
Tetrahydrobiopterin: Beyond Its Traditional Role as a Cofactor
 ,
,  ,
,
Abstract
:
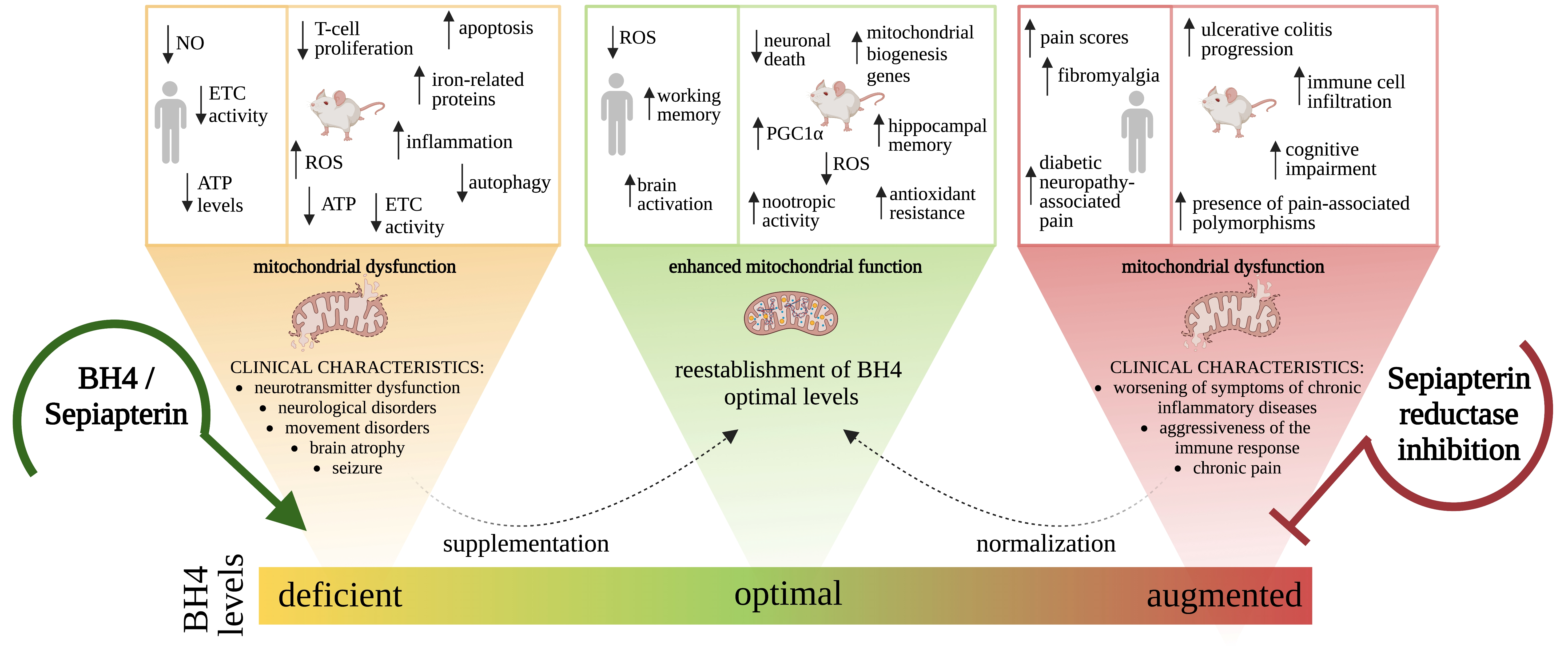
1. Tetrahydrobiopterin Biosynthesis
2. Levels of BH4 and Related Metabolites in Biological Fluids
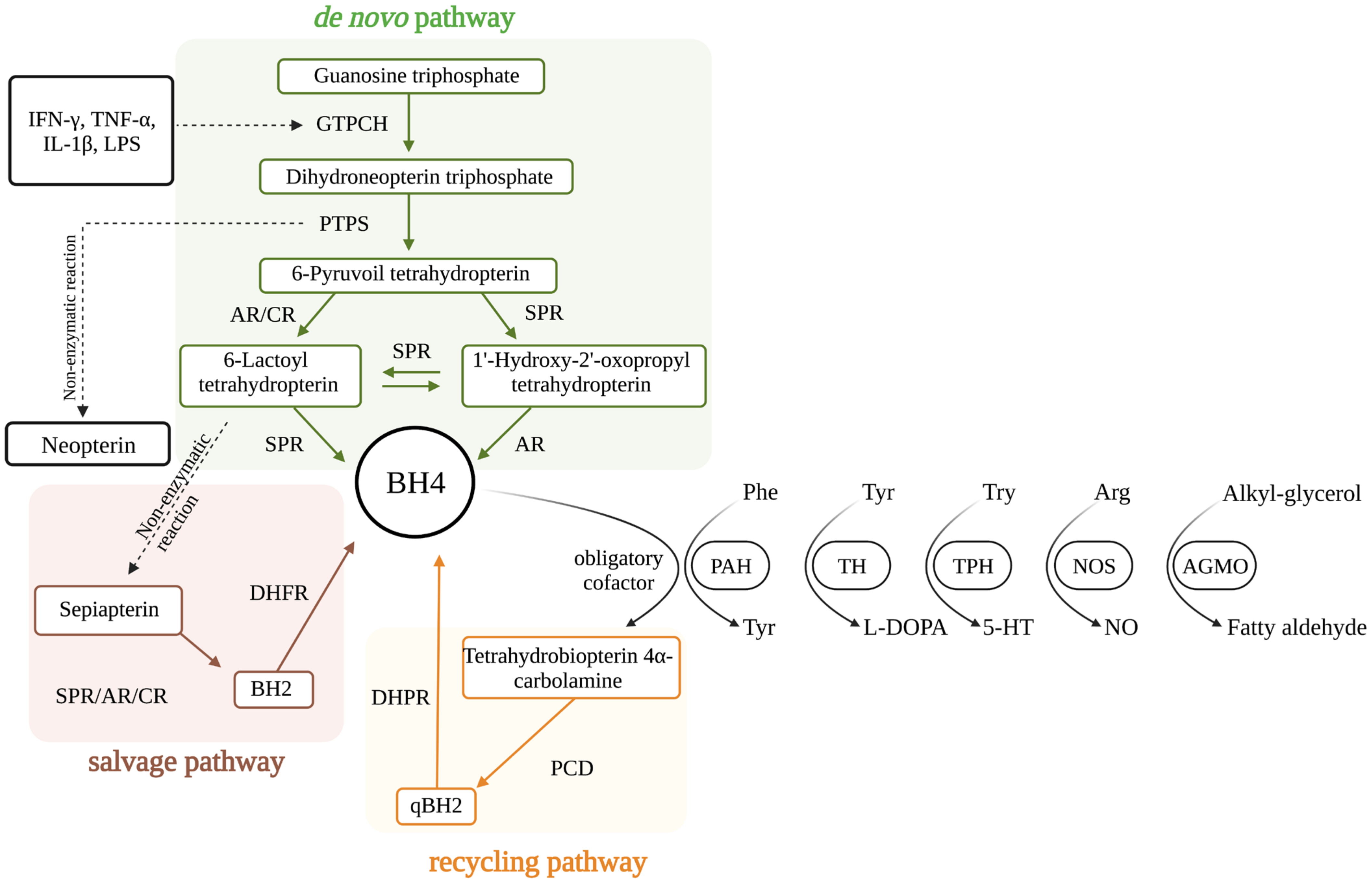
3. BH4 as an Enzyme Cofactor
3.1. PAH
3.2. TH
3.3. TPH
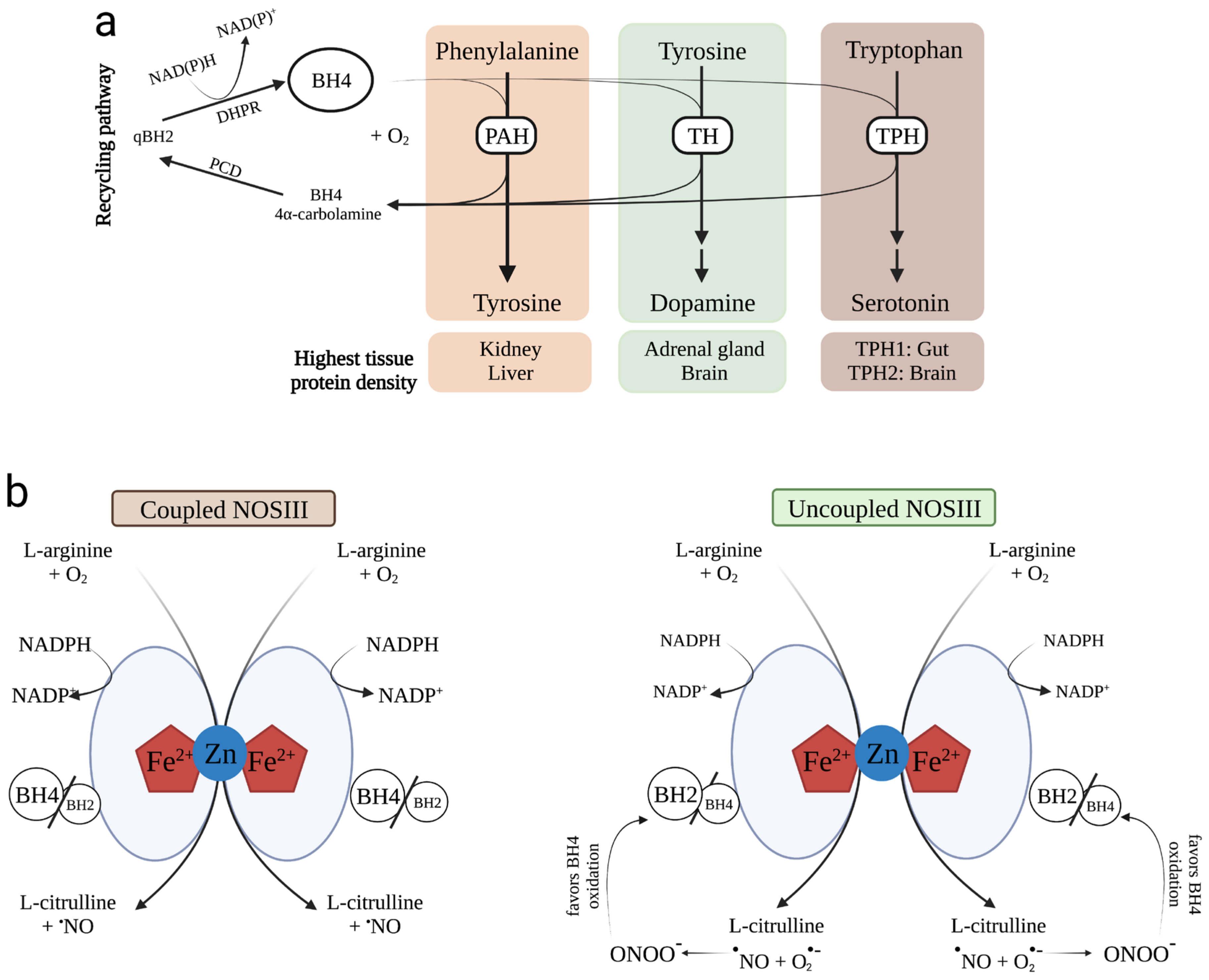
3.4. NOS

3.5. AGMO
4. Hereditary Deficiencies of BH4
4.1. GTPCH Deficiency
4.2. PTPS Deficiency
4.3. DHPR Deficiency
4.4. PCD Deficiency

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pterin Metabolism Defects with HPA | ||||||
|---|---|---|---|---|---|---|
| Disease Name | OMIM | Gene | Affected Enzyme | Symptoms | Diagnostic Based on Metabolites in CSF | First Line Treatment |
| Autosomal recessive GTP cyclohydrolase I deficiency | 233910 | GCH1 | GTPCH I | Developmental delay, hypotonia, hypertonia, dystonia, hypersalivation | ↓ HVA ↓ 5-HIAA ↓ /N 5-MTHF ↓ Biopterin ↓ Neopterin N Sepiapterin | Phe-reduced diet Sapropterin dihydrochloride L-Dopa/DC inhibitor 5-Hydroxytryptophan Folinic acid |
| 6-pyruvoyl-tetrahydropterin synthase deficiency | 261640 | PTS | PTPS | Developmental delay, hypotonia, hypertonia, epilepsy, cognitive impairment, low birth weight | ↓ HVA ↓ 5-HIAA ↓ /N 5-MTHF ↓ Biopterin ↑ Neopterin N Sepiapterin | Phe-reduced diet Sapropterin dihydrochloride L-Dopa/DC inhibitor 5-Hydroxytryptophan Folinic acid |
| Q-dihydropteridine reductase deficiency | 261630 | QDPR | DHPR | Developmental delay, hypotonia, hypertonia, epilepsy, microcephaly | ↓ HVA ↓ 5-HIAA ↓ /N 5-MTHF ↑ Biopterin ↓/N Neopterin N Sepiapterin | Phe-reduced diet L-Dopa/DC inhibitor 5-Hydroxytryptophan Folinic acid |
| Pterin-4-alpha-carbinolamine dehydratase deficiency | 264070 | PCBD1 | PCD | Developmental delay, hypotonia, hypomagnesemia, MODY3-like diabetes | NR HVA NR 5-HIAA NR 5-MTHF N Biopterin N Neopterin N Sepiapterin | Phe-reduced diet Sapropterin dihydrochloride L-Dopa/DC inhibitor 5-Hydroxytryptophan Folinic acid |
4.5. SPR Deficiency
| Pterin Metabolism Defects without HPA | ||||||
|---|---|---|---|---|---|---|
| Disease Name | OMIM | Gene | Affected Enzyme | Symptoms | Diagnostic Based on Metabolites in CSF | First Line Treatment |
| Autosomal dominant GTP cyclohydrolase I deficiency | 600225 | GCH1 | GTPCH I | Hypertonia, diurnal fluctuation of symptoms, dystonia, gait difficulties, hyperreflexia | ↓ /N HVA ↓ /N 5-HIAA NR 5-MTHF ↓ /N Biopterin ↓ /N Neopterin N Sepiapterin | Sapropterin dihydrochloride L-Dopa/DC inhibitor |
| Sepiapterin reductase deficiency | 182125 | SPR | SPR | Developmental delay, hypotonia, hypertonia, cognitive impairment, impaired speech development, dysarthria, diurnal fluctuation of symptoms, dystonia, oculogyric crises, dyskinesia/other involuntary movements, hypokinesia, hypersalivation, psychiatric and sleep problems | ↓ HVA ↓ 5-HIAA N 5-MTHF ↑ Biopterin N Neopterin ↑ Sepiapterin | Sapropterin dihydrochloride L-Dopa/DC inhibitor 5-Hydroxytryptophan |
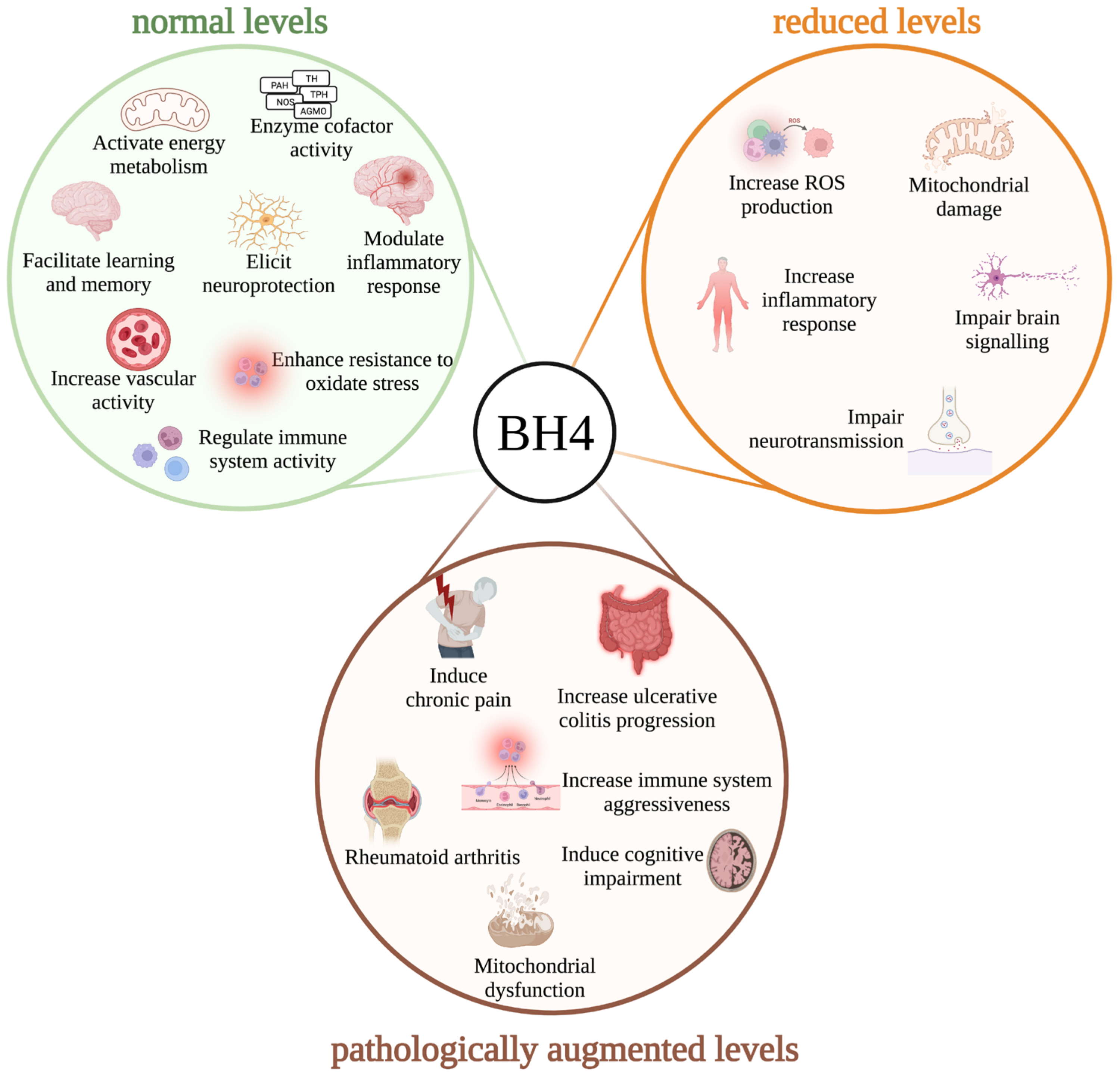
5. New Fundamental Roles of BH4 Metabolism Denoting Its Multifaceted Biological Functions
6. Biological Effects of Neopterin
7. Biological Effects of BH4
8. GTPCH and SPR Deficiencies Affect Energy Metabolism
9. Non-BH4-Linked Genetic Deficiencies of BH4 Metabolism
9.1. ASD
9.2. Human Rabies
9.3. Cerebral Malaria
9.4. PD
9.5. Alzheimer’s Disease (AD)
9.6. Fabry Disease
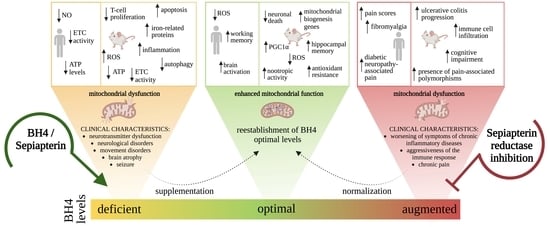
10. BH4 Administration as a New Therapeutic Horizon for Mitochondrial Diseases
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Werner, E.R.; Blau, N.; Thöny, B. Tetrahydrobiopterin: Biochemistry and Pathophysiology. Biochem. J. 2011, 438, 397–414. [Google Scholar] [CrossRef]
- Hirakawa, H.; Sawada, H.; Yamahama, Y.; Takikawa, S.-I.; Shintaku, H.; Hara, A.; Mase, K.; Kondo, T.; Iino, T. Expression Analysis of the Aldo-Keto Reductases Involved in the Novel Biosynthetic Pathway of Tetrahydrobiopterin in Human and Mouse Tissues. J. Biochem. 2009, 146, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Franscini, N.; Blau, N.; Walter, R.B.; Schaffner, A.; Schoedon, G. Critical Role of Interleukin-1β for Transcriptional Regulation of Endothelial 6-Pyruvoyltetrahydropterin Synthase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, e50-3. [Google Scholar] [CrossRef] [PubMed]
- D’Sa, C.; Hirayama, K.; West, A.; Hahn, M.; Zhu, M.; Kapatos, G. Tetrahydrobiopterin Biosynthesis in C6 Glioma Cells: Induction of GTP Cyclohydrolase I Gene Expression by Lipopolysaccharide and Cytokine Treatment. Brain Res. Mol. Brain Res. 1996, 41, 105–110. [Google Scholar] [CrossRef]
- Ishii, M.; Shimizu, S.; Wajima, T.; Hagiwara, T.; Negoro, T.; Miyazaki, A.; Tobe, T.; Kiuchi, Y. Reduction of GTP Cyclohydrolase I Feedback Regulating Protein Expression by Hydrogen Peroxide in Vascular Endothelial Cells. J. Pharmacol. Sci. 2005, 97, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.R.; Werner-Felmayer, G.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Yim, J.J.; Pfleiderer, W.; Wachter, H. Tetrahydrobiopterin Biosynthetic Activities in Human Macrophages, Fibroblasts, THP-1, and T 24 Cells. GTP-Cyclohydrolase I Is Stimulated by Interferon-Gamma, and 6-Pyruvoyl Tetrahydropterin Synthase and Sepiapterin Reductase Are Constitutively Present. J. Biol. Chem. 1990, 265, 3189–3192. [Google Scholar] [CrossRef] [PubMed]
- Ghisoni, K.; de Martins, R.P.; Barbeito, L.; Latini, A. Neopterin as a Potential Cytoprotective Brain Molecule. J. Psychiatr. Res. 2015, 71, 134–139. [Google Scholar] [CrossRef]
- Espíndola, G.; da Scheffer, D.L.; Latini, A. Commentary: Urinary Neopterin, a New Marker of the Neuroinflammatory Status in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 645694. [Google Scholar] [CrossRef]
