
Investigating the Link between Ketogenic Diet, NAFLD, Mitochondria, and Oxidative Stress: A Narrative Review


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. NAFLD (in Brief)
3. Ketogenic Diet: An Overview
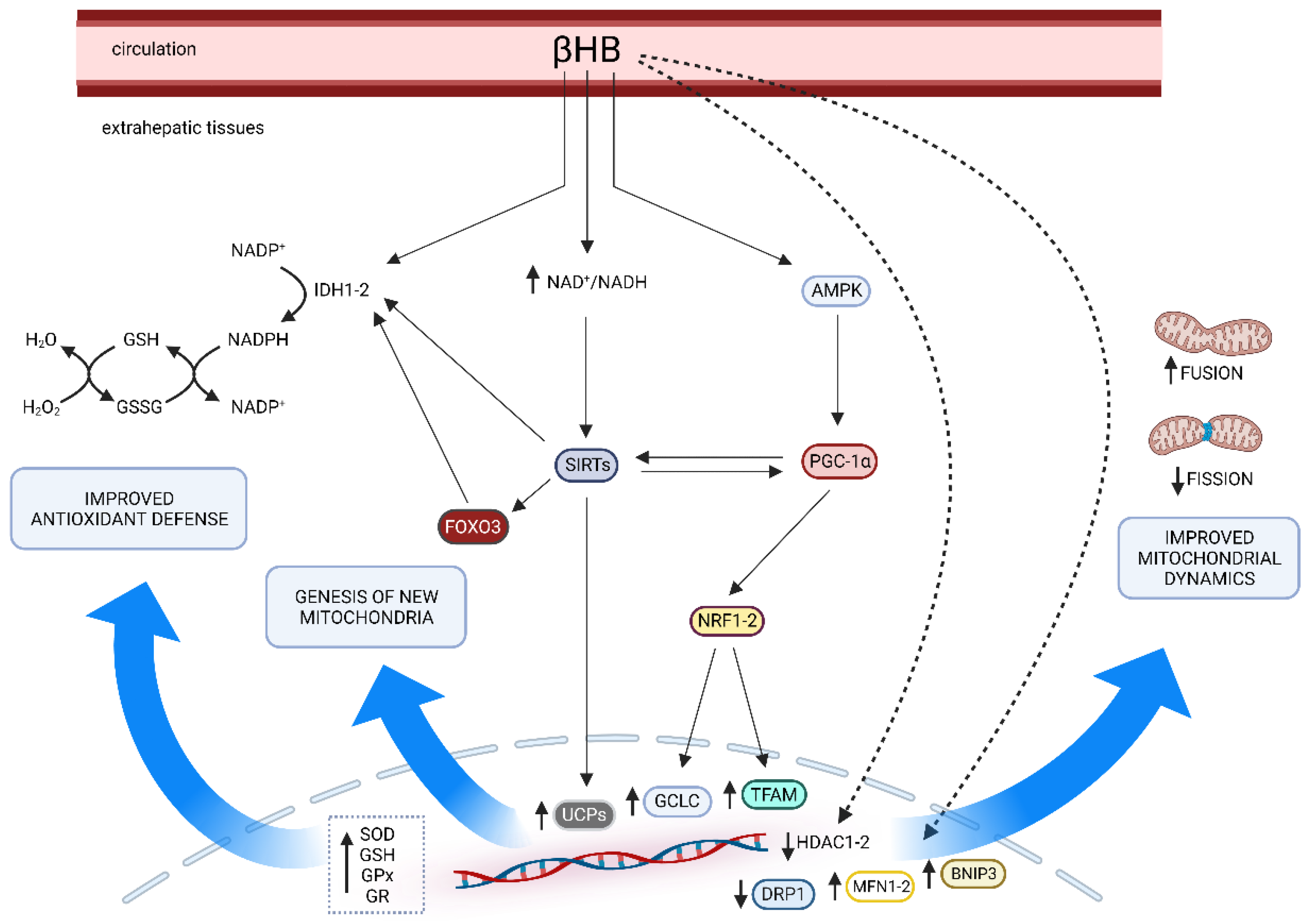
4. KD and Mitochondria
5. Effects of Ketosis on Oxidative Stress Pathways
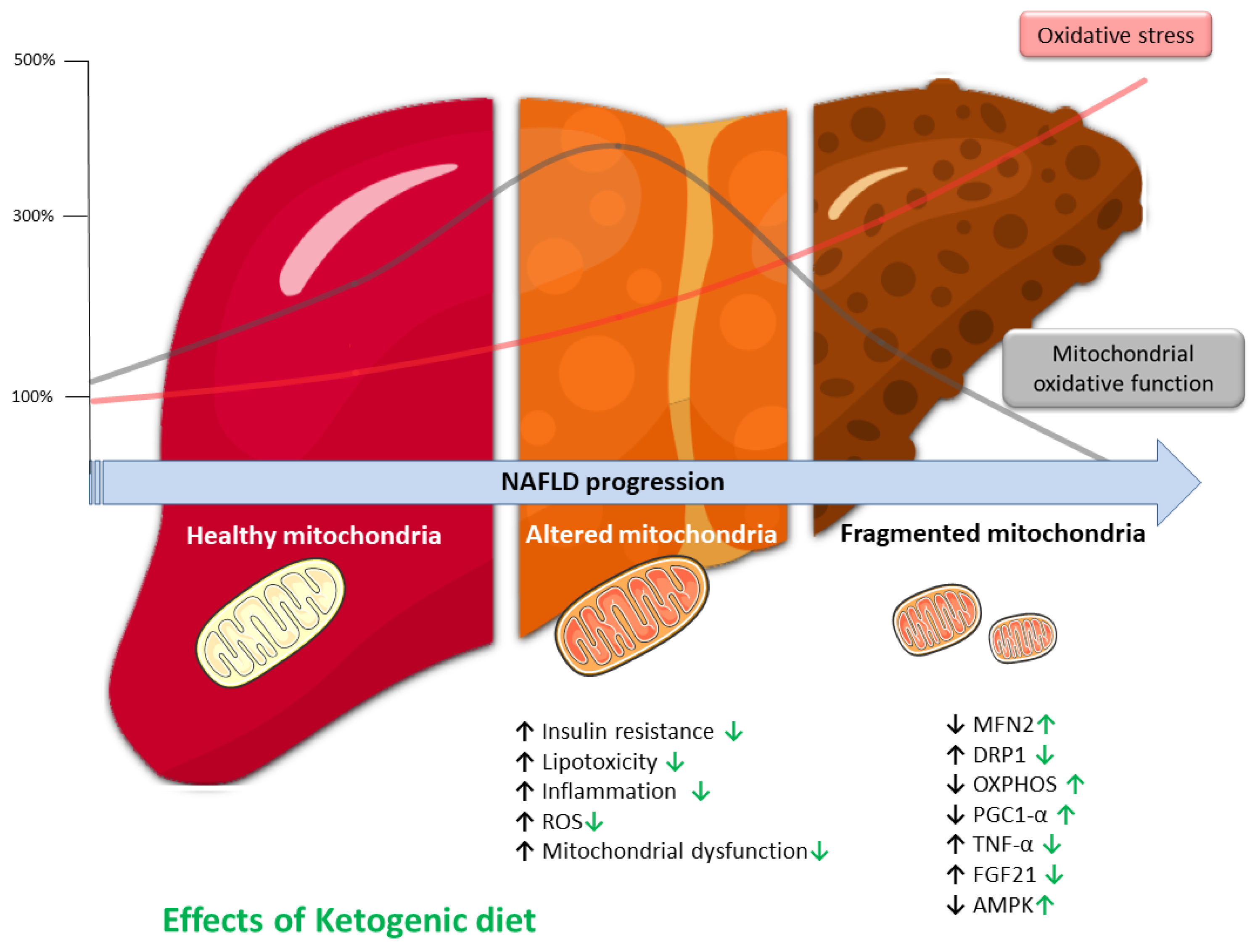
6. Liver and Mitochondria
7. Liver and Oxidative Stress
8. KD and Liver
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- EASL–EASD–EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xie, Z.; Song, Q.; Li, J. Mitochondria Homeostasis: Biology and Involvement in Hepatic Steatosis to NASH. Acta Pharm. Sin. 2022, 43, 1141–1155. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Gonzalez, A.; Huerta-Salgado, C.; Orozco-Aguilar, J.; Aguirre, F.; Tacchi, F.; Simon, F.; Cabello-Verrugio, C. Role of Oxidative Stress in Hepatic and Extrahepatic Dysfunctions during Nonalcoholic Fatty Liver Disease (NAFLD). Oxid. Med. Cell. Longev. 2020, 2020, 1617805. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Macchi, C.; Dongiovanni, P. Mitochondrial Dynamics and Nonalcoholic Fatty Liver Disease (NAFLD): New Perspectives for a Fairy-Tale Ending? Metabolism 2021, 117, 154708. [Google Scholar] [CrossRef]
- Cichoz-Lach, H.; Michalak, A. Oxidative Stress as a Crucial Factor in Liver Diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.-M.; Hakkarainen, A.; Lehtimäki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Järvinen, H. Effect of a Ketogenic Diet on Hepatic Steatosis and Hepatic Mitochondrial Metabolism in Nonalcoholic Fatty Liver Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef]
- Zhang, X.-J.; She, Z.-G.; Li, H. Time to Step-up the Fight against NAFLD. Hepatology 2018, 67, 2068–2071. [Google Scholar] [CrossRef]
- Parra-Vargas, M.; Rodriguez-Echevarria, R.; Jimenez-Chillaron, J.C. Nutritional Approaches for the Management of Nonalcoholic Fatty Liver Disease: An Evidence-Based Review. Nutrients 2020, 12, 3860. [Google Scholar] [CrossRef]
- Greco, T.; Glenn, T.C.; Hovda, D.A.; Prins, M.L. Ketogenic Diet Decreases Oxidative Stress and Improves Mitochondrial Respiratory Complex Activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef]
- Vidali, S.; Aminzadeh, S.; Lambert, B.; Rutherford, T.; Sperl, W.; Kofler, B.; Feichtinger, R.G. Mitochondria: The Ketogenic Diet—A Metabolism-Based Therapy. Int. J. Biochem. Cell Biol. 2015, 63, 55–59. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-Dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Mancin, L.; Piccini, F.; Paoli, A. Ketogenic Diet and NAFLD: A Great Therapeutic Opportunity? Acta Med. Mediterr. 2019, 35, 1909–1913. [Google Scholar]
- Mooli, R.G.R.; Ramakrishnan, S.K. Emerging Role of Hepatic Ketogenesis in Fatty Liver Disease. Front. Physiol. 2022, 13, 946474. [Google Scholar] [CrossRef]
- Sripongpun, P.; Churuangsuk, C.; Bunchorntavakul, C. Current Evidence Concerning Effects of Ketogenic Diet and Intermittent Fasting in Patients with Nonalcoholic Fatty Liver. J. Clin. Transl. Hepatol. 2022, 10, 730–739. [Google Scholar] [CrossRef]
- Watanabe, M.; Tozzi, R.; Risi, R.; Tuccinardi, D.; Mariani, S.; Basciani, S.; Spera, G.; Lubrano, C.; Gnessi, L. Beneficial Effects of the Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Comprehensive Review of the Literature. Obes. Rev. 2020, 21, e13024. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J. Past, Present and Future Perspectives in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Harrison, S.A.; Abdelmalek, M.F.; Anstee, Q.M.; Bedossa, P.; Castera, L.; Dimick-Santos, L.; Friedman, S.L.; Greene, K.; Kleiner, D.E.; et al. Case Definitions for Inclusion and Analysis of Endpoints in Clinical Trials for Nonalcoholic Steatohepatitis through the Lens of Regulatory Science. Hepatology 2018, 67, 2001–2012. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, Y.; Cai, J.; Li, H. Emerging Molecular Targets for Treatment of Nonalcoholic Fatty Liver Disease. Trends Endocrinol. Metab. 2019, 30, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef]
