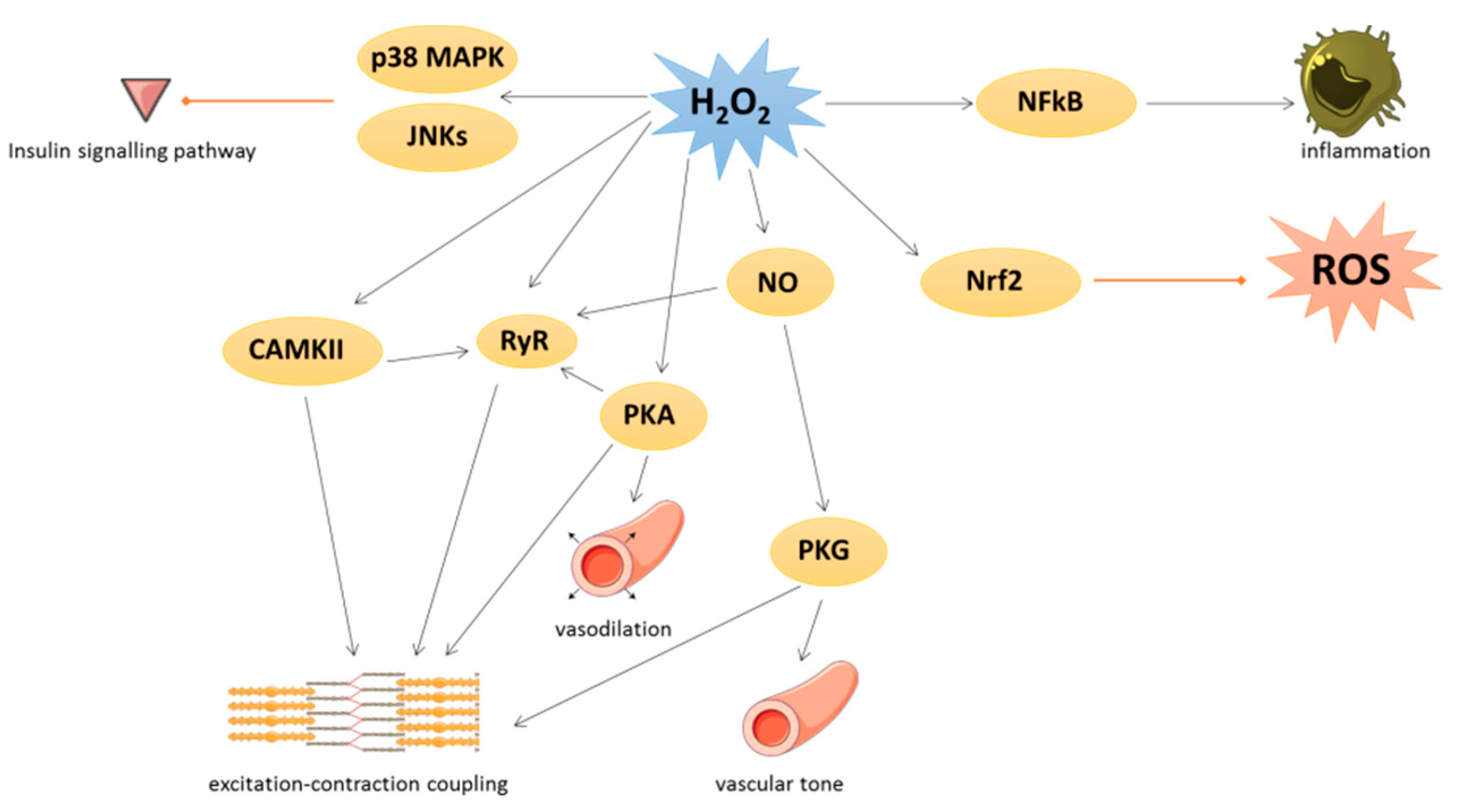
3. Physiological Roles of Oxidative Stress in Cardiovascular Tissues
4. Pathological Implications of Oxidative Stress in Cardiovascular Tissues
4.1. General Aspects
4.2. NOX-Dependent Effects
4.3. Endothelial Dysfunction
4.4. Mitochondrial Oxidative Stress
5. New Therapeutic Strategies
5.1. Vitamins
5.2. Polyphenols
5.3. Mitochondrial-Targeted Antioxidant
5.4. Clinical Trials
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Al Hariri, M.; Zibara, K.; Farhat, W.; Hashem, Y.; Soudani, N.; Al Ibrahim, F.; Hamade, E.; Zeidan, A.; Husari, A.; Kobeissy, F. Cigarette sImoking-induced cardiac hypertrophy, vascular inflammation and injury are attenuated by antioxidant supplementation in an animal model. Front. Pharmacol. 2016, 7, e397. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.; Itani, H.; Richmond, B.; Vergeade, A.; Rahman, S.M.J.; Boutaud, O.; Blackwell, T.; Massion, P.P.; Harrison, D.G.; Dikalova, A. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am. J. Physiol.-Heart Circ. Physiol. 2019, 316, H639–H646. [Google Scholar] [CrossRef]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for Cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal 2012, 16, 1295–1322. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, 2181–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Rad, M.; Kumar, N.V.A.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Fokou, P.V.T.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Cuvelliez, M.; Fiedler, J.; Charrier, H.; Mulder, P.; Hebbar, E.; Pfanne, A.; Beseme, O.; Chwastyniak, M.; Amouyel, P.; et al. MicroRNAs regulating superoxide dismutase 2 are new circulating biomarkers of heart failure. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Strassburger, M.; Bloch, W.; Sulyok, S.; Schüller, J.; Keist, A.F.; Schmidt, A.; Wenk, J.; Peters, T.; Wlaschek, M.; Krieg, T.; et al. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic. Biol. Med. 2005, 38, 1458–1470. [Google Scholar] [CrossRef]
- Li, X.; Lin, Y.; Wang, S.; Zhou, S.; Ju, J.; Wang, X.; Chen, Y.; Xia, M. Extracellular Superoxide Dismutase Is Associated With Left Ventricular Geometry and Heart Failure in Patients with Cardiovascular Disease. J. Am. Heart Assoc. 2020, 9, e016862. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, H.S.; Moosavi-Movahedi, A.A. Catalase and its mysteries. Prog. Biophys. Mol. Biol. 2018, 140, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Detienne, G.; De Haes, W.; Mergan, L.; Edwards, S.L.; Temmerman, L.; Van Bael, S. Beyond ROS clearance: Peroxiredoxins in stress signaling and aging. Ageing Res. Rev. 2018, 44, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Kuzuya, K.; Ichihara, S.; Suzuki, Y.; Inoue, C.; Ichihara, G.; Kurimoto, S.; Oikawa, S. Proteomics analysis identified peroxiredoxin 2 involved in early-phase left ventricular impairment in hamsters with cardiomyopathy. PLoS ONE 2018, 13, e0192624. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola, J.; Arrieta, V.; Sádaba, R.; Martinez-Martinez, E.; Garcia-Peña, A.; Alvarez, V.; Fernández-Celis, A.; Gainza, A.; Santamaría, E.; Fernández-Irigoyen, J.; et al. Galectin-3 down-regulates antioxidant peroxiredoxin-4 in human cardiac fibroblasts: A new pathway to induce cardiac damage. Clin. Sci. 2018, 132, 1471–1485. [Google Scholar] [CrossRef]