- Thony, B.; Auerbach, G.; Blau, N. Tetrahydrobiopterin Biosynthesis, Regeneration and Functions. Biochem. J. 2000, 347, 1. [Google Scholar] [CrossRef]
- Harada, T.; Kagamiyama, H.; Hatakeyama, K. Feedback Regulation Mechanisms for the Control of GTP Cyclohydrolase I Activity. Science 1993, 260, 1507–1510. [Google Scholar] [CrossRef]
- Maita, N.; Hatakeyama, K.; Okada, K.; Hakoshima, T. Structural Basis of Biopterin-Induced Inhibition of GTP Cyclohydrolase I by GFRP, Its Feedback Regulatory Protein. J. Biol. Chem. 2004, 279, 51534–51540. [Google Scholar] [CrossRef] [PubMed]
- Hyland, K.; Surtees, R.A.H.; Heales, S.J.R.; Bowron, A.; Howells, D.W.; Smith, I. Cerebrospinal Fluid Concentrations of Pterins and Metabolites of Serotonin and Dopamine in a Pediatric Reference Population. Pediatr. Res. 1993, 34, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.R.; Bichler, A.; Daxenbichler, G.; Fuchs, D.; Fuith, L.C.; Hausen, A.; Hetzel, H.; Reibnegger, G.; Wachter, H. Determination of Neopterin in Serum and Urine. Clin. Chem. 1987, 33, 62–66. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Hagberg, L.; Dotevall, L.; Norkrans, G.; Larsson, M.; Wachter, H.; Fuchs, D. Cerebrospinal Fluid Neopterin Concentrations in Central Nervous System Infection. J. Infect. Dis. 1993, 168, 1285–1288. [Google Scholar] [CrossRef]
- Kuehne, L.K.; Reiber, H.; Bechter, K.; Hagberg, L.; Fuchs, D. Cerebrospinal Fluid Neopterin Is Brain-Derived and Not Associated with Blood-CSF Barrier Dysfunction in Non-Inflammatory Affective and Schizophrenic Spectrum Disorders. J. Psychiatr. Res. 2013, 47, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- De Paula Martins, R.; Ghisoni, K.; Lim, C.K.; Aguiar, A.S.; Guillemin, G.J.; Latini, A. Neopterin Preconditioning Prevents Inflammasome Activation in Mammalian Astrocytes. Free Radic. Biol. Med. 2018, 115, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Latini, A.; de Bortoli da Silva, L.; da Luz Scheffer, D.; Pires, A.C.S.; de Matos, F.J.; Nesi, R.T.; Ghisoni, K.; de Paula Martins, R.; de Oliveira, P.A.; Prediger, R.D.; et al. Tetrahydrobiopterin Improves Hippocampal Nitric Oxide-Linked Long-Term Memory. Mol. Genet. Metab. 2018, 125, 104–111. [Google Scholar] [CrossRef]
- Guibal, P.; Lévêque, N.; Doummar, D.; Giraud, N.; Roze, E.; Rodriguez, D.; Couderc, R.; Billette De Villemeur, T.; Moussa, F. Simultaneous Determination of All Forms of Biopterin and Neopterin in Cerebrospinal Fluid. ACS Chem. Neurosci. 2014, 5, 533–541. [Google Scholar] [CrossRef]
- Kaufman, S. The Participation of Tetrahydrofolic Acid in the Enzymic Conversion of Phenylalanine to Tyrosine. Biochim. Biophys. Acta 1958, 27, 428–429. [Google Scholar] [CrossRef]
- Scriver, C.; Kaufman, S. Hyperphenylalaninemia: Phenylalanine Hydroxylase Deficiency. In The Metabolic and Molecular Bases of Inherited Disease; McGraw-Hill: New York, NY, USA, 2001; pp. 1667–1724. [Google Scholar]
- Dox, A.W. The Excretion of Phenylpyruvic Acid in the Urine as a Metabolic Anomaly in Connection with Imbecility. (Zeitschr. Physiol. Chem., Vol. Ccxxvii, Pp. 169–76, 1934.) Fölling, A. J. Ment. Sci. 1935, 81, 236. [Google Scholar] [CrossRef]
- Guthrie, R.; Susi, A. A Simples Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics 1963, 32, 338–343. [Google Scholar] [CrossRef]
- Burton, B.K.; Bausell, H.; Katz, R.; LaDuca, H.; Sullivan, C. Sapropterin Therapy Increases Stability of Blood Phenylalanine Levels in Patients with BH4-Responsive Phenylketonuria (PKU). Mol. Genet. Metab. 2010, 101, 110–114. [Google Scholar] [CrossRef]
- Levy, H.L.; Sarkissian, C.N.; Scriver, C.R. Phenylalanine Ammonia Lyase (PAL): From Discovery to Enzyme Substitution Therapy for Phenylketonuria. Mol. Genet. Metab. 2018, 124, 223–229. [Google Scholar] [CrossRef]
- Tada, K.; Yoshida, T.; Mochizuki, K.; Konno, T.; Nakagawa, H.; Yokoyama, Y.; Takada, G.; Arakawa, T. Two Siblings of Hyperphenylalaninemia: Suggestion to a Genetic Variant of Phenylketonuria. Tohoku J. Exp. Med. 1970, 100, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Kure, S.; Hou, D.C.; Ohura, T.; Iwamoto, H.; Suzuki, S.; Sugiyama, N.; Sakamoto, O.; Fujii, K.; Matsubara, Y.; Narisawa, K. Tetrahydrobiopterin-Responsive Phenylalanine Hydroxylase Deficiency. J. Pediatr. 1999, 135, 375–378. [Google Scholar] [CrossRef]
- Blau, N.; Thöny, B.; Cotton, R.G.H.; Hyland, K. Disorders of Tetrahydrobiopterin and Related Biogenic Amines. In The Online Metabolic and Molecular Bases of Inherited Disease; McGraw-Hill: New York, NY, USA, 2019; pp. 1725–1776. [Google Scholar]
- Kaufman, S. Tyrosine Hydroxylase. In Advances in Enzymology and Related Areas of Molecular Biology; John Wiley & Sons: Hoboken, NJ, USA, 1995; Volume 70, pp. 103–220. [Google Scholar]
- Nagatsu, T.; Nagatsu, I. Tyrosine Hydroxylase (TH), Its Cofactor Tetrahydrobiopterin (BH4), Other Catecholamine-Related Enzymes, and Their Human Genes in Relation to the Drug and Gene Therapies of Parkinson’s Disease (PD): Historical Overview and Future Prospects. J. Neural Transm. 2016, 123, 1255–1278. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Dickson, P.W. Tyrosine Hydroxylase Phosphorylation In Vivo. J. Neurochem. 2019, 149, 706–728. [Google Scholar] [CrossRef] [PubMed]
- Nyhan, W.L.; Hoffmann, G.F. Atlas of Inherited Metabolic Diseases; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Lloyd, K.G.; Davidson, L.; Hornykiewicz, O. The Neurochemistry of Parkinson’s Disease: Effect of L-Dopa Therapy. J. Pharmacol. Exp. Ther. 1975, 195, 453–464. [Google Scholar] [PubMed]
