Inactivation of Carbonyl-Detoxifying Enzymes by H2O2 Is a Trigger to Increase Carbonyl Load for Initiating Programmed Cell Death in Plants


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of Cells
2.2. Cell Viability Assay
2.3. Determination of C1LP and C3LP Activities
2.4. Analysis of Glutathione
2.5. Analysis of Ascorbate
2.6. Detection and Quantification of Carbonyls Using HPLC
2.7. Determination of Enzymatic Activity
3. Results
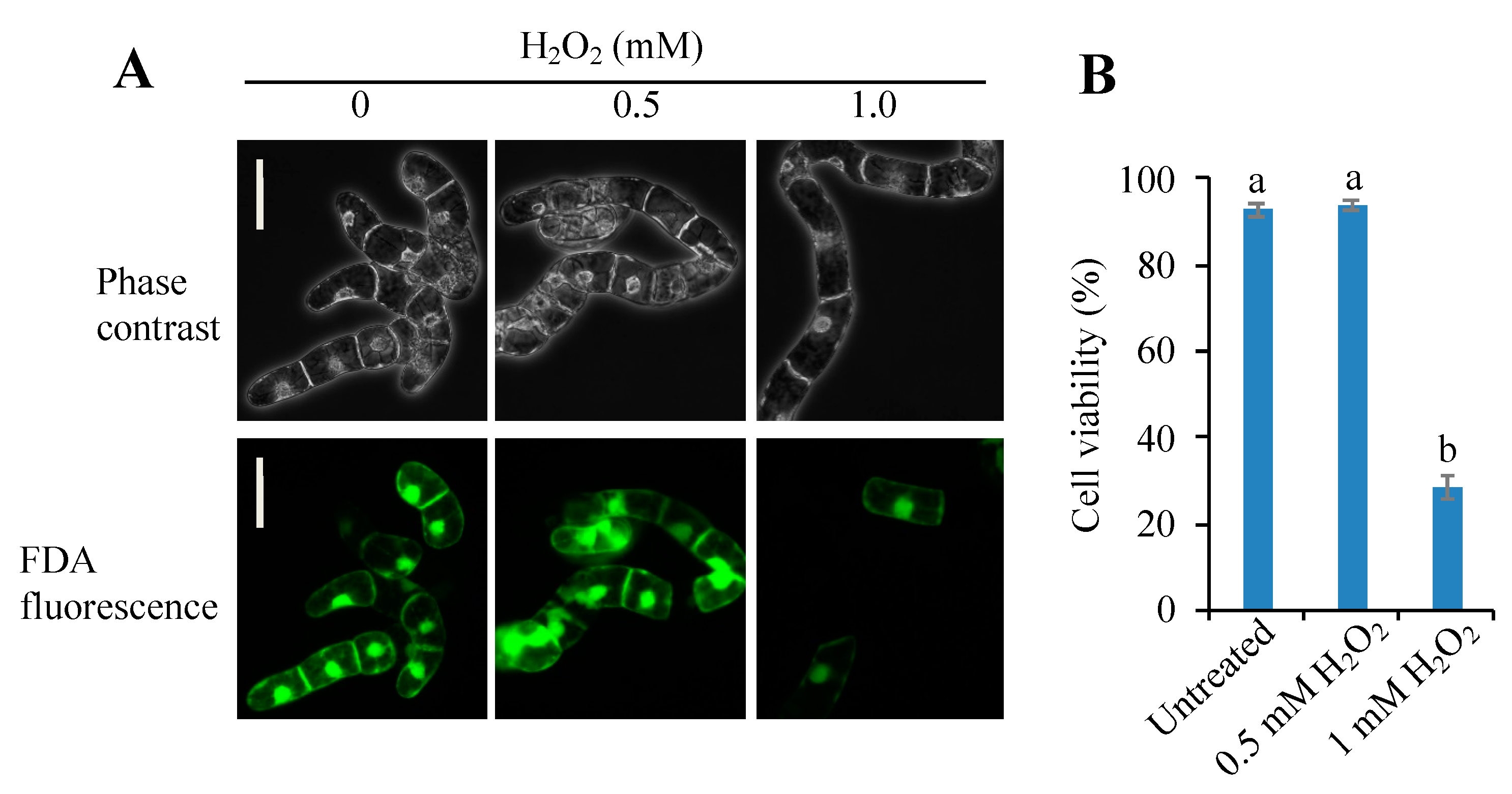
3.1. Threshold Level of Hydrogen Peroxide for Inducing PCD
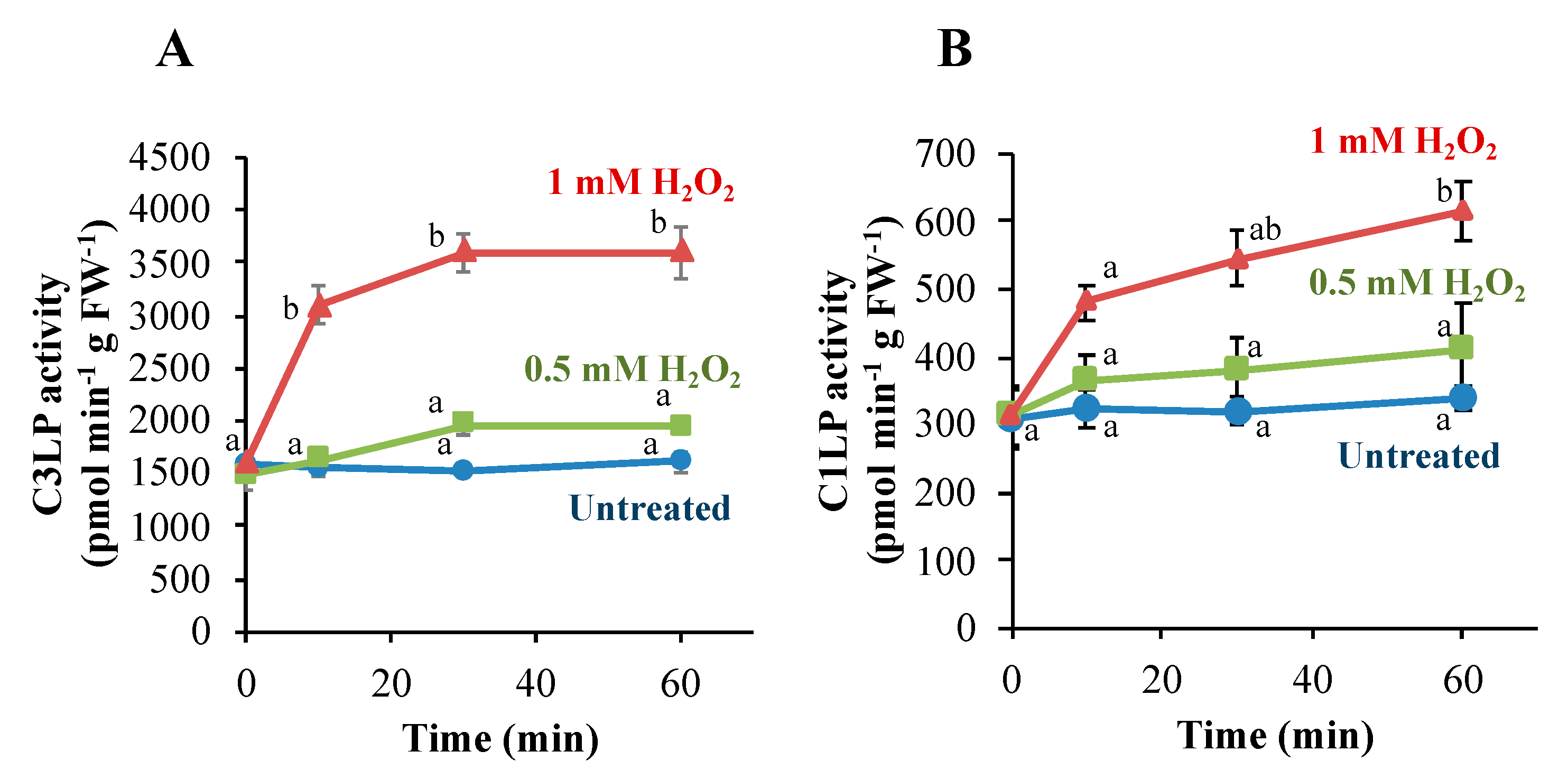
3.2. Caspase-Like Proteases in Tobacco BY-2 Cells Are Activated Only When H2O2 Induces PCD
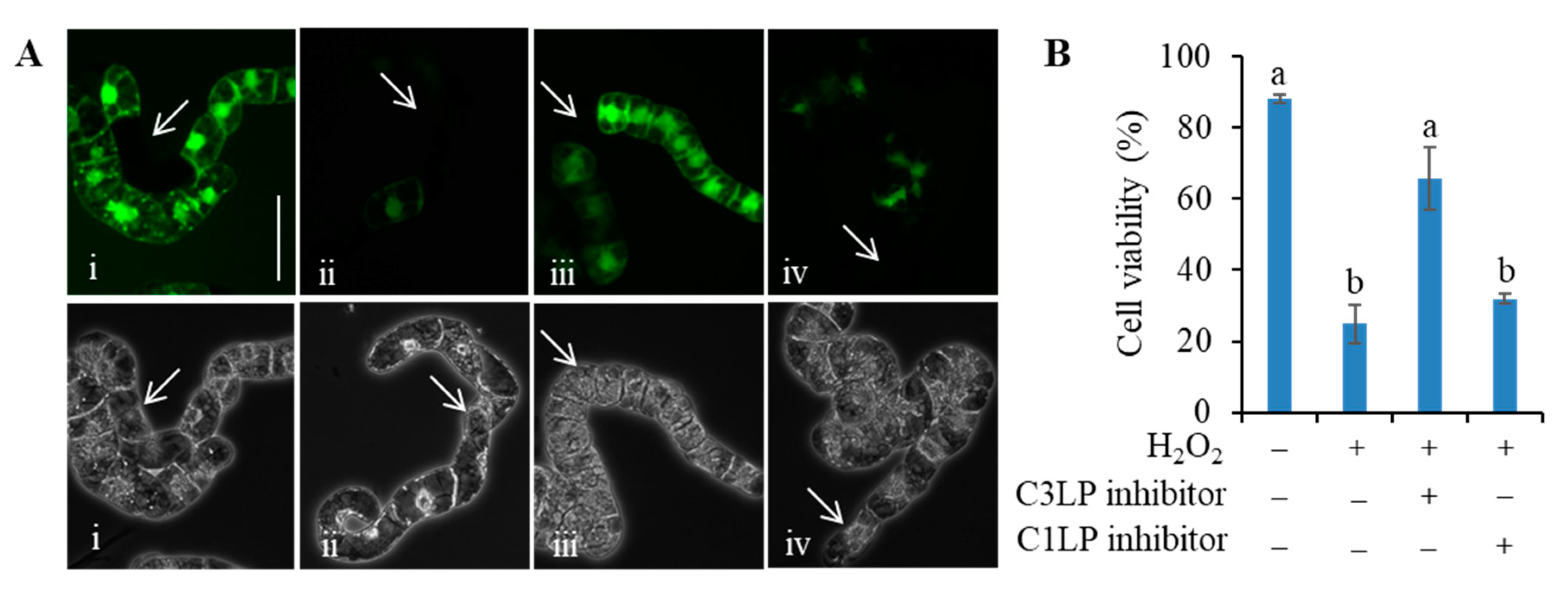
3.3. Activation of C3LP, Rather Than C1LP, Triggers PCD
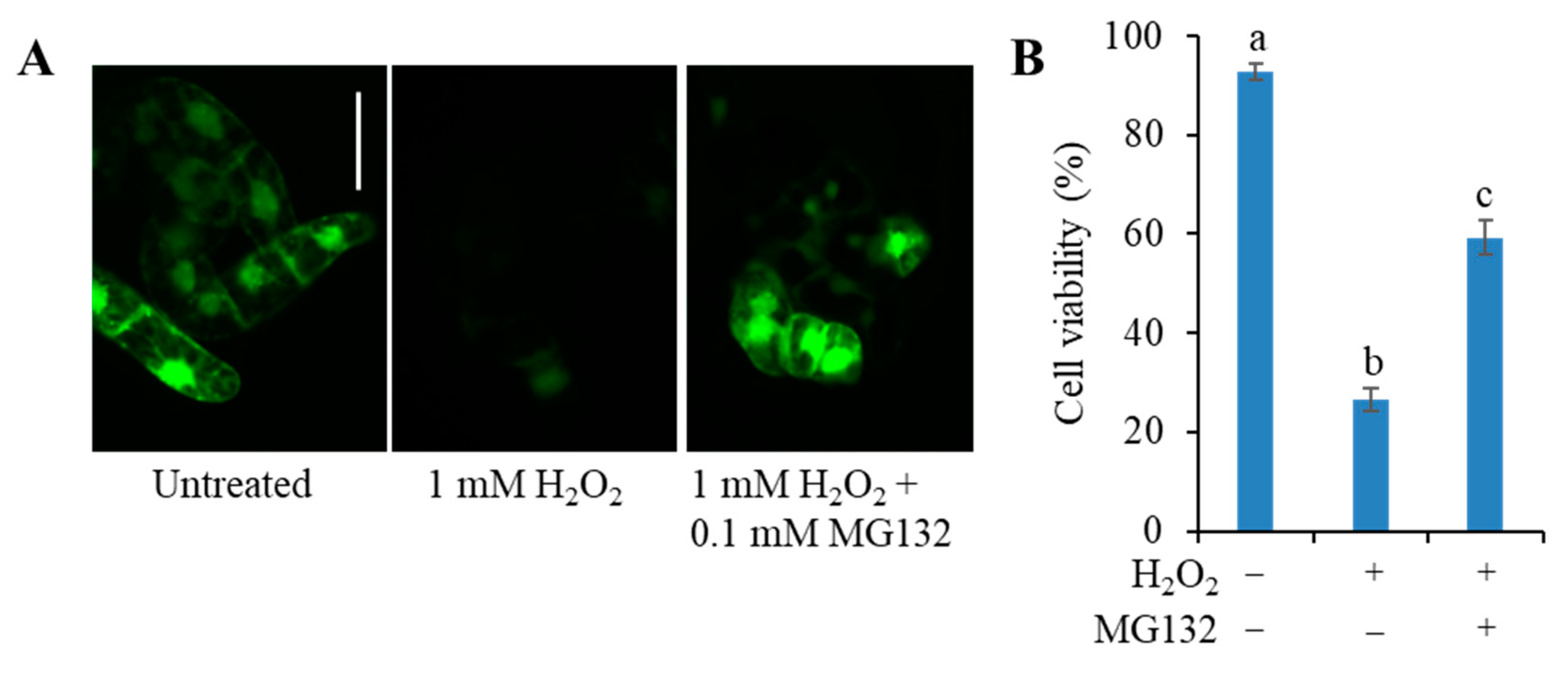
3.4. The C3LP Activity of the 20S Proteasome Is Responsible for the H2O2-Initiated PCD
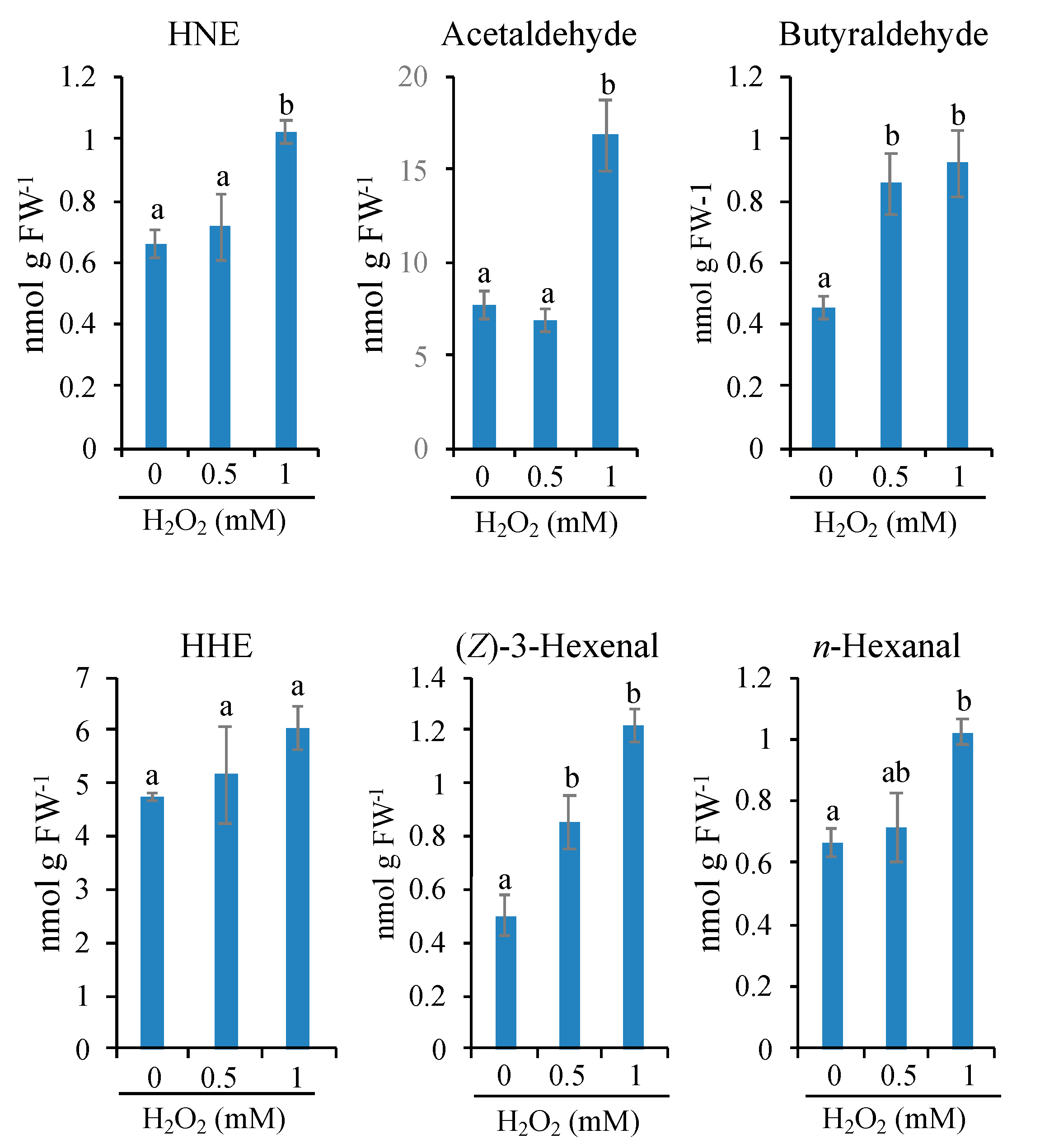
3.5. Limited Types of Oxylipin Carbonyls Are Increased in Very Early Stages of PCD
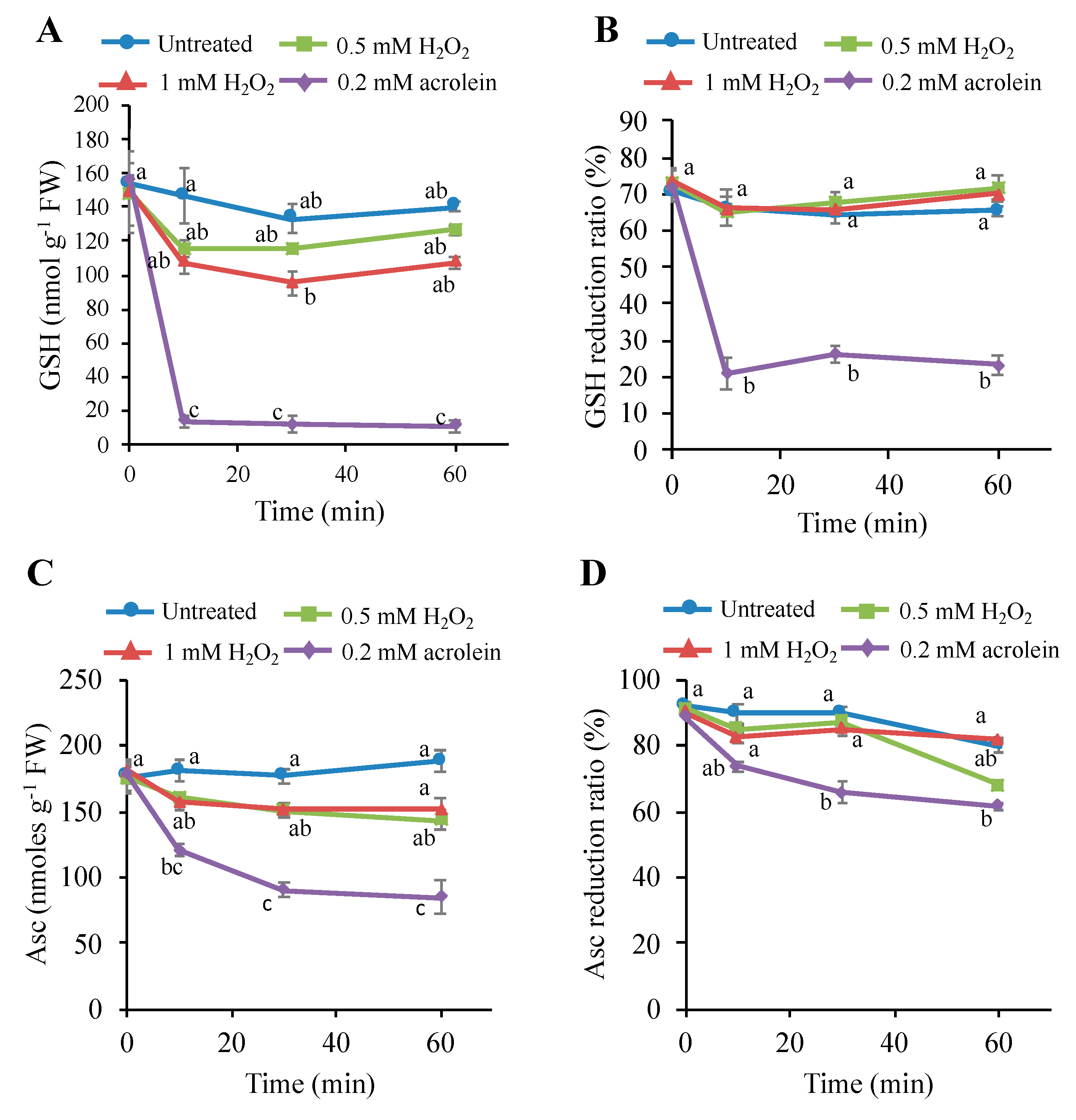
3.6. Effects of H2O2 on the Intracellular Glutathione and Ascorbate Levels
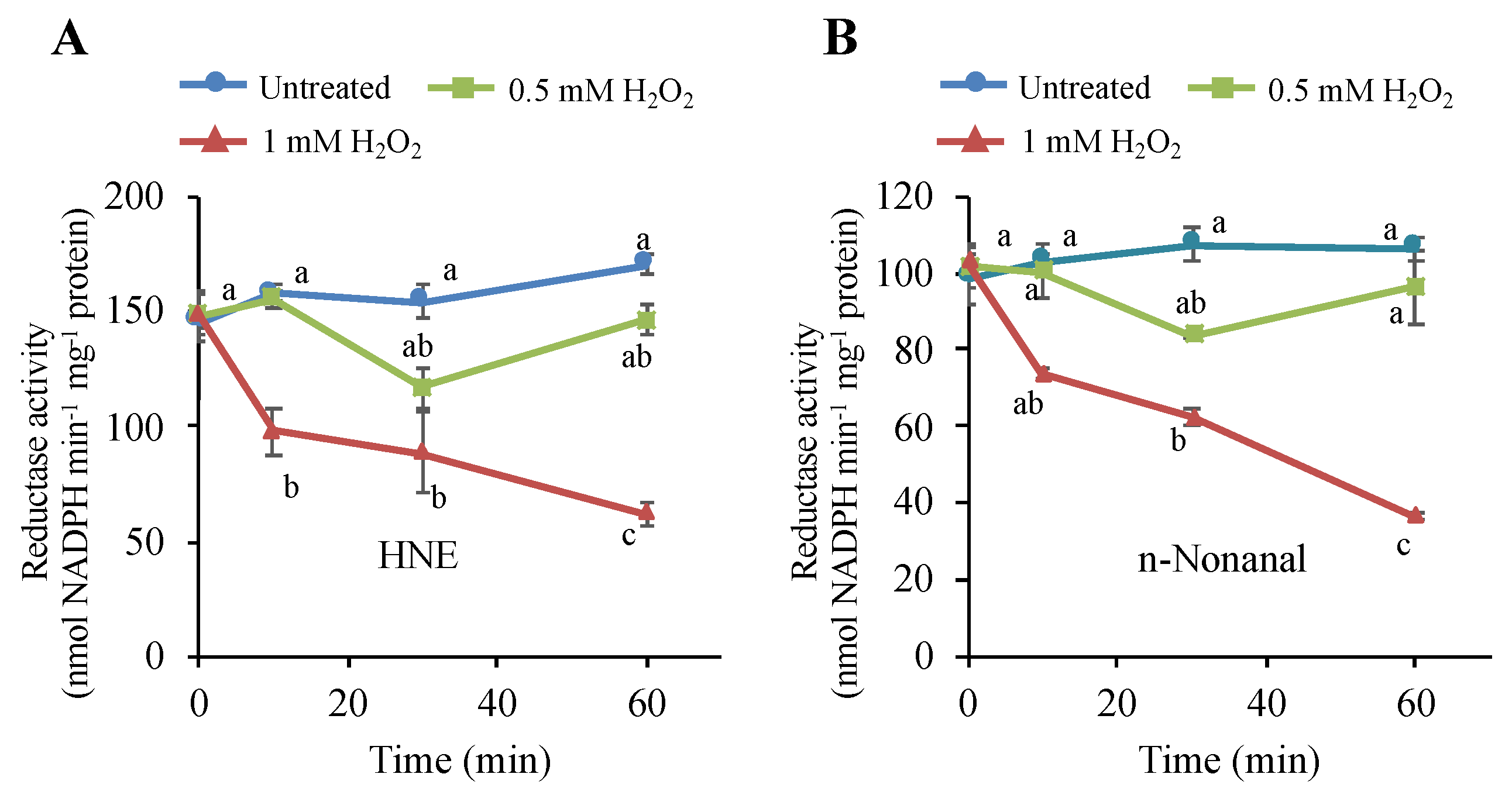
3.7. H2O2 Stimulus Regulates a Carbonyl-Scavenging Capacity in Tobacco BY-2 Cells
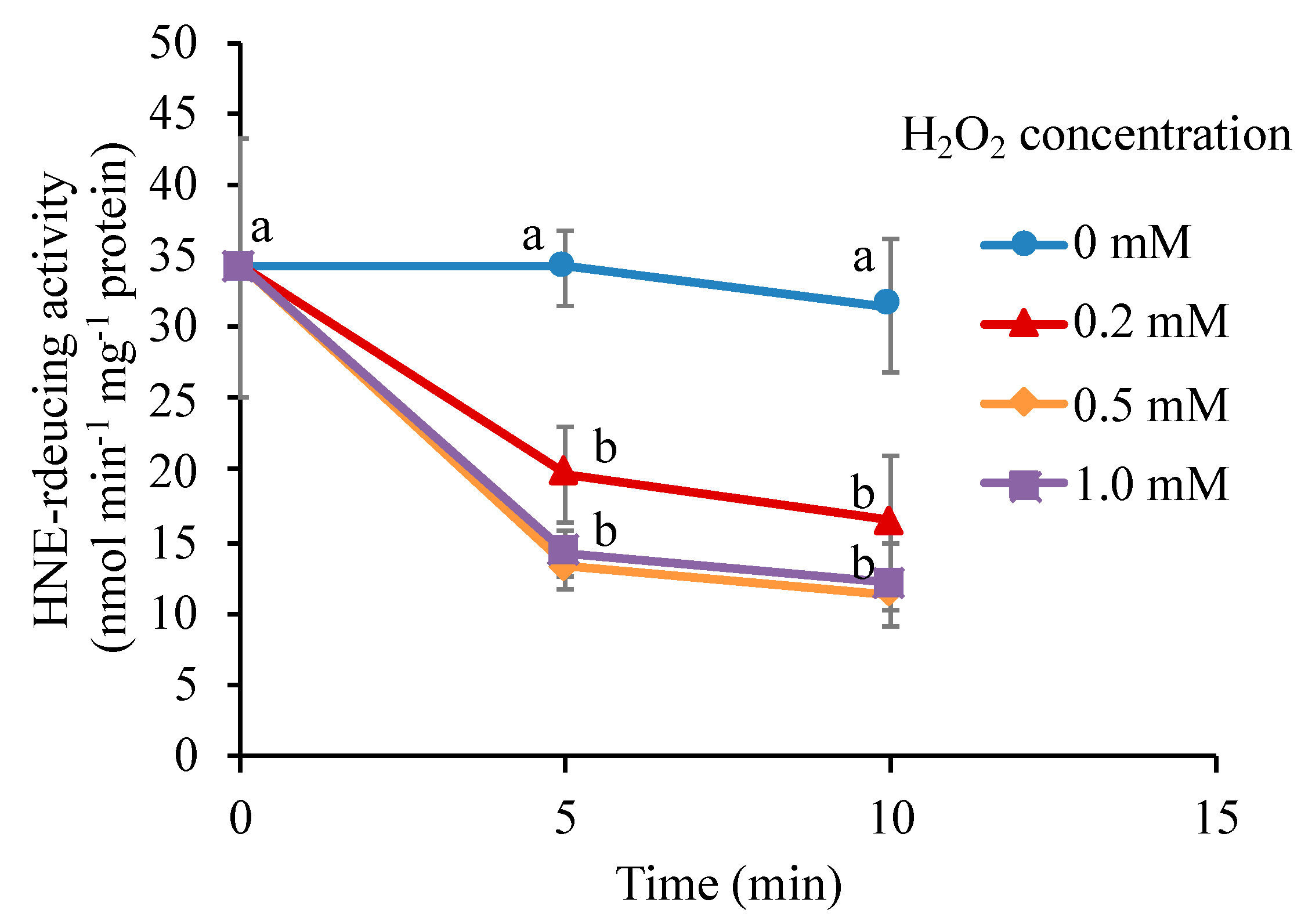
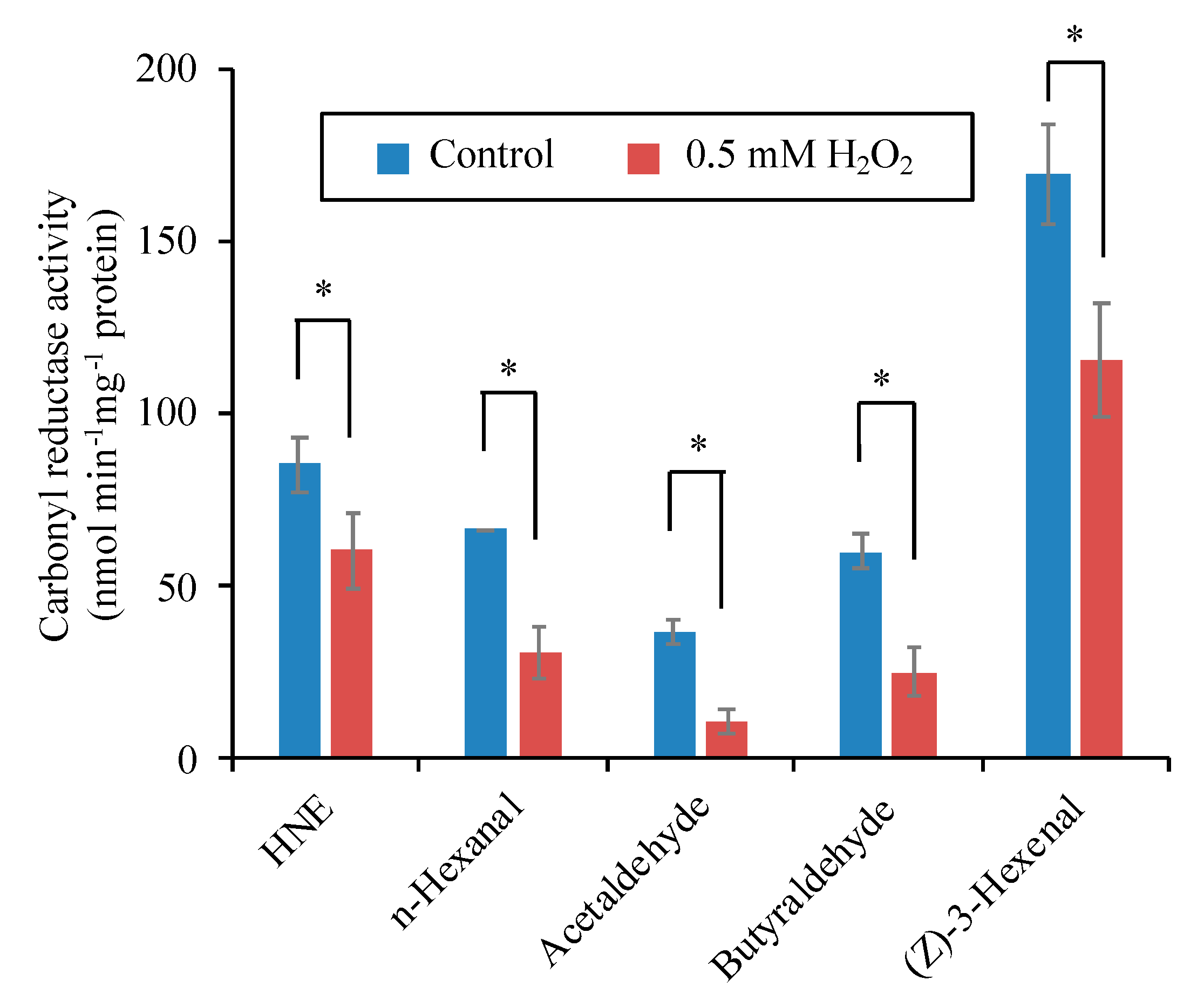
3.8. H2O2 Directly Inactivates the Carbonyl Reductase
4. Discussion
4.1. H2O2-Induced Inactivation of Carbonyl-Detoxifying Enzyme(s) Enhances the HNE Level and Triggers PCD
4.2. Consumption of Antioxidants Is Not a Major Cause of PCD