- Cieniewski-Bernard, C.; Mulder, P.; Henry, J.-P.; Drobecq, H.; Dubois, E.; Pottiez, G.; Thuillez, C.; Amouyel, P.; Richard, V.; Pinet, F. Proteomic Analysis of Left Ventricular Remodeling in an Experimental Model of Heart Failure. J. Proteome Res. 2008, 7, 5004–5016. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol. 2013, 1, 244–257. [Google Scholar] [CrossRef] [Green Version]
- Park, T.J.; Park, J.H.; Lee, G.S.; Lee, J.Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.J.; Han, B.S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res. 2008, 81, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Sack, M.N.; Fyhrquist, F.Y.; Saijonmaa, O.J.; Fuster, V.; Kovacic, J.C. Basic Biology of Oxidative Stress and the Cardiovascular System: Part 1 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 196–211. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Moris, D.; Spartalis, M.; Tzatzaki, E.; Spartalis, E.; Karachaliou, G.S.; Triantafyllis, A.S.; Karaolanis, G.I.; Tsilimigras, D.I.; Theocharis, S. The role of reactive oxygen species in myocardial redox signaling and regulation. Ann. Transl. Med. 2017, 5, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal hydrogen peroxide metabolism and signaling in health and disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef] [Green Version]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- Loyer, X.; Heymes, C.; Samuel, J.L. Constitutive nitric oxide synthases in the heart from hypertrophy to failure. Clin. Exp. Pharmacol. Physiol. 2008, 35, 483–488. [Google Scholar] [CrossRef]
- Hammond, J.; Balligand, J.L. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: From contractility to remodeling. J. Mol. Cell. Cardiol. 2012, 52, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Lancel, S.; Qin, F.; Lennon, S.L.; Zhang, J.; Tong, X.; Mazzini, M.J.; Kang, Y.J.; Siwik, D.A.; Cohen, R.A.; Colucci, W.S. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ. Res. 2010, 107, 228–232. [Google Scholar] [CrossRef]
- Steinberg, S.F. Oxidative stress and sarcomeric proteins. Circ. Res. 2013, 112, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Hermida, N.; Michel, L.; Esfahani, H.; Dubois-Deruy, E.; Hammond, J.; Bouzin, C.; Markl, A.; Colin, H.; Van Steenbergen, A.; De Meester, C.; et al. Cardiac myocyte β3-adrenergic receptors prevent myocardial fibrosis by modulating oxidant stress-dependent paracrine signaling. Eur. Heart J. 2018, 39, 888–897. [Google Scholar] [CrossRef]
- Serpillon, S.; Floyd, B.C.; Gupte, R.S.; George, S.; Kozicky, M.; Neito, V.; Recchia, F.; Stanley, W.; Wolin, M.S.; Gupte, S.A. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H153. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-Specific Derangements in Mitochondrial Metabolism and Redox Balance in the Atrium of the Type 2 Diabetic Human Heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortuño, A.; José, G.S.; Moreno, M.U.; Beloqui, O.; Díez, J.; Zalba, G. Phagocytic NADPH oxidase overactivity underlies oxidative stress in metabolic syndrome. Diabetes 2006, 55, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.L.; Elezaby, A.; Qin, F.; Behring, J.B.; Luptak, I.; Calamaras, T.D.; Siwik, D.A.; Miller, E.J.; Liesa, M.; Shirihai, O.S.; et al. Mitochondrial reactive oxygen species mediate cardiac structural, functional, and mitochondrial consequences of diet-induced metabolic heart disease. J. Am. Heart Assoc. 2016, 5, e002555. [Google Scholar] [CrossRef] [Green Version]