- Bence, K.K.; Birnbaum, M.J. Metabolic Drivers of Non-Alcoholic Fatty Liver Disease. Mol. Metab. 2021, 50, 101143. [Google Scholar] [CrossRef]
- Ziolkowska, S.; Binienda, A.; Jabłkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 11128. [Google Scholar] [CrossRef]
- Phinney, S.D.; Bistrian, B.R.; Evans, W.J.; Gervino, E.; Blackburn, G.L. The Human Metabolic Response to Chronic Ketosis without Caloric Restriction: Preservation of Submaximal Exercise Capability with Reduced Carbohydrate Oxidation. Metabolism 1983, 32, 769–776. [Google Scholar] [CrossRef]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond Weight Loss: A Review of the Therapeutic Uses of Very-Low-Carbohydrate (Ketogenic) Diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef]
- Ashtary-Larky, D.; Bagheri, R.; Bavi, H.; Baker, J.S.; Moro, T.; Mancin, L.; Paoli, A. Ketogenic Diets, Physical Activity and Body Composition: A Review. Br. J. Nutr. 2022, 127, 1898–1920. [Google Scholar] [CrossRef]
- Paoli, A. Ketogenic Diet for Obesity: Friend or Foe? Int. J. Environ. Res. Public Health 2014, 11, 2092–2107. [Google Scholar] [CrossRef]
- Goldenberg, J.Z.; Johnston, B.C. Low and Very Low Carbohydrate Diets for Diabetes Remission. BMJ 2021, 373, n262. [Google Scholar] [CrossRef]
- Harvey, C.J.D.C.; Schofield, G.M.; Zinn, C.; Thornley, S.J.; Crofts, C.; Merien, F.L.R. Low-Carbohydrate Diets Differing in Carbohydrate Restriction Improve Cardiometabolic and Anthropometric Markers in Healthy Adults: A Randomised Clinical Trial. PeerJ 2019, 7, e6273. [Google Scholar] [CrossRef]
- Ilyas, Z.; Perna, S.; Alalwan, T.A.; Zahid, M.N.; Spadaccini, D.; Gasparri, C.; Peroni, G.; Faragli, A.; Alogna, A.; la Porta, E.; et al. The Ketogenic Diet: Is It an Answer for Sarcopenic Obesity? Nutrients 2022, 14, 620. [Google Scholar] [CrossRef]
- Robinson, P.J.; Rapoport, S.I. Glucose Transport and Metabolism in the Brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1986, 250, R127–R136. [Google Scholar] [CrossRef]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F. Brain Metabolism during Fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef]
- Mitchell, R.W.; Hatch, G.M. Fatty Acid Transport into the Brain: Of Fatty Acid Fables and Lipid Tails. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 293–302. [Google Scholar] [CrossRef]
- McGarry, J.D.; Foster, D.W. Ketogenesis and Its Regulation. Am. J. Med. 1976, 61, 9–13. [Google Scholar] [CrossRef]
- Veech, R.L. The Therapeutic Implications of Ketone Bodies: The Effects of Ketone Bodies in Pathological Conditions: Ketosis, Ketogenic Diet, Redox States, Insulin Resistance, and Mitochondrial Metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Leino, R.L.; Gerhart, D.Z.; Duelli, R.; Enerson, B.E.; Drewes, L.R. Diet-Induced Ketosis Increases Monocarboxylate Transporter (MCT1) Levels in Rat Brain. Neurochem. Int. 2001, 38, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Cenci, L.; Grimaldi, K.A. Effect of Ketogenic Mediterranean Diet with Phytoextracts and Low Carbohydrates/High-Protein Meals on Weight, Cardiovascular Risk Factors, Body Composition and Diet Compliance in Italian Council Employees. Nutr. J. 2011, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A. The Regulation of the Release of Ketone Bodies by the Liver. Adv. Enzym. Regul. 1966, 4, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Bianco, A.; Grimaldi, K.A. The Ketogenic Diet and Sport. Exerc. Sport Sci. Rev. 2015, 43, 153–162. [Google Scholar] [CrossRef]
- Pathak, S.J.; Baar, K. Ketogenic Diets and Mitochondrial Function: Benefits for Aging But Not for Athletes. Exerc. Sport Sci. Rev. 2023, 51, 27–33. [Google Scholar] [CrossRef]
- Akbari, M.; Kirkwood, T.B.L.; Bohr, V.A. Mitochondria in the Signaling Pathways That Control Longevity and Health Span. Ageing Res. Rev. 2019, 54, 100940. [Google Scholar] [CrossRef]