4.3. Candidates of the H2O2-Sensitive Carbonyl Reductase
4.4. Mechanism of the Inactivation of the Carbonyl Reductase by H2O2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Petrov, V.; Hille, J.; Mueller-Roeber, B.; Gechev, T.S. ROS-mediated abiotic stress-induced programmed cell death in plants. Front. Plant Sci. 2015, 6, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittler, R. ROS are good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukagoshi, H.; Busch, W.; Benfey, P.N. Transcriptional regulation of ROS controls transition from proliferation to differentiation in the root. Cell 2010, 143, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunand, C.; Penel, C. Localization of superoxide in the root apex of Arabidopsis. Plant Signal. Behav. 2007, 2, 131–132. [Google Scholar] [CrossRef] [Green Version]
- Leymarie, J.; Vitkauskaité, G.; Hoang, H.H.; Gendreau, E.; Chazoule, V.; Meimoun, P.; Corbineau, F.; El-Maarouf-Bouteau, H.; Bailly, C. Role of reactive oxygen species in the regulation of Arabidopsis seed dormancy. Plant Cell Physiol. 2012, 53, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Bethke, P.C.; Jones, R.L. Cell death of barley aleurone protoplasts is mediated by reactive oxygen species. Plant J. 2001, 25, 19–29. [Google Scholar] [CrossRef]
- Van Breusegem, F.; Dat, J.F. Reactive oxygen species in plant cell death. Plant Physiol. 2006, 141, 384–390. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.N.; Hailstones, D.L.; Wilkes, M.; Sutton, B.G. Drought-induced oxidative conditions in rice anthers leading to a programmed cell death and pollen abortion. J. Agron. Crop Sci. 2009, 195, 157–164. [Google Scholar] [CrossRef]