- Ludecke, B.; Dworniczak, B.; Bartholomé, K. A Point Mutation in the Tyrosine Hydroxylase Gene Associated with Segawa’s Syndrome. Hum. Genet. 1995, 95, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, M.A.; Verbeek, M.M.; Kamsteeg, E.-J.; de Rijk-van Andel, J.F.; Aeby, A.; Blau, N.; Burlina, A.; Donati, M.A.; Geurtz, B.; Grattan-Smith, P.J.; et al. Tyrosine Hydroxylase Deficiency: A Treatable Disorder of Brain Catecholamine Biosynthesis. Brain 2010, 133, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Swaans, R.J.; Rondot, P.; Renier, W.O.; Van Den Heuvel, L.P.; Steenbergen-Spanjers, G.C.; Wevers, R.A. Four Novel Mutations in the Tyrosine Hydroxylase Gene in Patients with Infantile Parkinsonism. Ann. Hum. Genet. 2000, 64, 25–31. [Google Scholar] [CrossRef]
- Kurosaki, H.; Yamaguchi, K.; Man-yoshi, K.; Muramatsu, S.; Hara, S.; Ichinose, H. Administration of Tetrahydrobiopterin Restored the Decline of Dopamine in the Striatum Induced by an Acute Action of MPTP. Neurochem. Int. 2019, 125, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-W.; Jeon, B.S. Clinical Spectrum of Dopa-Responsive Dystonia and Related Disorders. Curr. Neurol. Neurosci. Rep. 2014, 14, 461. [Google Scholar] [CrossRef]
- Davis, M.D.; Kaufman, S. Evidence for the Formation of the 4a-Carbinolamine during the Tyrosine-Dependent Oxidation of Tetrahydrobiopterin by Rat Liver Phenylalanine Hydroxylase. J. Biol. Chem. 1989, 264, 8585–8596. [Google Scholar] [CrossRef]
- Kuhn, D.M.; Sakowski, S.A.; Geddes, T.J.; Wilkerson, C.; Haycock, J.W. Phosphorylation and Activation of Tryptophan Hydroxylase 2: Identification of Serine-19 as the Substrate Site for Calcium, Calmodulin-Dependent Protein Kinase II. J. Neurochem. 2007, 103, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Lindseth, G.; Helland, B.; Caspers, J. The Effects of Dietary Tryptophan on Affective Disorders. Arch. Psychiatr. Nurs. 2015, 29, 102–107. [Google Scholar] [CrossRef]
- Mawe, G.M.; Hoffman, J.M. Serotonin Signalling in the Gut—Functions, Dysfunctions and Therapeutic Targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486. [Google Scholar] [CrossRef]
- Walther, D.J.; Bader, M. A Unique Central Tryptophan Hydroxylase Isoform. Biochem. Pharmacol. 2003, 66, 1673–1680. [Google Scholar] [CrossRef]
- Knowles, R.G.; Moncada, S. Nitric Oxide Synthases in Mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef]
- Bredt, D.S.; Hwang, P.M.; Glatt, C.E.; Lowenstein, C.; Reed, R.R.; Snyder, S.H. Cloned and Expressed Nitric Oxide Synthase Structurally Resembles Cytochrome P-450 Reductase. Nature 1991, 351, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.P.; Lange, R.; Gorren, A.C.F.; Werner, E.R.; Mayer, B.; Andersson, K.K. Formation of a Protonated Trihydrobiopterin Radical Cation in the First Reaction Cycle of Neuronal and Endothelial Nitric Oxide Synthase Detected by Electron Paramagnetic Resonance Spectroscopy. JBIC J. Biol. Inorg. Chem. 2001, 6, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Alp, N.J.; Channon, K.M. Regulation of Endothelial Nitric Oxide Synthase by Tetrahydrobiopterin in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Sueta, G.; Radi, R. Chemical Biology of Peroxynitrite: Kinetics, Diffusion, and Radicals. ACS Chem. Biol. 2009, 4, 161–177. [Google Scholar] [CrossRef]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric Oxide Synthase Isozymes. Characterization, Purification, Molecular Cloning, and Functions. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, D.-Y. Neuronal Nitric Oxide Synthase: Structure, Subcellular Localization, Regulation, and Clinical Implications. Nitric Oxide 2009, 20, 223–230. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-Derived Relaxing Factor Produced and Released from Artery and Vein Is Nitric Oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Kim, H.K.; Han, J. Tetrahydrobiopterin in Energy Metabolism and Metabolic Diseases. Pharmacol. Res. 2020, 157, 104827. [Google Scholar] [CrossRef] [PubMed]
- Tietz, A.; Lindenberg, M.; Kennedy, E.P. A New Pteridine-Requiring Ezyme System for the Oxidation of Glyceryl Ethers. J. Biol. Chem. 1964, 239, 4081–4090. [Google Scholar] [CrossRef]
- Watschinger, K.; Keller, M.A.; Golderer, G.; Hermann, M.; Maglione, M.; Sarg, B.; Lindner, H.H.; Hermetter, A.; Werner-Felmayer, G.; Konrat, R.; et al. Identification of the Gene Encoding Alkylglycerol Monooxygenase Defines a Third Class of Tetrahydrobiopterin-Dependent Enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 13672–13677. [Google Scholar] [CrossRef] [PubMed]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong Association of De Novo Copy Number Mutations with Autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Awadalla, P.; Gauthier, J.; Myers, R.A.; Casals, F.; Hamdan, F.F.; Griffing, A.R.; Côté, M.; Henrion, E.; Spiegelman, D.; Tarabeux, J.; et al. Direct Measure of the De Novo Mutation Rate in Autism and Schizophrenia Cohorts. Am. J. Hum. Genet. 2010, 87, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Doan, R.N.; Bae, B.-I.; Cubelos, B.; Chang, C.; Hossain, A.A.; Al-Saad, S.; Mukaddes, N.M.; Oner, O.; Al-Saffar, M.; Balkhy, S.; et al. Mutations in Human Accelerated Regions Disrupt Cognition and Social Behavior. Cell 2016, 167, 341–354.e12. [Google Scholar] [CrossRef]
- Eto, I.; Bandy, M.D.; Butterworth, C.E. Plasma and Urinary Levels of Biopterin, Neopterin, and Related Pterins and Plasma Levels of Folate in Infantile Autism. J. Autism Dev. Disord. 1992, 22, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Klaiman, C.; Huffman, L.; Masaki, L.; Elliott, G.R. Tetrahydrobiopterin as a Treatment for Autism Spectrum Disorders: A Double-Blind, Placebo-Controlled Trial. J. Child Adolesc. Psychopharmacol. 2013, 23, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.; Berlow, S.; Summer, G.K.; Milstien, S.; Schulman, J.D.; Orloff, S.; Spielberg, S.; Pueschel, S. Hyperphenylalaninemia Due to a Deficiency of Biopterin. N. Engl. J. Med. 1978, 299, 673–679. [Google Scholar] [CrossRef]