- Jeong, E.M.; Chung, J.; Liu, H.; Go, Y.; Gladstein, S.; Farzaneh-Far, A.; Lewandowski, E.D.; Dudley, S.C. Role of Mitochondrial Oxidative Stress in Glucose Tolerance, Insulin Resistance, and Cardiac Diastolic Dysfunction. J. Am. Heart Assoc. 2016, 5, e003046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemann, B.; Chen, Y.; Teschner, M.; Li, L.; Silber, R.E.; Rohrbach, S. Obesity induces signs of premature cardiac aging in younger patients: The role of mitochondria. J. Am. Coll. Cardiol. 2011, 57, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-González, S.; Marín-Royo, G.; Jurado-López, R.; Bartolomé, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef] [Green Version]
- Li, J.M.; Gall, N.P.; Grieve, D.J.; Chen, M.; Shah, A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002, 40, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W.; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and gαq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Hsieh, E.J.; Liu, Y.; Chen, T.; Beyer, R.P.; Chin, M.T.; MacCoss, M.J.; Rabinovitch, P.S. Mitochondrial proteome remodelling in pressure overload-induced heart failure: The role of mitochondrial oxidative stress. Cardiovasc. Res. 2012, 93, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.I.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Merabet, N.; Bellien, J.; Glevarec, E.; Nicol, L.; Lucas, D.; Remy-Jouet, I.; Bounoure, F.; Dreano, Y.; Thuillez, C.; Mulder, P. Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J. Mol. Cell. Cardiol. 2012, 52, 660–666. [Google Scholar] [CrossRef]
- Santillo, M.; Colantuoni, A.; Mondola, P.; Guida, B.; Damiano, S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front. Physiol. 2015, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.; Yu, L.; Liang, P.; Li, L.; Dai, X.; Zhou, B.; Wu, X.; Xu, H.; Fang, M.; Chen, Q.; et al. A crosstalk between chromatin remodeling and histone H3K4 methyltransferase complexes in endothelial cells regulates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2015, 82, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, J.R.; Mei-ling, A.J.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A Dynamic Pathway for Calcium-Independent Activation of CaMKII by Methionine Oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Rodríguez Guzmán, R.; Centofanti, F.; Doldo, E.; Miranda, E.M.C.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; Van Der Laarse, A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol. Cell. Biochem. 2010, 333, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Wang, K.; Shang, J.; Cao, C.; Zhen, P.; Liu, X.; Wang, W.; Zhang, H.; Du, Y.; Liu, H. Anti-peroxynitrite treatment ameliorated vasorelaxation of resistance arteries in aging rats: Involvement with NO-sGC-cGKs pathway. PLoS ONE 2014, 9, e104788. [Google Scholar] [CrossRef]