- Negro, M.; Cerullo, G.; Parimbelli, M.; Ravazzani, A.; Feletti, F.; Berardinelli, A.; Cena, H.; D’Antona, G. Exercise, Nutrition, and Supplements in the Muscle Carnitine Palmitoyl-Transferase II Deficiency: New Theoretical Bases for Potential Applications. Front. Physiol. 2021, 12, 704290. [Google Scholar] [CrossRef]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial Dynamics, Mitophagy and Cardiovascular Disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial Dysfunction in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharm. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Rai, S.N.; Singh, C.; Singh, A.; Singh, M.P.; Singh, B.K. Mitochondrial Dysfunction: A Potential Therapeutic Target to Treat Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 3075–3088. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.v.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Cardiovascular Disease: A Brief Review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. PGC-1α-Mediated Regulation of Mitochondrial Function and Physiological Implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and Sirtuins in Aging and Disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Kyriazis, I.; Vassi, E.; Alvanou, M.; Angelakis, C.; Skaperda, Z.; Tekos, F.; Garikipati, V.; Spandidos, D.; Kouretas, D. The Impact of Diet upon Mitochondrial Physiology (Review). Int. J. Mol. Med. 2022, 50, 135. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone Bodies: From Enemy to Friend and Guardian Angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Miller, V.J.; LaFountain, R.A.; Barnhart, E.; Sapper, T.S.; Short, J.; Arnold, W.D.; Hyde, P.N.; Crabtree, C.D.; Kackley, M.L.; Kraemer, W.J.; et al. A Ketogenic Diet Combined with Exercise Alters Mitochondrial Function in Human Skeletal Muscle While Improving Metabolic Health. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E995–E1007. [Google Scholar] [CrossRef]
- Qu, C.; Keijer, J.; Adjobo-Hermans, M.J.W.; van de Wal, M.; Schirris, T.; van Karnebeek, C.; Pan, Y.; Koopman, W.J.H. The Ketogenic Diet as a Therapeutic Intervention Strategy in Mitochondrial Disease. Int. J. Biochem. Cell Biol. 2021, 138, 106050. [Google Scholar] [CrossRef]
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional Ketosis and Mitohormesis: Potential Implications for Mitochondrial Function and Human Health. J. Nutr. Metab. 2018, 2018, 5157645. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The Ketogenic Diet Increases Mitochondrial Uncoupling Protein Levels and Activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Wolkow, C.A.; Iser, W.B. Uncoupling Protein Homologs May Provide a Link between Mitochondria, Metabolism and Lifespan. Ageing Res. Rev. 2006, 5, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res. 2019, 44, 22–37. [Google Scholar] [CrossRef]
- Abduraman, M.A.; Azizan, N.A.; Teoh, S.H.; Tan, M.L. Ketogenesis and SIRT1 as a Tool in Managing Obesity. Obes. Res. Clin. Pract. 2021, 15, 10–18. [Google Scholar] [CrossRef]
- Tozzi, R.; Cipriani, F.; Masi, D.; Basciani, S.; Watanabe, M.; Lubrano, C.; Gnessi, L.; Mariani, S. Ketone Bodies and SIRT1, Synergic Epigenetic Regulators for Metabolic Health: A Narrative Review. Nutrients 2022, 14, 3145. [Google Scholar] [CrossRef]
- Wallace, M.A.; Aguirre, N.W.; Marcotte, G.R.; Marshall, A.G.; Baehr, L.M.; Hughes, D.C.; Hamilton, K.L.; Roberts, M.N.; Lopez-Dominguez, J.A.; Miller, B.F.; et al. The Ketogenic Diet Preserves Skeletal Muscle with Aging in Mice. Aging Cell 2021, 20, e13322. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S.; Sandhu, M.A. PGC-1 α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Carelli, V.; Maresca, A.; Caporali, L.; Trifunov, S.; Zanna, C.; Rugolo, M. Mitochondria: Biogenesis and Mitophagy Balance in Segregation and Clonal Expansion of Mitochondrial DNA Mutations. Int. J. Biochem. Cell Biol. 2015, 63, 21–24. [Google Scholar] [CrossRef]
- Popov, L. Mitochondrial Biogenesis: An Update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Jurczak, M.J.; Lee, H.-Y.; Birkenfeld, A.L.; Frederick, D.W.; Zhang, D.; Zhang, X.-M.; Samuel, V.T.; Shulman, G.I. A High-Fat, Ketogenic Diet Causes Hepatic Insulin Resistance in Mice, despite Increasing Energy Expenditure and Preventing Weight Gain. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E808–E815. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Kelty, T.J.; Boccardi, L.R.; Nguyen, N.Y.; Booth, F.W.; Rector, R.S. Ketogenic Diet in Combination with Voluntary Exercise Impacts Markers of Hepatic Metabolism and Oxidative Stress in Male and Female Wistar Rats. Appl. Physiol. Nutr. Metab. 2020, 45, 35–44. [Google Scholar] [CrossRef]