- Blau, N.; Thony, B.; Spada, M.; Ponzone, A. Tetrahydrobiopterin and Inherited Hyperphenylalaninemias. Turk. J. Pediatr. 1996, 38, 19–35. [Google Scholar]
- Van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The Complete European Guidelines on Phenylketonuria: Diagnosis and Treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef]
- Opladen, T.; Hoffmann, G.F.; Blau, N. An International Survey of Patients with Tetrahydrobiopterin Deficiencies Presenting with Hyperphenylalaninaemia. J. Inherit. Metab. Dis. 2012, 35, 963–973. [Google Scholar] [CrossRef]
- Kikuchi, A.; Takeda, A.; Fujihara, K.; Kimpara, T.; Shiga, Y.; Tanji, H.; Nagai, M.; Ichinose, H.; Urano, F.; Okamura, N.; et al. Arg(184)His Mutant GTP Cyclohydrolase I, Causing Recessive Hyperphenylalaninemia, Is Responsible for Dopa-Responsive Dystonia with Parkinsonism: A Case Report. Mov. Disord. 2004, 19, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Opladen, T.; López-Laso, E.; Cortès-Saladelafont, E.; Pearson, T.S.; Sivri, H.S.; Yildiz, Y.; Assmann, B.; Kurian, M.A.; Leuzzi, V.; Heales, S.; et al. Consensus Guideline for the Diagnosis and Treatment of Tetrahydrobiopterin (BH4) Deficiencies. Orphanet J. Rare Dis. 2020, 15, 126. [Google Scholar] [CrossRef] [PubMed]
- Thöny, B.; Blau, N. Mutations in the BH 4 -Metabolizing Genes GTP Cyclohydrolase I, 6-Pyruvoyl-Tetrahydropterin Synthase, Sepiapterin Reductase, Carbinolamine-4a-Dehydratase, and Dihydropteridine Reductase. Hum. Mutat. 2006, 27, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Woody, R.C.; Brewster, M.A.; Glasier, C. Progressive Intracranial Calcification in Dihydropteridine Reductase Deficiency Prior to Folinic Acid Therapy. Neurology 1989, 39, 673. [Google Scholar] [CrossRef]
- Gudinchet, F.; Maeder, P.; Meuli, R.A.; Deonna, T.; Mathieu, J.M. Cranial CT and MRI in Malignant Phenylketonuria. Pediatr. Radiol. 1992, 22, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Ullrich, K.; Korinthenberg, R.; Peters, P.E. Basal Ganglion Calcification in Hyperphenylalaninemia Due to Deficiency of Dihydropteridine Reductase. Pediatr. Radiol. 1988, 19, 54–56. [Google Scholar] [CrossRef]
- De Sanctis, L.; Alliaudi, C.; Spada, M.; Farrugia, R.; Cerone, R.; Biasucci, G.; Meli, C.; Zammarchi, E.; Coskun, T.; Blau, N.; et al. Genotype-Phenotype Correlation in Dihydropteridine Reductase Deficiency. J. Inherit. Metab. Dis. 2000, 23, 333–337. [Google Scholar] [CrossRef]
- Thöny, B.; Neuheiser, F.; Kierat, L.; Rolland, M.O.; Guibaud, P.; Schlüter, T.; Germann, R.; Heidenreich, R.A.; Duran, M.; de Klerk, J.B.C.; et al. Mutations in the Pterin-4α-Carbinolamine Dehydratase (PCBD) Gene Cause a Benign Form of Hyperphenylalaninemia. Hum. Genet. 1998, 103, 162–167. [Google Scholar] [CrossRef]
- Himmelreich, N.; Blau, N.; Thöny, B. Molecular and Metabolic Bases of Tetrahydrobiopterin (BH4) Deficiencies. Mol. Genet. Metab. 2021, 133, 123–136. [Google Scholar] [CrossRef]
- Burlina, A.; van Spronsen, F.J.; Blau, N. Disorders of Phenylalanine and Tetrahydrobiopterin Metabolism. In Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases; Springer: Cham, Switzerland, 2022; pp. 331–351. [Google Scholar]
- Hevel, J.M.; Stewart, J.A.; Gross, K.L.; Ayling, J.E. Can the DCoHα Isozyme Compensate in Patients with 4a-Hydroxy-Tetrahydrobiopterin Dehydratase/DCoH Deficiency? Mol. Genet. Metab. 2006, 88, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Blau, N.; van Spronsen, F.J. Disorders of Phenylalanine and Tetrahydrobiopterin Metabolism. In Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–21. [Google Scholar]
- Bonafé, L.; Thöny, B.; Penzien, J.M.; Czarnecki, B.; Blau, N. Mutations in the Sepiapterin Reductase Gene Cause a Novel Tetrahydrobiopterin-Dependent Monoamine-Neurotransmitter Deficiency without Hyperphenylalaninemia. Am. J. Hum. Genet. 2001, 69, 269–277. [Google Scholar] [CrossRef]
- Blau, N.; Bonafé, L.; Thöny, B. Tetrahydrobiopterin Deficiencies without Hyperphenylalaninemia: Diagnosis and Genetics of DOPA-Responsive Dystonia and Sepiapterin Reductase Deficiency. Mol. Genet. Metab. 2001, 74, 172–185. [Google Scholar] [CrossRef]
- Longo, N. Disorders of Biopterin Metabolism. J. Inherit. Metab. Dis. 2009, 32, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Abeling, N.; Duran, M.; Bakker, H.; Strommer, L.; Thony, B.; Blau, N.; Booij, J.; Pollthe, B. Sepiapterin Reductase Deficiency an Autosomal Recessive DOPA-Responsive Dystonia. Mol. Genet. Metab. 2006, 89, 116–120. [Google Scholar] [CrossRef]
- Neville, B.G.R.; Parascandalo, R.; Farrugia, R.; Felice, A. Sepiapterin Reductase Deficiency: A Congenital Dopa-Responsive Motor and Cognitive Disorder. Brain 2005, 128, 2291–2296. [Google Scholar] [CrossRef]
- Ghisoni, K.; Latini, A.; Kuehne, L.K.; Reiber, H.; Bechter, K.; Hagberg, L.; Fuchs, D. Cerebrospinal fluid neopterin is brain-derived and not associated with blood-CSF barrier dysfunction in non-inflammatory affective and schizophrenic spectrum disorders. Letter to the Editor. J. Psychiatr. Res. 2015, 63, 141–142. [Google Scholar] [CrossRef]
- Cronin, S.J.F.; Seehus, C.; Weidinger, A.; Talbot, S.; Reissig, S.; Seifert, M.; Pierson, Y.; McNeill, E.; Longhi, M.S.; Turnes, B.L.; et al. The Metabolite BH4 Controls T Cell Proliferation in Autoimmunity and Cancer. Nature 2018, 563, 564–568. [Google Scholar] [CrossRef]