- Monetti, E.; Kadono, T.; Tran, D.; Azzarello, E.; Arbelet-Bonnin, D.; Biligui, B.; Briand, J.; Kawano, T.; Mancuso, S.; Bouteau, F. Deciphering early events involved in hyperosmotic stress-induced programmed cell death in tobacco BY-2 cells. J. Exp. Bot. 2014, 65, 1361–1375. [Google Scholar] [CrossRef] [Green Version]
- Locato, V.; Gadaleta, C.; De Gara, L.; de Pinto, M.C. Production of reactive species and modulation of antioxidant network in response to heat shock: a critical balance for cell fate. Plant Cell Environ. 2008, 31, 1606–1619. [Google Scholar] [CrossRef]
- Yoda, H.; Yamaguchi, Y.; Sano, H. Induction of hypersensitive cell death by hydrogen peroxide produced through polyamine degradation in tobacco plants. Plant Physiol. 2003, 132, 1973–1981. [Google Scholar] [CrossRef] [Green Version]
- Biswas, M.S.; Mano, J. Lipid peroxide-derived short-chain carbonyls mediate hydrogen peroxide-induced and salt-induced programmed cell death in plants. Plant Physiol. 2015, 168, 885–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mano, J. Reactive carbonyl species: Their production from lipid peroxides, action in environmental stress, and the detoxification mechanism. Plant Physiol. Biochem. 2012, 59, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Mano, J.; Biswas, M.S.; Sugimoto, K. Reactive carbonyl species: a missing link in ROS signaling. Plants 2019, 8, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, Y.; Kunishima, M.; Mizutani, M.; Sugimoto, Y. Reactive short-chain leaf volatiles act as powerful