- Milder, J.B.; Liang, L.-P.; Patel, M. Acute Oxidative Stress and Systemic Nrf2 Activation by the Ketogenic Diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Sidarala, V.; Zhu, J.; Levi-D’Ancona, E.; Pearson, G.L.; Reck, E.C.; Walker, E.M.; Kaufman, B.A.; Soleimanpour, S.A. Mitofusin 1 and 2 Regulation of Mitochondrial DNA Content Is a Critical Determinant of Glucose Homeostasis. Nat. Commun. 2022, 13, 2340. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef]
- Williams, M.; Caino, M.C. Mitochondrial Dynamics in Type 2 Diabetes and Cancer. Front. Endocrinol. 2018, 9, 211. [Google Scholar] [CrossRef]
- Thai, P.N.; Seidlmayer, L.K.; Miller, C.; Ferrero, M.; Dorn, G.W.; Schaefer, S.; Bers, D.M.; Dedkova, E.N. Mitochondrial Quality Control in Aging and Heart Failure: Influence of Ketone Bodies and Mitofusin-Stabilizing Peptides. Front. Physiol. 2019, 10, 382. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, C.; Shang, F.-F.; Luo, M.; You, Y.; Zhai, Q.; Xia, Y.; Suxin, L. Ketogenic Diet Ameliorates Cardiac Dysfunction via Balancing Mitochondrial Dynamics and Inhibiting Apoptosis in Type 2 Diabetic Mice. Aging Dis. 2020, 11, 229. [Google Scholar] [CrossRef]
- Guo, M.; Wang, X.; Zhao, Y.; Yang, Q.; Ding, H.; Dong, Q.; Chen, X.; Cui, M. Ketogenic Diet Improves Brain Ischemic Tolerance and Inhibits NLRP3 Inflammasome Activation by Preventing Drp1-Mediated Mitochondrial Fission and Endoplasmic Reticulum Stress. Front. Mol. Neurosci. 2018, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Newell, C.; Shutt, T.E.; Ahn, Y.; Hittel, D.S.; Khan, A.; Rho, J.M.; Shearer, J. Tissue Specific Impacts of a Ketogenic Diet on Mitochondrial Dynamics in the BTBRT+tf/j Mouse. Front. Physiol. 2016, 7, 654. [Google Scholar] [CrossRef] [PubMed]
- Haces, M.L.; Hernández-Fonseca, K.; Medina-Campos, O.N.; Montiel, T.; Pedraza-Chaverri, J.; Massieu, L. Antioxidant Capacity Contributes to Protection of Ketone Bodies against Oxidative Damage Induced during Hypoglycemic Conditions. Exp. Neurol. 2008, 211, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, Ketone Bodies, and Mitochondrial Energy Transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef]
- Veech, R.L. The Determination of the Redox States and Phosphorylation Potential in Living Tissues and Their Relationship to Metabolic Control of Disease Phenotypes. Biochem. Mol. Biol. Educ. 2006, 34, 168–179. [Google Scholar] [CrossRef]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a Dietary Ketone Ester on Hippocampal Glycolytic and Tricarboxylic Acid Cycle Intermediates and Amino Acids in a 3xTgAD Mouse Model of Alzheimer’s Disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef]
- Xin, L.; Ipek, Ö.; Beaumont, M.; Shevlyakova, M.; Christinat, N.; Masoodi, M.; Greenberg, N.; Gruetter, R.; Cuenoud, B. Nutritional Ketosis Increases NAD+/NADH Ratio in Healthy Human Brain: An in Vivo Study by 31P-MRS. Front. Nutr. 2018, 5, 62. [Google Scholar] [CrossRef]
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 Mediates Neuroprotection of Ketones against Ischemic Stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1783–1789. [Google Scholar] [CrossRef]
- Yin, J.; Nielsen, M.; Li, S.; Shi, J. Ketones Improves Apolipoprotein E4-Related Memory Deficiency via Sirtuin 3. Aging 2019, 11, 4579–4586. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; King, M.T.; Veech, R.L. Substrate Signaling by Insulin. Am. J. Cardiol. 1997, 80, 50A–64A. [Google Scholar] [CrossRef]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones Inhibit Mitochondrial Production of Reactive Oxygen Species Production Following Glutamate Excitotoxicity by Increasing NADH Oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef]
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate Regulates Energy Metabolism and Induces BDNF Expression in Cerebral Cortical Neurons. J. Neurochem. 2016, 139, 769–781. [Google Scholar] [CrossRef]