- Latremoliere, A.; Latini, A.; Andrews, N.; Cronin, S.J.; Fujita, M.; Gorska, K.; Hovius, R.; Romero, C.; Chuaiphichai, S.; Painter, M.; et al. Reduction of Neuropathic and Inflammatory Pain through Inhibition of the Tetrahydrobiopterin Pathway. Neuron 2015, 86, 1393–1406. [Google Scholar] [CrossRef]
- Fujita, M.; da Scheffer, D.L.; Turnes, B.L.; Cronin, S.J.F.; Latrémolière, A.; Costigan, M.; Woolf, C.J.; Latini, A.; Andrews, N.A. Sepiapterin Reductase Inhibition Leading to Selective Reduction of Inflammatory Joint Pain in Mice and Increased Urinary Sepiapterin Levels in Humans and Mice. Arthritis Rheumatol. 2020, 72, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Staats Pires, A.; Heng, B.; Tan, V.X.; Latini, A.; Russo, M.A.; Santarelli, D.M.; Bailey, D.; Wynne, K.; O’Brien, J.A.; Guillemin, G.J.; et al. Kynurenine, Tetrahydrobiopterin, and Cytokine Inflammatory Biomarkers in Individuals Affected by Diabetic Neuropathic Pain. Front. Neurosci. 2020, 14, 890. [Google Scholar] [CrossRef]
- Staats Pires, A.; Tan, V.X.; Heng, B.; Guillemin, G.J.; Latini, A. Kynurenine and Tetrahydrobiopterin Pathways Crosstalk in Pain Hypersensitivity. Front. Neurosci. 2020, 14, 620. [Google Scholar] [CrossRef]
- Woolf, C.; Latini, A.; Andrews, N.; Costigan, M.; Latremoliere, A. Methods and Assays Relating to Sepiapterin Reductase Inhibition. U.S. Patent 10,365,267, 30 July 2016. [Google Scholar]
- Weiss, G.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Werner, E.R.; Werner-Felmayer, G.; Semenitz, E.; Dierich, M.P.; Wachter, H. Neopterin Modulates Toxicity Mediated by Reactive Oxygen and Chloride Species. FEBS Lett. 1993, 321, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Baier-Bitterlich, G.; Fuchs, D.; Murr, C.; Reibnegger, G.; Werner-Felmayer, G.; Sgonc, R.; Böck, G.; Dierich, M.P.; Wachter, H. Effect of Neopterin and 7,8-Dihydroneopterin on Tumor Necrosis Factor-α Induced Programmed Cell Death. FEBS Lett. 1995, 364, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Schobersberger, W.; Hoffmann, G.; Hobisch-Hagen, P.; Böck, G.; Völkl, H.; Baier-Bitterlich, G.; Wirleitner, B.; Wachter, H.; Fuchs, D. Neopterin and 7,8-Dihydroneopterin Induce Apoptosis in the Rat Alveolar Epithelial Cell Line L2. FEBS Lett. 1996, 397, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.; Schobersberger, W.; Frede, S.; Pelzer, L.; Fandrey, J.; Wachter, H.; Fuchs, D.; Grote, J. Neopterin Activates Transcription Factor Nuclear Factor-ΚB in Vascular Smooth Muscle Cells. FEBS Lett. 1996, 391, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.; Rieder, J.; Smolny, M.; Seibel, M.; Wirleitner, B.; Fuchs, D.; Schobersberger, W. Neopterin-Induced Expression of Intercellular Adhesion Molecule-1 (ICAM-1) in Type II-like Alveolar Epithelial Cells. Clin. Exp. Immunol. 1999, 118, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, P.; Pacileo, M.; De Rosa, S.; Calabro, P.; Gargiulo, A.; Angri, V.; Granato-Corigliano, F.; Fiorentino, I.; Prevete, N.; De Palma, R.; et al. Neopterin Induces Pro-atherothrombotic Phenotype in Human Coronary Endothelial Cells. J. Thromb. Haemost. 2006, 4, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Matiollo, C.; de Rateke, E.C.M.; de Oliveira, K.G.; Turnes, B.L.; da Silva, T.E.; Maccali, C.; Latini, A.S.; Narciso-Schiavon, J.L.; Schiavon, L. Elevated Neopterin Levels Are Associated with Acute-on-Chronic Liver Failure and Mortality in Patients with Liver Cirrhosis. Dig. Liver Dis. 2020, 52, 753–760. [Google Scholar] [CrossRef]
- Ghisoni, K.; Aguiar, A.S.; de Oliveira, P.A.; Matheus, F.C.; Gabach, L.; Perez, M.; Carlini, V.P.; Barbeito, L.; Mongeau, R.; Lanfumey, L.; et al. Neopterin Acts as an Endogenous Cognitive Enhancer. Brain. Behav. Immun. 2016, 56, 156–164. [Google Scholar] [CrossRef]
- Izquierdo, I.; Medina, J.H. Memory Formation: The Sequence of Biochemical Events in the Hippocampus and Its Connection to Activity in Other Brain Structures. Neurobiol. Learn. Mem. 1997, 68, 285–316. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Kim, K.-M.; Kim, S.W.; Hwang, O.; Choi, H.J. Bromocriptine Activates NQO1 via Nrf2-PI3K/Akt Signaling: Novel Cytoprotective Mechanism against Oxidative Damage. Pharmacol. Res. 2008, 57, 325–331. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.; da Silva, R.A.; da Luz Scheffer, D.; Penteado, R.; Solano, A.; Barros, L.; Budde, H.; Trostchansky, A.; Latini, A. Physical-Exercise-Induced Antioxidant Effects on the Brain and Skeletal Muscle. Antioxidants 2022, 11, 826. [Google Scholar] [CrossRef]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial Transcription Factor A Regulates Mitochondrial Transcription Initiation, DNA Packaging, and Genome Copy Number. Biochim. Biophys. Acta-Gene Regul. Mech. 2012, 1819, 921–929. [Google Scholar] [CrossRef]
- Falkenberg, M.; Larsson, N.-G.; Gustafsson, C.M. DNA Replication and Transcription in Mammalian Mitochondria. Annu. Rev. Biochem. 2007, 76, 679–699. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Christ, S.E.; Moffitt, A.J.; Peck, D.; White, D.A. The Effects of Tetrahydrobiopterin (BH4) Treatment on Brain Function in Individuals with Phenylketonuria. NeuroImage Clin. 2013, 3, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.B.; Stratford, M.R.L.; Wardman, P.; Everett, S.A. Oxidation of Tetrahydrobiopterin by Biological Radicals and Scavenging of the Trihydrobiopterin Radical by Ascorbate. Free Radic. Biol. Med. 2002, 32, 203–211. [Google Scholar] [CrossRef]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of Peroxynitrite, Tetrahydrobiopterin, Ascorbic Acid, and Thiols. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef]