inducers of abiotic stress-related gene expression. Sci. Rep. 2015, 5, 8030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Brychkova, C.; Yarmolinsky, D.; Soltabayeva, A.; Samani, T.; Sagi, M. Aldehyde oxidase 4 plays a critical role in delaying silique senescence by catalyzing aldehyde detoxification. Plant Physiol. 2017, 173, 1977–1997. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.M.; Ye, W.; Matsushima, D.; Munemasa, S.; Okuma, E.; Nakamura, Y.; Biswas, M.S.; Mano, J.; Murata, Y. Reactive carbonyl species mediate ABA signaling in guard cells. Plant Cell Physiol. 2016, 57, 2552–2563. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.M.; Ye, W.; Matsushima, D.; Rhaman, M.S.; Munemasa, S.; Okuma, E.; Nakamura, Y.; Biswas, M.S.; Mano, J.; Murata, Y. Reactive carbonyl species function as signal mediators downstream of H2O2 production and regulate [Ca2+]cyt elevation in ABA signal pathway in Arabidopsis guard cells. Plant Cell Physiol. 2019, 60, 1146–1159. [Google Scholar] [CrossRef]
- Biswas, M.S.; Fukaki, H.; Mori, I.C.; Nakahara, K.; Mano, J. Reactive oxygen species and reactive carbonyl species constitute a feed-forward loop in auxin signaling for lateral root formation. Plant J. 2019, 100, 536–548. [Google Scholar] [CrossRef]
- Biswas, M.S.; Mano, J. Reactive carbonyl species activate caspase-3-like protease to initiate programmed cell death in plants. Plant Cell Physiol. 2016, 57, 1432–1442. [Google Scholar] [CrossRef] [Green Version]