- Hirst, J.; King, M.S.; Pryde, K.R. The Production of Reactive Oxygen Species by Complex I. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, Function, and Post-Translational Regulation of the Catalytic and Modifier Subunits of Glutamate Cysteine Ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 Regulatory Network Provides an Interface between Redox and Intermediary Metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of Activation of the Transcription Factor Nrf2 by Redox Stressors, Nutrient Cues, and Energy Status and the Pathways through Which It Attenuates Degenerative Disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef]
- Miller, A.L. Therapeutic Considerations of L-Glutamine: A Review of the Literature. Altern. Med. Rev. 1999, 4, 239–248. [Google Scholar]
- Pasyukova, E.G.; Vaiserman, A.M. HDAC Inhibitors: A New Promising Drug Class in Anti-Aging Research. Mech. Ageing Dev. 2017, 166, 6–15. [Google Scholar] [CrossRef]
- Wang, X.; Wu, X.; Liu, Q.; Kong, G.; Zhou, J.; Jiang, J.; Wu, X.; Huang, Z.; Su, W.; Zhu, Q. Ketogenic Metabolism Inhibits Histone Deacetylase (HDAC) and Reduces Oxidative Stress After Spinal Cord Injury in Rats. Neuroscience 2017, 366, 36–43. [Google Scholar] [CrossRef]
- Li, B.; Yu, Y.; Liu, K.; Zhang, Y.; Geng, Q.; Zhang, F.; Li, Y.; Qi, J. β-Hydroxybutyrate Inhibits Histone Deacetylase 3 to Promote Claudin-5 Generation and Attenuate Cardiac Microvascular Hyperpermeability in Diabetes. Diabetologia 2021, 64, 226–239. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; Pedraza-Chaverri, J.; Tapia, E. Ketone Bodies, Stress Response, and Redox Homeostasis. Redox Biol. 2020, 29, 101395. [Google Scholar] [CrossRef] [PubMed]
- Chriett, S.; Dąbek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent Action of Butyrate over β-Hydroxybutyrate as Histone Deacetylase Inhibitor, Transcriptional Modulator and Anti-Inflammatory Molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Weng, H.; Ma, Y.; Chen, L.; Cai, G.; Chen, Z.; Zhang, S.; Ye, Q. A New Vision of Mitochondrial Unfolded Protein Response to the Sirtuin Family. Curr. Neuropharmacol. 2020, 18, 613–623. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.; Zhao, S.; Xiong, Y. Tumour Suppressor SIRT3 Deacetylates and Activates Manganese Superoxide Dismutase to Scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.; Krüger, A.; Tauqeer Alam, M.; et al. The Return of Metabolism: Biochemistry and Physiology of the Pentose Phosphate Pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- White, K.; Someya, S. The Roles of NADPH and Isocitrate Dehydrogenase in Cochlear Mitochondrial Antioxidant Defense and Aging. Hear. Res. 2023, 427, 108659. [Google Scholar] [CrossRef]
- Ziegler, D.R.; Ribeiro, L.C.; Hagenn, M.; Siqueira, I.R.; Araújo, E.; Torres, I.L.S.; Gottfried, C.; Netto, C.A.; Gonçalves, C. Ketogenic Diet Increases Glutathione Peroxidase Activity in Rat Hippocampus. Neurochem. Res. 2003, 28, 1793–1797. [Google Scholar] [CrossRef]
- Vogel, R.; Wiesinger, H.; Hamprecht, B.; Dringen, R. The Regeneration of Reduced Glutathione in Rat Forebrain Mitochondria Identifies Metabolic Pathways Providing the NADPH Required. Neurosci. Lett. 1999, 275, 97–100. [Google Scholar] [CrossRef]
- Garcia, J.; Han, D.; Sancheti, H.; Yap, L.-P.; Kaplowitz, N.; Cadenas, E. Regulation of Mitochondrial Glutathione Redox Status and Protein Glutathionylation by Respiratory Substrates. J. Biol. Chem. 2010, 285, 39646–39654. [Google Scholar] [CrossRef]
- Noh, M.R.; Kong, M.J.; Han, S.J.; Kim, J.I.; Park, K.M. Isocitrate Dehydrogenase 2 Deficiency Aggravates Prolonged High-Fat Diet Intake-Induced Hypertension. Redox Biol. 2020, 34, 101548. [Google Scholar] [CrossRef]
- Charitou, P.; Rodriguez-Colman, M.; Gerrits, J.; Triest, M.; Groot Koerkamp, M.; Hornsveld, M.; Holstege, F.; Verhoeven-Duif, N.M.; Burgering, B.M. FOXOs Support the Metabolic Requirements of Normal and Tumor Cells by Promoting IDH1 Expression. EMBO Rep. 2015, 16, 456–466. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone Bodies Mimic the Life Span Extending Properties of Caloric Restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Guerrieri, F.; Nicoletti, C.; Adorisio, E.; Caraccio, G.; Leonetti, P.; Zanotti, F.; Cantatore, P. Correlation between Decreased Expression of Mitochondrial F0F1-ATP Synthase and Low Regenerating Capability of the Liver after Partial Hepatectomy in Hypothyroid Rats. J. Bioenerg. Biomembr. 2000, 32, 183–191. [Google Scholar] [CrossRef]