- Mollnau, H.; Oelze, M.; August, M.; Wendt, M.; Daiber, A.; Schulz, E.; Baldus, S.; Kleschyov, A.L.; Materne, A.; Wenzel, P.; et al. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2554–2559. [Google Scholar] [CrossRef] [Green Version]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of mitochondrial reverse electron transport in ROS signaling: Potential roles in health and disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Mazat, J.P.; Devin, A.; Ransac, S. Modelling mitochondrial ROS production by the respiratory chain. Cell. Mol. Life Sci. 2020, 77, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Rohrbach, S.; Miller, M.R.; Newby, D.E.; Fuster, V.; Kovacic, J.C. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 230–251. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Rababa’h, A.M.; Guillory, A.N.; Mustafa, R.; Hijjawi, T. Oxidative Stress and Cardiac Remodeling: An Updated Edge. Curr. Cardiol. Rev. 2018, 14, 53–59. [Google Scholar] [CrossRef]
- Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications. Ann. Med. 2011, 43, 259–272. [Google Scholar] [CrossRef]
- Stamler, J.S.; Osborne, J.A.; Jaraki, O.; Rabbani, L.E.; Mullins, M.; Singel, D.; Loscalzo, J. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J. Clin. Investig. 1993, 91, 308–318. [Google Scholar] [CrossRef]
- McCully, K.S. Homocystinuria, Arteriosclerosis, Methylmalonic Aciduria, and Methyltransferase Deficiency: A Key Case Revisited. Nutr. Rev. 1992, 50, 7–12. [Google Scholar] [CrossRef]
- Cianciolo, G.; De Pascalis, A.; Di Lullo, L.; Ronco, C.; Zannini, C.; La Manna, G. Folic acid and homocysteine in chronic kidney disease and cardiovascular disease progression: Which comes first? CardioRenal Med. 2017, 7, 255–266. [Google Scholar] [CrossRef]
- Stroes, E.S.G.; Van Faassen, E.E.; Yo, M.; Martasek, P.; Boer, P.; Govers, R.; Rabelink, T.J. Folic acid reverts dysfunction of endothelial nitric oxide synthase. Circ. Res. 2000, 86, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Hagar, H.H. Folic acid and vitamin B12 supplementation attenuates isoprenaline-induced myocardial infarction in experimental hyperhomocysteinemic rats. Pharmacol. Res. 2002, 46, 213–219. [Google Scholar] [CrossRef]
- Farhangi, M.A.; Nameni, G.; Hajiluian, G.; Mesgari-Abbasi, M. Cardiac tissue oxidative stress and inflammation after vitamin D administrations in high fat- diet induced obese rats. BMC Cardiovasc. Disord. 2017, 17, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leme Goto, P.; Cinato, M.; Merachli, F.; Vons, B.; Jimenez, T.; Marsal, D.; Todua, N.; Loi, H.; Santin, Y.; Cassel, S.; et al. In vitro and in vivo cardioprotective and metabolic efficacy of vitamin E TPGS/Apelin. J. Mol. Cell. Cardiol. 2020, 138, 165–174. [Google Scholar] [CrossRef]
- Wallert, M.; Ziegler, M.; Wang, X.; Maluenda, A.; Xu, X.; Yap, M.L.; Witt, R.; Giles, C.; Kluge, S.; Hortmann, M.; et al. α-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. 2019, 26, 101292. [Google Scholar] [CrossRef]
- Banez, M.J.; Geluz, M.I.; Chandra, A.; Hamdan, T.; Biswas, O.S.; Bryan, N.S.; Von Schwarz, E.R. A systemic review on the antioxidant and anti-inflammatory effects of resveratrol, curcumin, and dietary nitric oxide supplementation on human cardiovascular health. Nutr. Res. 2020, 78, 11–26. [Google Scholar] [CrossRef]
- Agouni, A.; Lagrue-Lak-Hal, A.H.; Mostefai, H.A.; Tesse, A.; Mulder, P.; Rouet, P.; Desmoulin, F.; Heymes, C.; Martínez, M.C.; Andriantsitohaina, R. Red wine polyphenols prevent metabolic and cardiovascular alterations associated with obesity in Zucker fatty rats (Fa/Fa). PLoS ONE 2009, 4, e5557. [Google Scholar] [CrossRef] [Green Version]
- Farkhondeh, T.; Folgado, S.L.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Samarghandian, S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomed. Pharmacother. 2020, 127, 110234. [Google Scholar] [CrossRef]