- Hatsugai, N.; Kuroyanagi, M.; Yamada, K.; Meshi, T.; Tsuda, S.; Kondo, M.; Mikio Nishimura, M.; Hara-Nishimura, I. A plant vacuolar protease, VPE, mediates virus-induced hypersensitive cell death. Science 2004, 305, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Hatsugai, N.; Yamada, K.; Goto-Yamada, S.; Hara-Nishimura, I. Vacuolar processing enzyme in plant programmed cell death. Front. Plant Sci. 2015, 6, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, S.; Huang, W.; Pan, C.; Zhan, J.; He, L.-F. Caspase-like proteases regulate aluminum-induced programmed cell death in peanut. Plant Cell Tissue Organ Cult. 2016, 127, 691–703. [Google Scholar] [CrossRef]
- Ye, Y.; Li, Z.; Xing, D. Nitric oxide promotes MPK6-mediated caspase-3-like activation in cadmium-induced Arabidopsis thaliana programmed cell death. Plant Cell Environ. 2013, 36, 1–15. [Google Scholar] [CrossRef]
- Hatsugai, N.; Iwasaki, S.; Tamura, K.; Kondo, M.; Fuji, K.; Ogasawara, K.; Nishimura, M.; Hara-Nishimura, I. A novel membrane fusion-mediated plant immunity against bacterial pathogens. Genes Dev. 2009, 23, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Han, J.J.; Lin, W.; Oda, Y.; Cui, K.M.; Fukuda, H. The proteasome is responsible for caspase-3-like activity during xylem development. Plant J. 2012, 72, 129–141. [Google Scholar] [CrossRef]
- Ge, Y.; Cai, Y.; Bonneau, L.; Rotari, V.; Danon, A.; McKenzie, E.A.; McLellan, H.; Mach, L.; Gallois, P. Inhibition of cathepsin B by caspase-3 inhibitors blocks programmed cell death in Arabidopsis. Cell Death Differ. 2016, 25, 1532. [Google Scholar] [CrossRef]
- Nagata, T.; Sakamoto, K.; Shimizu, T. Tobacco BY-2 cells: The present and beyond. Vitr. Cell Dev. Biol. Plant 2004, 40, 163–166. [Google Scholar] [CrossRef]