- Mashek, D.G. Hepatic Fatty Acid Trafficking: Multiple Forks in the Road. Adv. Nutr. 2013, 4, 697–710. [Google Scholar] [CrossRef]
- Barrows, B.R.; Parks, E.J. Contributions of Different Fatty Acid Sources to Very Low-Density Lipoprotein-Triacylglycerol in the Fasted and Fed States. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef]
- Passarella, S.; de Bari, L.; Valenti, D.; Pizzuto, R.; Paventi, G.; Atlante, A. Mitochondria and L-Lactate Metabolism. FEBS Lett. 2008, 582, 3569–3576. [Google Scholar] [CrossRef]
- Paventi, G.; Pizzuto, R.; Passarella, S. The Occurrence of L-Lactate Dehydrogenase in the Inner Mitochondrial Compartment of Pig Liver. Biochem. Biophys. Res. Commun. 2017, 489, 255–261. [Google Scholar] [CrossRef]
- Tamura, Y.; Kawano, S.; Endo, T. Lipid Homeostasis in Mitochondria. Biol. Chem. 2020, 401, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Roden, M. Mitochondrial Alterations in Fatty Liver Diseases. J. Hepatol. 2023, 78, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, Y.; Yu, M.; Qin, J.; Ye, B.; Wang, Q. Mitochondrial Dysfunction and Chronic Liver Disease. Curr. Issues Mol. Biol. 2022, 44, 3156–3165. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef]
- Grattagliano, I.; de Bari, O.; Bernardo, T.C.; Oliveira, P.J.; Wang, D.Q.-H.; Portincasa, P. Role of Mitochondria in Nonalcoholic Fatty Liver Disease-from Origin to Propagation. Clin. Biochem. 2012, 45, 610–618. [Google Scholar] [CrossRef]
- Muriel, P.; Gordillo, K.R. Role of Oxidative Stress in Liver Health and Disease. Oxid. Med. Cell. Longev. 2016, 2016, 9037051. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, Oxidative Stress and Cell Death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in Non-Alcoholic Fatty Liver Disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99. [Google Scholar] [CrossRef]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-Mediated Reactive Oxygen Species Generation: Modulation by Cytochrome c and NAD(P)+ Oxidation-Reduction State. Biochem. J. 2002, 368, 545. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of Oxidative Stress and Inflammation in Neurodegenerative Disease: Role of TNF. Oxid. Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. The Connection between the Dynamic Remodeling of the Mitochondrial Network and the Regulation of Muscle Mass. Cell. Mol. Life Sci. 2021, 78, 1305–1328. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. Implications of Mitochondrial Fusion and Fission in Skeletal Muscle Mass and Health. Semin. Cell Dev. Biol. 2023, 143, 46–53. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.-X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef]
- Zuo, J.; Zhang, Z.; Luo, M.; Zhou, L.; Nice, E.C.; Zhang, W.; Wang, C.; Huang, C. Redox Signaling at the Crossroads of Human Health and Disease. MedComm 2022, 3, e127. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Figueira, T.R.; Barros, M.H.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.B.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.C.; Vercesi, A.E. Mitochondria as a Source of Reactive Oxygen and Nitrogen Species: From Molecular Mechanisms to Human Health. Antioxid. Redox Signal. 2013, 18, 2029–2074. [Google Scholar] [CrossRef]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wong, H.S. Are Mitochondria the Main Contributor of Reactive Oxygen Species in Cells? J. Exp. Biol. 2021, 224, jeb221606. [Google Scholar] [CrossRef] [PubMed]
- del Río, L.A.; López-Huertas, E. ROS Generation in Peroxisomes and Its Role in Cell Signaling. Plant Cell Physiol. 2016, 57, pcw076. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Henríquez-Olguín, C.; Boronat, S.; Cabello-Verrugio, C.; Jaimovich, E.; Hidalgo, E.; Jensen, T.E. The Emerging Roles of Nicotinamide Adenine Dinucleotide Phosphate Oxidase 2 in Skeletal Muscle Redox Signaling and Metabolism. Antioxid. Redox Signal. 2019, 31, 1371–1410. [Google Scholar] [CrossRef]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-Induced Oxidative Stress: Friend or Foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef]