- Foxton, R.H.; Land, J.M.; Heales, S.J.R. Tetrahydrobiopterin Availability in Parkinson’s and Alzheimer’s Disease; Potential Pathogenic Mechanisms. Neurochem. Res. 2007, 32, 751–756. [Google Scholar] [CrossRef]
- Xue, J.; Yu, C.; Sheng, W.; Zhu, W.; Luo, J.; Zhang, Q.; Yang, H.; Cao, H.; Wang, W.; Zhou, J.; et al. The Nrf2/GCH1/BH4 Axis Ameliorates Radiation-Induced Skin Injury by Modulating the ROS Cascade. J. Investig. Dermatol. 2017, 137, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Nakanishi, N.; Akino, M. Stoichiometric Studies on the Oxidation of Tetrahydropterin with Ferri-Cytochrome C. J. Biochem. 1978, 84, 499–506. [Google Scholar] [CrossRef]
- Davis, M.D.; Kaugman, S.; Milstien, S. The Auto-Oxidation of Tetrahydrobiopterin. Eur. J. Biochem. 1988, 173, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Jeon, J.; Song, I.-S.; Heo, H.J.; Jeong, S.H.; Long, L.T.; Thu, V.T.; Ko, T.H.; Kim, M.; Kim, N.; et al. Tetrahydrobiopterin Enhances Mitochondrial Biogenesis and Cardiac Contractility via Stimulation of PGC1α Signaling. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 165524. [Google Scholar] [CrossRef]
- Porkert, M.; Sher, S.; Reddy, U.; Cheema, F.; Niessner, C.; Kolm, P.; Jones, D.P.; Hooper, C.; Taylor, W.R.; Harrison, D.; et al. Tetrahydrobiopterin: A Novel Antihypertensive Therapy. J. Hum. Hypertens. 2008, 22, 401–407. [Google Scholar] [CrossRef]
- Robbins, I.M.; Hemnes, A.R.; Simon Gibbs, J.; Christman, B.W.; Howard, L.; Meehan, S.; Cabrita, I.; Gonzalez, R.; Oyler, T.; Zhao, L.; et al. Safety of Sapropterin Dihydrochloride (6r–Bh4) in Patients with Pulmonary Hypertension. Exp. Lung Res. 2011, 37, 26–34. [Google Scholar] [CrossRef]
- Frye, R.E.; DeLaTorre, R.; Taylor, H.B.; Slattery, J.; Melnyk, S.; Chowdhury, N.; James, S.J. Metabolic Effects of Sapropterin Treatment in Autism Spectrum Disorder: A Preliminary Study. Transl. Psychiatry 2013, 3, e237. [Google Scholar] [CrossRef] [PubMed]
- De Maria, R.; Campolo, J.; Frontali, M.; Taroni, F.; Federico, A.; Inzitari, D.; Tavani, A.; Romano, S.; Puca, E.; Orzi, F.; et al. Effects of Sapropterin on Endothelium-Dependent Vasodilation in Patients with CADASIL. Stroke 2014, 45, 2959–2966. [Google Scholar] [CrossRef]
- Tobin, J.E.; Cui, J.; Wilk, J.B.; Latourelle, J.C.; Laramie, J.M.; McKee, A.C.; Guttman, M.; Karamohamed, S.; DeStefano, A.L.; Myers, R.H. Sepiapterin Reductase Expression Is Increased in Parkinson’s Disease Brain Tissue. Brain Res. 2007, 1139, 42–47. [Google Scholar] [CrossRef]
- Sawada, M.; Hirata, Y.; Arai, H.; Iizuka, R.; Nagatsu, T. Tyrosine Hydroxylase, Tryptophan Hydroxylase, Biopterin, and Neopterin in the Brains of Normal Controls and Patients with Senile Dementia of Alzheimer Type. J. Neurochem. 1987, 48, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Romanowicz, J.; Leonetti, C.; Dhari, Z.; Korotcova, L.; Ramachandra, S.D.; Saric, N.; Morton, P.D.; Bansal, S.; Cheema, A.; Gallo, V.; et al. Treatment with Tetrahydrobiopterin Improves White Matter Maturation in a Mouse Model for Prenatal Hypoxia in Congenital Heart Disease. J. Am. Heart Assoc. 2019, 8, e012711. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Vásquez-Vivar, J.; Jiang, R.; Luo, K.; Derrick, M.; Tan, S. Developmental Susceptibility of Neurons to Transient Tetrahydrobiopterin Insufficiency and Antenatal Hypoxia–Ischemia in Fetal Rabbits. Free Radic. Biol. Med. 2014, 67, 426–436. [Google Scholar] [CrossRef]
- Kim, H.K.; Ha, S.H.; Han, J. Potential Therapeutic Applications of Tetrahydrobiopterin: From Inherited Hyperphenylalaninemia to Mitochondrial Diseases. Ann. N. Y. Acad. Sci. 2010, 1201, 177–182. [Google Scholar] [CrossRef]
- Raphael, S. Tetrahydrobiopterin Concentrations in Normal and Coronary Artery Diseased Heart Tissue. J. Cardiovasc. Med. Cardiol. 2016, 3, 014–017. [Google Scholar] [CrossRef]
- Remor, A.P.; da Silva, R.A.; de Matos, F.J.; Glaser, V.; de Paula Martins, R.; Ghisoni, K.; da Luz Scheffer, D.; Andia, D.C.; Portinho, D.; de Souza, A.P.; et al. Chronic Metabolic Derangement-Induced Cognitive Deficits and Neurotoxicity Are Associated with REST Inactivation. Mol. Neurobiol. 2018, 56, 1539–1557. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Scarpulla, R.C. PGC-1-Related Coactivator, a Novel, Serum-Inducible Coactivator of Nuclear Respiratory Factor 1-Dependent Transcription in Mammalian Cells. Mol. Cell. Biol. 2001, 21, 3738–3749. [Google Scholar] [CrossRef]
- Rambold, A.S.; Lippincott-Schwartz, J. Mechanisms of Mitochondria and Autophagy Crosstalk. Cell Cycle 2011, 10, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.S.; Suk, J.; Choi, J.H.; Yang, S.; Kim, J.W.; Sohn, S.; Chung, J.H.; Hong, Y.H.; Lee, D.H.; Ahn, J.K.; et al. Autophagy Induction by Tetrahydrobiopterin Deficiency. Autophagy 2011, 7, 1323–1334. [Google Scholar] [CrossRef]
- Delgado-Esteban, M.; Almeida, A.; Medina, J.M. Tetrahydrobiopterin Deficiency Increases Neuronal Vulnerability to Hypoxia. J. Neurochem. 2002, 82, 1148–1159. [Google Scholar] [CrossRef]
- Tani, Y.; Fernell, E.; Watanabe, Y.; Kanai, T.; Långström, B. Decrease in 6R-5,6,7,8-Tetrahydrobiopterin Content in Cerebrospinal Fluid of Autistic Patients. Neurosci. Lett. 1994, 181, 169–172. [Google Scholar] [CrossRef]
- Danfors, T.; von Knorring, A.-L.; Hartvig, P.; Langstrom, B.; Moulder, R.; Stromberg, B.; Torstenson, R.; Wester, U.; Watanabe, Y.; Eeg-Olofsson, O. Tetrahydrobiopterin in the Treatment of Children with Autistic Disorder. J. Clin. Psychopharmacol. 2005, 25, 485–489. [Google Scholar] [CrossRef]