- de Pinto, M.C.; Francis, D.; De Gara, L. The redox state of the ascorbate–dehydroascorbate pair as a specific sensor of cell division in tobacco BY-2 cells. Protoplasma 1999, 209, 90–97. [Google Scholar] [CrossRef]
- Kampfenkel, K.; Montagu, M.V.; Inzé, D. Extraction and determination of ascorbate and dehydroascorbate from plant tissue. Anal. Biochem. 1995, 225, 165–167. [Google Scholar] [CrossRef]
- Yin, L.; Mano, J.; Wang, S.; Tsuji, W.; Tanaka, K. The involvement of lipid peroxide-derived aldehydes in aluminum toxicity of tobacco roots. Plant Physiol. 2010, 152, 1406–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mano, J.; Biswas, M.S. Analysis of reactive carbonyl species generated under oxidative stress. In Plant Programmed Cell Death: Methods and Protocols; De Gara, L., Locato, V., Eds.; Springer: New York, NY, USA, 2018; pp. 117–124. [Google Scholar]
- Cai, Y.-M.; Yu, J.; Ge, Y.; Mironov, A.; Gallois, P. Two proteases with caspase-3-like activity, cathepsin B and proteasome, antagonistically control ER-stress-induced programmed cell death in Arabidopsis. New Phytol. 2018, 218, 1143–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacca, R.A.; Valenti, D.; Bobba, A.; de Pinto, M.C.; Merafina, R.S.; De Gara, L.; Passarella, S.; Marra, E. Proteasome function is required for activation of programmed cell death in heat shocked tobacco Bright-Yellow 2 cells. FEBS Lett. 2007, 581, 917–922. [Google Scholar] [CrossRef] [Green Version]
- Esterbauer, H.; Zollner, H.; Scholz, N. Reaction of glutathione with conjugated carbonyls. Z. Naturforsch. C 1975, 30, 466–473. [Google Scholar] [CrossRef]