- Henriquez-Olguin, C.; Meneses-Valdes, R.; Jensen, T.E. Compartmentalized Muscle Redox Signals Controlling Exercise Metabolism–Current State, Future Challenges. Redox Biol. 2020, 35, 101473. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Zheng, W.; Sun, Q.; Li, L.; Cheng, Y.; Chen, Y.; Lv, M.; Xiang, X. Role of Endoplasmic Reticulum Stress in Hepatic Glucose and Lipid Metabolism and Therapeutic Strategies for Metabolic Liver Disease. Int. Immunopharmacol. 2022, 113, 109458. [Google Scholar] [CrossRef]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic Reticulum Stress Signalling and the Pathogenesis of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic Reticulum Stress and Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Herranz-Itúrbide, M.; Peñuelas-Haro, I.; Espinosa-Sotelo, R.; Bertran, E.; Fabregat, I. The TGF-β/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells 2021, 10, 2312. [Google Scholar] [CrossRef]
- Matuz-Mares, D.; Vázquez-Meza, H.; Vilchis-Landeros, M.M. NOX as a Therapeutic Target in Liver Disease. Antioxidants 2022, 11, 2038. [Google Scholar] [CrossRef]
- de Medeiros, I.C.; de Lima, J.G. Is Nonalcoholic Fatty Liver Disease an Endogenous Alcoholic Fatty Liver Disease?—A Mechanistic Hypothesis. Med. Hypotheses 2015, 85, 148–152. [Google Scholar] [CrossRef]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity during Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10. [Google Scholar] [CrossRef]
- Zhai, L.; Pei, H.; Yang, Y.; Zhu, Y.; Ruan, S. NOX4 Promotes Kupffer Cell Inflammatory Response via ROS-NLRP3 to Aggravate Liver Inflammatory Injury in Acute Liver Injury. Aging 2022, 14, 6905–6916. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Brenner, D.A. Mechanisms of Liver Injury. I. TNF-Alpha-Induced Liver Injury: Role of IKK, JNK, and ROS Pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G583–G589. [Google Scholar] [CrossRef]
- Jeon, S.-M. Regulation and Function of AMPK in Physiology and Diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- von Loeffelholz, C.; Coldewey, S.M.; Birkenfeld, A.L. A Narrative Review on the Role of AMPK on De Novo Lipogenesis in Non-Alcoholic Fatty Liver Disease: Evidence from Human Studies. Cells 2021, 10, 1822. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of Nonalcoholic Fatty Liver Disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.W.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Görgün, C.Z.; Carling, D.; et al. Tumor Necrosis Factor α-Induced Skeletal Muscle Insulin Resistance Involves Suppression of AMP-Kinase Signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; Wong, K.I.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK–Caspase-6 Axis Controls Liver Damage in Nonalcoholic Steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J. Effects of H2O2 on Insulin Signaling the Glucose Transport System in Mammalian Skeletal Muscle. Methods Enzym. 2013, 528, 269–278. [Google Scholar]
- Blanquicett, C.; Kang, B.-Y.; Ritzenthaler, J.D.; Jones, D.P.; Hart, C.M. Oxidative Stress Modulates PPARγ in Vascular Endothelial Cells. Free Radic. Biol. Med. 2010, 48, 1618–1625. [Google Scholar] [CrossRef]
- Tendler, D.; Lin, S.; Yancy, W.S.; Mavropoulos, J.; Sylvestre, P.; Rockey, D.C.; Westman, E.C. The Effect of a Low-Carbohydrate, Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Pilot Study. Dig. Dis. Sci. 2007, 52, 589–593. [Google Scholar] [CrossRef]
- Cunha, G.M.; Guzman, G.; Correa De Mello, L.L.; Trein, B.; Spina, L.; Bussade, I.; Marques Prata, J.; Sajoux, I.; Countinho, W. Efficacy of a 2-Month Very Low-Calorie Ketogenic Diet (VLCKD) Compared to a Standard Low-Calorie Diet in Reducing Visceral and Liver Fat Accumulation in Patients with Obesity. Front. Endocrinol. 2020, 11, 607. [Google Scholar] [CrossRef]
- Ministrini, S.; Calzini, L.; Nulli Migliola, E.; Ricci, M.A.; Roscini, A.R.; Siepi, D.; Tozzi, G.; Daviddi, G.; Martorelli, E.-E.; Paganelli, M.T.; et al. Lysosomal Acid Lipase as a Molecular Target of the Very Low Carbohydrate Ketogenic Diet in Morbidly Obese Patients: The Potential Effects on Liver Steatosis and Cardiovascular Risk Factors. J. Clin. Med. 2019, 8, 621. [Google Scholar] [CrossRef]