- Dyck, G.J.B.; Raj, P.; Zieroth, S.; Dyck, J.R.B.; Ezekowitz, J.A. The effects of resveratrol in patients with cardiovascular disease and heart failure: A narrative review. Int. J. Mol. Sci. 2019, 20, 904. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.P.; Hsu, S.C.; Li, D.E.; Chen, K.H.; Kuo, C.Y.; Hung, L.M. Resveratrol mitigates high-fat diet-induced vascular dysfunction by activating the Akt/eNOS/NO and Sirt1/ER pathway. J. Cardiovasc. Pharmacol. 2018, 72, 231–241. [Google Scholar] [CrossRef]
- Chan, A.Y.M.; Dolinsky, V.W.; Soltys, C.-L.M.; Viollet, B.; Baksh, S.; Light, P.E.; Dyck, J.R.B. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J. Biol. Chem. 2008, 283, 24194–24201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, M.M.; Das, S.K.; Levasseur, J.; Byrne, N.J.; Fung, D.; Kim, T.T.; Masson, G.; Boisvenue, J.; Soltys, C.L.; Oudit, G.Y.; et al. Resveratrol treatment of mice with pressure-overloadinduced heart failure improves diastolic function and cardiac energy metabolism. Circ. Heart Fail. 2015, 8, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, M.; Kuno, A.; Yano, T.; Miura, T.; Hisahara, S.; Ishikawa, S.; Shimamoto, K.; Horio, Y. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J. Biol. Chem. 2010, 285, 8375–8382. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, H.N. Targeting mitochondrial dysfunction in the treatment of heart failure. Expert Rev. Cardiovasc. Ther. 2016, 14, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Tissier, S.L.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Song, J.; Ernst, P.; Latimer, M.N.; Ha, C.M.; Goh, K.Y.; Ma, W.; Rajasekaran, N.S.; Zhang, J.; Liu, X.; et al. MitoQ regulates redox-related noncoding RNAs to preserve mitochondrial network integrity in pressure-overload heart failure. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H682–H695. [Google Scholar] [CrossRef]
- Rao, V.A.; Klein, S.R.; Bonar, S.J.; Zielonka, J.; Mizuno, N.; Dickey, J.S.; Keller, P.W.; Joseph, J.; Kalyanaraman, B.; Shacter, E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J. Biol. Chem. 2010, 285, 34447–34459. [Google Scholar] [CrossRef] [Green Version]
- Doughan, A.K.; Dikalov, S.I. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid. Redox Signal 2007, 9, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Pokrzywinski, K.L.; Biel, T.G.; Kryndushkin, D.; Rao, V.A. Therapeutic targeting of the mitochondria initiates excessive superoxide production and mitochondrial depolarization causing decreased mtDNA integrity. PLoS ONE 2016, 11, e0168283. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.M.; Cook, N.R.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.A.E.; Hennekens, C.H.; Buring, J.E. Vitamin E in the primary prevention of cardiovascular disease and cancer. The women’s health study: A randomized controlled trial. J. Am. Med. Assoc. 2005, 294, 56–65. [Google Scholar] [CrossRef]
- Toole, J.F.; Malinow, M.R.; Chambless, L.E.; Spence, J.D.; Pettigrew, L.C.; Howard, V.J.; Sides, E.G.; Wang, C.H.; Stampfer, M. Lowering Homocysteine in Patients with Ischemic Stroke to Prevent Recurrent Stroke, Myocardial Infarction, and Death: The Vitamin Intervention for Stroke Prevention (VISP) Randomized Controlled Trial. J. Am. Med. Assoc. 2004, 291, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Lonn, E. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: A randomized controlled trial. J. Am. Med. Assoc. 2005, 293, 1338–1347. [Google Scholar]