- Mano, J.; Miyatake, F.; Hiraoka, E.; Tamoi, M. Evaluation of the toxicity of stress related aldehydes to photosynthesis in chloroplasts. Planta 2009, 230, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Mano, J.; Torii, Y.; Hayashi, S.; Takimoto, K.; Matsui, K.; Nakamura, K.; Inzé, D.; Babiychuk, E.; Kushnir, S.; Asada, K. The NADPH: quinone oxidoreductase P1-ζ-crystallin in Arabidopsis catalyzes the α,β-hydrogenation of 2-alkenals: Detoxication of the lipid peroxide-derived reactive aldehydes. Plant Cell Physiol. 2002, 43, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Mano, J.; Belles-Boix, E.; Babiychuk, E.; Inzé, D.; Torii, Y.; Hiraoka, H.; Takimoto, K.; Slooten, L.; Asada, K.; Kushnir, S. Protection against photooxidative injury of tobacco leaves by 2-alkenal reductase. Detoxication of lipid peroxide-derived reactive carbonyls. Plant Physiol. 2005, 139, 1773–1783. [Google Scholar] [CrossRef] [Green Version]
- Yalcinkaya, T.; Uzilday, B.; Ozgur, R.; Turkan, I.; Mano, J. Lipid peroxidation-derived reactive carbonyl species (RCS): their interaction with ROS and cellular redox during environmental stresses. Environ. Exp. Bot. 2019, 165, 139–149. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Hasegawa, A.; Taninaka, A.; Mizutani, M.; Sugimoto, Y. NADPH-dependent reductases involved in the detoxification of reactive carbonyls in plants. J. Biol. Chem. 2011, 286, 6999–7009. [Google Scholar] [CrossRef] [Green Version]