- Bian, H.; Hakkarainen, A.; Lundbom, N.; Yki-Järvinen, H. Effects of Dietary Interventions on Liver Volume in Humans. Obesity 2014, 22, 989–995. [Google Scholar] [CrossRef]
- Yu, H.; Jia, W.; Guo, Z. Reducing Liver Fat by Low Carbohydrate Caloric Restriction Targets Hepatic Glucose Production in Non-Diabetic Obese Adults with Non-Alcoholic Fatty Liver Disease. J. Clin. Med. 2014, 3, 1050–1063. [Google Scholar] [CrossRef]
- Pérez-Guisado, J.; Muñoz-Serrano, A. The Effect of the Spanish Ketogenic Mediterranean Diet on Nonalcoholic Fatty Liver Disease: A Pilot Study. J. Med. Food 2011, 14, 677–680. [Google Scholar] [CrossRef]
- Paoli, A.; Tinsley, G.; Bianco, A.; Moro, T. The Influence of Meal Frequency and Timing on Health in Humans: The Role of Fasting. Nutrients 2019, 11, 719. [Google Scholar] [CrossRef]
- Kirk, E.; Reeds, D.N.; Finck, B.N.; Mayurranjan, M.S.; Patterson, B.W.; Klein, S. Dietary Fat and Carbohydrates Differentially Alter Insulin Sensitivity During Caloric Restriction. Gastroenterology 2009, 136, 1552–1560. [Google Scholar] [CrossRef]
- Browning, J.D.; Baker, J.A.; Rogers, T.; Davis, J.; Satapati, S.; Burgess, S.C. Short-Term Weight Loss and Hepatic Triglyceride Reduction: Evidence of a Metabolic Advantage with Dietary Carbohydrate Restriction. Am. J. Clin. Nutr. 2011, 93, 1048–1052. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Julia Xu, X.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A Long-Standing Partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef]
- Draznin, B.; Wang, C.; Adochio, R.; Leitner, J.; Cornier, M.-A. Effect of Dietary Macronutrient Composition on AMPK and SIRT1 Expression and Activity in Human Skeletal Muscle. Horm. Metab. Res. 2012, 44, 650–655. [Google Scholar] [CrossRef]
- Jani, S.; da Eira, D.; Stefanovic, M.; Ceddia, R.B. The Ketogenic Diet Prevents Steatosis and Insulin Resistance by Reducing Lipogenesis, Diacylglycerol Accumulation and Protein Kinase C Activity in Male Rat Liver. J. Physiol. 2022, 600, 4137–4151. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Wu, H.; Bjornson, E.; Zhang, C.; Hakkarainen, A.; Räsänen, S.M.; Lee, S.; Mancina, R.M.; Bergentall, M.; Pietiläinen, K.H.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571.e5. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Jia, W. Fibroblast Growth Factor 21: A Novel Metabolic Regulator from Pharmacology to Physiology. Front. Med. 2013, 7, 25–30. [Google Scholar] [CrossRef]
- Li, H.; Fang, Q.; Gao, F.; Fan, J.; Zhou, J.; Wang, X.; Zhang, H.; Pan, X.; Bao, Y.; Xiang, K.; et al. Fibroblast Growth Factor 21 Levels Are Increased in Nonalcoholic Fatty Liver Disease Patients and Are Correlated with Hepatic Triglyceride. J. Hepatol. 2010, 53, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Dushay, J.; Chui, P.C.; Gopalakrishnan, G.S.; Varela–Rey, M.; Crawley, M.; Fisher, F.M.; Badman, M.K.; Martinez–Chantar, M.L.; Maratos–Flier, E. Increased Fibroblast Growth Factor 21 in Obesity and Nonalcoholic Fatty Liver Disease. Gastroenterology 2010, 139, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Bueno, N.B.; de Melo, I.S.V.; de Oliveira, S.L.; da Rocha Ataide, T. Very-Low-Carbohydrate Ketogenic Diet v. Low-Fat Diet for Long-Term Weight Loss: A Meta-Analysis of Randomised Controlled Trials. Br. J. Nutr. 2013, 110, 1178–1187. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paoli, A.; Cerullo, G. Investigating the Link between Ketogenic Diet, NAFLD, Mitochondria, and Oxidative Stress: A Narrative Review. Antioxidants 2023, 12, 1065. https://doi.org/10.3390/antiox12051065
Paoli A, Cerullo G. Investigating the Link between Ketogenic Diet, NAFLD, Mitochondria, and Oxidative Stress: A Narrative Review. Antioxidants. 2023; 12(5):1065. https://doi.org/10.3390/antiox12051065
Chicago/Turabian StylePaoli, Antonio, and Giuseppe Cerullo. 2023. "Investigating the Link between Ketogenic Diet, NAFLD, Mitochondria, and Oxidative Stress: A Narrative Review" Antioxidants 12, no. 5: 1065. https://doi.org/10.3390/antiox12051065