- Fernell, E.; Watanabe, Y.; Adolfsson, I.; Tani, Y.; Bergström, M.; Lilja, A.; von Knorring, A.-L.; Gillberg, C.; Lángström, B. Possible Effects of Tetrahydrobiopterin Treatment in Six Children with Autism-Clinical and Positron Emission Tomography Data: A Pilot Study. Dev. Med. Child Neurol. 2008, 39, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Naruse, H.; Takesada, M.; Nagahata, M.; Kazamatsuri, H.; Nakane, Y.; Yamazaki, K. An Open Clinical Study of Apropterin Hydrochloride (R-Tetrahydrobiopterin SUN 0588) in Infantile Autism–Clinical Study Using a Rating Scale for Abnormal Behaviors in Children. Rinsho Iyaku 1990, 6, 1859–1875. (In Japanese) [Google Scholar]
- Naruse, H.; Hayashi, T.; Takesada, M. A Preliminary Study on Clinical Effect of Tetrahydrobiopterin in Infantile Autism; Rep. 1983 New Drug Dev; Ministry of Health and Welfare: Tokyo, Japan, 1984; pp. 71–81. (In Japanese) [Google Scholar]
- Naruse, H.; Takesada, M.; Nakane, Y.; Yamazaki, K.; Uchiyama, T.; Kaihara, S.; Ohashi, T. Clinical Evaluation of R-Tetrahydrobiopterin (SUN 0588) on Infantile Autism–A Double-Blind Comparative Study Using Placebo as a Control. Rinsho Iyaku 1990, 6, 1343–1368. [Google Scholar]
- Takesada, M.; Naruse, H.; Nagahata, M. An Open Clinical Study of Sapropterin Hydrochloride (R-Tetrahydrobiopterin, R-THBP) in Infantile Autism: Clinical Effects and Long-Term Follow-Up. In Proceedings of the International Symposium on Neurobiology of Infantile Autism, Tokyo, Japan, 10–11 November 1990; Excerpta Medica: Tokyo, Japan, 1992; pp. 355–358. [Google Scholar]
- Nakane, Y.; Asuo, T.; Shimogawa, S.; Fujiwara, T.; Kawabata, Y.; Kubota, J. Clinical Efficacy and Effects on Physical Development of Long-Term Treatment of R-Tetrahydrobiopterin (R-THBP, SUN 0588) for Autism. Kiso Rinshou 1990, 24, 4579–4598. [Google Scholar]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic Biomarkers of Increased Oxidative Stress and Impaired Methylation Capacity in Children with Autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Trivedi, M.H.; Garver, C.R.; Grannemann, B.D.; Andrews, A.A.; Savla, J.S.; Johnson, D.G.; Mehta, J.A.; Schroeder, J.L. The Pattern of Sensory Processing Abnormalities in Autism. Autism 2006, 10, 480–494. [Google Scholar] [CrossRef]
- Koshimura, K.; Murakami, Y.; Tanaka, J.; Kato, Y. Self-Protection of PC12 Cells by 6R-Tetrahydrobiopterin from Nitric Oxide Toxicity. J. Neurosci. Res. 1998, 54, 664–672. [Google Scholar] [CrossRef]
- İnci, A.; Özaslan, A.; Okur, İ.; Biberoğlu, G.; Güney, E.; Ezgü, F.S.; Tümer, L.; İşeri, E. Autism: Screening of Inborn Errors of Metabolism and Unexpected Results. Autism Res. 2021, 14, 887–896. [Google Scholar] [CrossRef]
- Schnetz-Boutaud, N.C.; Anderson, B.M.; Brown, K.D.; Wright, H.H.; Abramson, R.K.; Cuccaro, M.L.; Gilbert, J.R.; Pericak-Vance, M.A.; Haines, J.L. Examination of Tetrahydrobiopterin Pathway Genes in Autism. Genes Brain Behav. 2009, 8, 753–757. [Google Scholar] [CrossRef]
- Willoughby, R.E.; Opladen, T.; Maier, T.; Rhead, W.; Schmiedel, S.; Hoyer, J.; Drosten, C.; Rupprecht, C.E.; Hyland, K.; Hoffmann, G.F. Tetrahydrobiopterin Deficiency in Human Rabies. J. Inherit. Metab. Dis. 2009, 32, 65–72. [Google Scholar] [CrossRef]
- Willoughby, R.E.; Tieves, K.S.; Hoffman, G.M.; Ghanayem, N.S.; Amlie-Lefond, C.M.; Schwabe, M.J.; Chusid, M.J.; Rupprecht, C.E. Survival after Treatment of Rabies with Induction of Coma. N. Engl. J. Med. 2005, 352, 2508–2514. [Google Scholar] [CrossRef]
- Rubach, M.P.; Mukemba, J.; Florence, S.; Lopansri, B.K.; Hyland, K.; Volkheimer, A.D.; Yeo, T.W.; Anstey, N.M.; Weinberg, J.B.; Mwaikambo, E.D.; et al. Impaired Systemic Tetrahydrobiopterin Bioavailability and Increased Oxidized Biopterins in Pediatric Falciparum Malaria: Association with Disease Severity. PLoS Pathog. 2015, 11, e1004655. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Thuma, P.E.; Biemba, G.; Mabeza, G.; Werner, E.R.; Gordeuk, V.R. Cerebrospinal Fluid Levels of Biopterin, Nitric Oxide Metabolites, and Immune Activation Markers and the Clinical Course of Human Cerebral Malaria. J. Infect. Dis. 1998, 177, 1064–1068. [Google Scholar] [CrossRef]
- Shang, T.; Kotamraju, S.; Kalivendi, S.V.; Hillard, C.J.; Kalyanaraman, B. 1-Methyl-4-Phenylpyridinium-Induced Apoptosis in Cerebellar Granule Neurons Is Mediated by Transferrin Receptor Iron-Dependent Depletion of Tetrahydrobiopterin and Neuronal Nitric-Oxide Synthase-Derived Superoxide. J. Biol. Chem. 2004, 279, 19099–19112. [Google Scholar] [CrossRef] [PubMed]
- Lovenberg, W.; Levine, R.A.; Robinson, D.S.; Ebert, M.; Williams, A.C.; Calne, D.B. Hydroxylase Cofactor Activity in Cerebrospinal Fluid of Normal Subjects and Patients with Parkinson’s Disease. Science 1979, 204, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Yamaguchi, T.; Kato, T.; Sugimoto, T.; Matsuura, S.; Akino, M.; Nagatsu, I.; Iizuka, R.; Narabayashi, H. Biopterin in Human Brain and Urine from Controls and Parkinsonian Patients: Application of a New Radioimmunoassay. Clin. Chim. Acta 1981, 109, 305–311. [Google Scholar] [CrossRef]
- Da Scheffer, D.L.; Freitas, F.C.; Aguiar Jr, A.S.; Ward, C.; Guglielmo, L.G.A.; Prediger, R.D.; Cronin, S.J.F.; Walz, R.; Andrews, N.A.; Latini, A. Impaired Dopamine Metabolism Is Linked to Fatigability in Mice and Fatigue in Parkinson’s Disease Patients. Brain Commun. 2021, 3, fcab116. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2021 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Barford, P.A.; Blair, J.A.; Eggar, C.; Hamon, C.; Morar, C.; Whitburn, S.B. Tetrahydrobiopterin Metabolism in the Temporal Lobe of Patients Dying with Senile Dementia of Alzheimer Type. J. Neurol. Neurosurg. Psychiatry 1984, 47, 736–738. [Google Scholar] [CrossRef] [PubMed]