- Mano, J.; Kanameda, S.; Kuramitsu, R.; Matsuura, N.; Yamauchi, Y. Detoxification of Reactive Carbonyl Species by Glutathione Transferase Tau Isozymes. Front. Plant Sci. 2019, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- Wible, R.S.; Sutter, T.R. Soft cysteine signaling network: the functional significance of cysteine in protein function and the soft acids/bases thiol chemistry that facilitates cysteine modification. Chem. Res. Toxicol. 2017, 30, 729–762. [Google Scholar] [CrossRef] [PubMed]
- Kullik, I.; Toledano, M.B.; Tartaglia, L.A.; Storz, G. Mutational analysis of the redox-sensitive transcriptional regulator OxyR: regions important for oxidation and transcriptional activation. J. Bacteriol. 1995, 177, 1275–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karplus, P.A. A primer on peroxiredoxin biochemistry. Free Radic. Biol. Med. 2015, 80, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, M.S.; Terada, R.; Mano, J. Inactivation of Carbonyl-Detoxifying Enzymes by H2O2 Is a Trigger to Increase Carbonyl Load for Initiating Programmed Cell Death in Plants. Antioxidants 2020, 9, 141. https://doi.org/10.3390/antiox9020141
Biswas MS, Terada R, Mano J. Inactivation of Carbonyl-Detoxifying Enzymes by H2O2 Is a Trigger to Increase Carbonyl Load for Initiating Programmed Cell Death in Plants. Antioxidants. 2020; 9(2):141. https://doi.org/10.3390/antiox9020141
Chicago/Turabian StyleBiswas, Md. Sanaullah, Ryota Terada, and Jun’ichi Mano. 2020. "Inactivation of Carbonyl-Detoxifying Enzymes by H2O2 Is a Trigger to Increase Carbonyl Load for Initiating Programmed Cell Death in Plants" Antioxidants 9, no. 2: 141. https://doi.org/10.3390/antiox9020141