- Bønaa, K.H.; Njølstad, I.; Ueland, P.M.; Schirmer, H.; Tverdal, A.; Steigen, T.; Wang, H.; Nordrehaug, J.E.; Arnesen, E.; Rasmussen, K. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N. Engl. J. Med. 2006, 354, 1578–1588. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Mankowski, R.T.; You, L.; Buford, T.W.; Leeuwenburgh, C.; Manini, T.M.; Schneider, S.; Qiu, P.; Anton, S.D. Higher dose of resveratrol elevated cardiovascular disease risk biomarker levels in overweight older adults–A pilot study. Exp. Gerontol. 2020, 131, 110821. [Google Scholar] [CrossRef]
- Tomé-Carneiro, J.; Gonzálvez, M.; Larrosa, M.; Yáñez-Gascón, M.J.; García-Almagro, F.J.; Ruiz-Ros, J.A.; García-Conesa, M.T.; Tomás-Barberán, F.A.; Espín, J.C. One-year consumption of a grape nutraceutical containing resveratrol improves the inflammatory and fibrinolytic status of patients in primary prevention of cardiovascular disease. Am. J. Cardiol. 2012, 110, 356–363. [Google Scholar] [CrossRef]
- Macedo, R.C.S.; Vieira, A.; Marin, D.P.; Otton, R. Effects of chronic resveratrol supplementation in military firefighters undergo a physical fitness test-A placebo-controlled, double blind study. Chem. Biol. Interact. 2015, 227, 89–95. [Google Scholar] [CrossRef]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; Van De Weijer, T.; Goossens, G.H.; Hoeks, J.; Van Der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujitaka, K.; Otani, H.; Jo, F.; Jo, H.; Nomura, E.; Iwasaki, M.; Nishikawa, M.; Iwasaka, T.; Das, D.K. Modified resveratrol Longevinex improves endothelial function in adults with metabolic syndrome receiving standard treatment. Nutr. Res. 2011, 31, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Yamaguchi, T.; Nagayama, D.; Saiki, A.; Shirai, K.; Tatsuno, I. Resveratrol ameliorates arterial stiffness assessed by cardio-ankle vascular index in patients with type 2 diabetes mellitus. Int. Heart J. 2017, 58, 577–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Made, S.M.; Plat, J.; Mensink, R.P. Resveratrol does not influence metabolic risk markers related to cardiovascular health in overweight and slightly obese subjects: A randomized, placebo-controlled crossover trial. PLoS ONE 2015, 10, e0118393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, J.; Gliemann, L.; Biensø, R.; Schmidt, J.; Hellsten, Y.; Pilegaard, H. Exercise training, but not resveratrol, improves metabolic and inflammatory status in skeletal muscle of aged men. J. Physiol. 2014, 592, 1873–1886. [Google Scholar] [CrossRef]
- Agarwal, B.; Campen, M.J.; Channell, M.M.; Wherry, S.J.; Varamini, B.; Davis, J.G.; Baur, J.A.; Smoliga, J.M. Resveratrol for primary prevention of atherosclerosis: Clinical trial evidence for improved gene expression in vascular endothelium. Int. J. Cardiol. 2013, 166, 246–248. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. https://doi.org/10.3390/antiox9090864
Dubois-Deruy E, Peugnet V, Turkieh A, Pinet F. Oxidative Stress in Cardiovascular Diseases. Antioxidants. 2020; 9(9):864. https://doi.org/10.3390/antiox9090864
Chicago/Turabian StyleDubois-Deruy, Emilie, Victoriane Peugnet, Annie Turkieh, and Florence Pinet. 2020. "Oxidative Stress in Cardiovascular Diseases" Antioxidants 9, no. 9: 864. https://doi.org/10.3390/antiox9090864