- Fanet, H.; Tournissac, M.; Leclerc, M.; Caron, V.; Tremblay, C.; Vancassel, S.; Calon, F. Tetrahydrobiopterin Improves Recognition Memory in the Triple-Transgenic Mouse Model of Alzheimer’s Disease, Without Altering Amyloid-β and Tau Pathologies. J. Alzheimer’s Dis. 2021, 79, 709–727. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic Defect in Fabry’s Disease. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef]
- Desnick, R.J.; Ioannou, Y.A.; Eng, C.M. α-Galactosidase A Deficiency: Fabry Disease. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw Hill: New York, NY, USA, 2019. [Google Scholar]
- Chinnery, P.F.; Turnbull, D.M. Epidemiology and Treatment of Mitochondrial Disorders. Am. J. Med. Genet. 2001, 106, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A Mitochondrial Bioenergetic Etiology of Disease. J. Clin. Investig. 2013, 123, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Vargas, C.; Wajner, M.; Sirtori, L.; Goulart, L.; Chiochetta, M.; Coelho, D.; Latini, A.; Llesuy, S.; Bello-Klein, A.; Giugliani, R.; et al. Evidence That Oxidative Stress Is Increased in Patients with X-Linked Adrenoleukodystrophy. Biochim. Biophys. Acta-Mol. Basis Dis. 2004, 1688, 26–32. [Google Scholar] [CrossRef]
- Wajner, M. Neurological Manifestations of Organic Acidurias. Nat. Rev. Neurol. 2019, 15, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Seminotti, B.; Amaral, A.U.; Ribeiro, R.T.; Rodrigues, M.D.N.; Colín-González, A.L.; Leipnitz, G.; Santamaría, A.; Wajner, M. Oxidative Stress, Disrupted Energy Metabolism, and Altered Signaling Pathways in Glutaryl-CoA Dehydrogenase Knockout Mice: Potential Implications of Quinolinic Acid Toxicity in the Neuropathology of Glutaric Acidemia Type I. Mol. Neurobiol. 2016, 53, 6459–6475. [Google Scholar] [CrossRef] [PubMed]
- Stiles, A.R.; Simon, M.T.; Stover, A.; Eftekharian, S.; Khanlou, N.; Wang, H.L.; Magaki, S.; Lee, H.; Partynski, K.; Dorrani, N.; et al. Mutations in TFAM, Encoding Mitochondrial Transcription Factor A, Cause Neonatal Liver Failure Associated with MtDNA Depletion. Mol. Genet. Metab. 2016, 119, 91–99. [Google Scholar] [CrossRef]
- Simon, M.T.; Eftekharian, S.S.; Stover, A.E.; Osborne, A.F.; Braffman, B.H.; Chang, R.C.; Wang, R.Y.; Steenari, M.R.; Tang, S.; Hwu, P.W.-L.; et al. Novel Mutations in the Mitochondrial Complex I Assembly Gene NDUFAF5 Reveal Heterogeneous Phenotypes. Mol. Genet. Metab. 2019, 126, 53–63. [Google Scholar] [CrossRef]
- Latini, A.; Larovere, L.; De Kremer, D.R. Purines, Lactate and Myo-Inositol in CSF Might Reflect Excitotoxicity in Inherited Metabolic Disorders. In Purine and Pyrimidine Metabolism in Man X; Springer: Berlin/Heidelberg, Germany, 2000; Volume 486. [Google Scholar]
- Wajner, M.; Latini, A.; Wyse, A.T.S.; Dutra-Filho, C.S. The Role of Oxidative Damage in the Neuropathology of Organic Acidurias: Insights from Animal Studies. J. Inherit. Metab. Dis. 2004, 27, 427–448. [Google Scholar] [CrossRef] [PubMed]
- De Paula Martins, R.; Glaser, V.; Aguiar Jr, A.S.; de Paula Ferreira, P.M.; Ghisoni, K.; da Luz Scheffer, D.; Lanfumey, L.; Raisman-Vozari, R.; Corti, O.; De Paul, A.L.; et al. De Novo Tetrahydrobiopterin Biosynthesis Is Impaired in the Inflammed Striatum of Parkin (−/−) Mice. Cell Biol. Int. 2018, 42, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Muntau, A.C.; Burlina, A.; Eyskens, F.; Freisinger, P.; Leuzzi, V.; Sivri, H.S.; Gramer, G.; Pazdírková, R.; Cleary, M.; Lotz-Havla, A.S.; et al. Long-Term Efficacy and Safety of Sapropterin in Patients Who Initiated Sapropterin at < 4 Years of Age with Phenylketonuria: Results of the 3-Year Extension of the SPARK Open-Label, Multicentre, Randomised Phase IIIb Trial. Orphanet J. Rare Dis. 2021, 16, 341. [Google Scholar] [CrossRef] [PubMed]




| CSF | Urine | ||||
|---|---|---|---|---|---|
| Age | BH4 (nmol/L) | Biopterin (nmol/L) | Neopterin (nmol/L) | Biopterin (mmol/mol creatinine) | Neopterin (mmol/mol creatinine) |
| Newborns | 25–121 | 20–70 | 15–35 | 0.5–3.0 | 1.1–4.0 |
| 0–1 year | 24–59 | 15–40 | 12–30 | 0.5–3.0 | 1.1–4.0 |
| 2–4 year | 20–61 | 10–30 | 9–20 | 0.5–3.0 | 1.1–4.0 |
| 5–10 years | 20–49 | 10–30 | 9–20 | 0.5–3.0 | 1.1–4.0 |
| 11–16 years | 20–49 | 10–30 | 9–20 | 0.5–2.7 | 0.2–1.7 |
| Serum | Dried blood spot | ||||
| Biopterin (nmol/L) | Neopterin (nmol/L) | Biopterin (nmol/L Hb) | Neopterin (nmol/g Hb) | ||
| All ages | 4–18 | 3–11 | 0.08–1.20 | 0.19–2.93 | |
| Neopterin Levels | |
|---|---|
| Age | CSF (nmol/L) |
| 19–75 | 4.2 ± 1.0 |
| Age | Urine (μmol/mol creatinine) |
| 19–25 | 125 ± 32 |
| 26–35 | 112 ± 33 |
| 36–45 | 124 ± 33 |
| 46–55 | 126 ± 34 |
| 56–65 | 137 ± 37 |
| >65 | 142 ± 39 |
| Age | Serum (nmol/L) |
| 19–75 | 5.3 ± 2.7 |
| >75 | 9.7 ± 5.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eichwald, T.; da Silva, L.d.B.; Staats Pires, A.C.; Niero, L.; Schnorrenberger, E.; Filho, C.C.; Espíndola, G.; Huang, W.-L.; Guillemin, G.J.; Abdenur, J.E.; et al. Tetrahydrobiopterin: Beyond Its Traditional Role as a Cofactor. Antioxidants 2023, 12, 1037. https://doi.org/10.3390/antiox12051037
Eichwald T, da Silva LdB, Staats Pires AC, Niero L, Schnorrenberger E, Filho CC, Espíndola G, Huang W-L, Guillemin GJ, Abdenur JE, et al. Tetrahydrobiopterin: Beyond Its Traditional Role as a Cofactor. Antioxidants. 2023; 12(5):1037. https://doi.org/10.3390/antiox12051037
Chicago/Turabian StyleEichwald, Tuany, Lucila de Bortoli da Silva, Ananda Christina Staats Pires, Laís Niero, Erick Schnorrenberger, Clovis Colpani Filho, Gisele Espíndola, Wei-Lin Huang, Gilles J. Guillemin, José E. Abdenur, and et al. 2023. "Tetrahydrobiopterin: Beyond Its Traditional Role as a Cofactor" Antioxidants 12, no. 5: 1037. https://doi.org/10.3390/antiox12051037