

EGCG Attenuates CA1 Neuronal Death by Regulating GPx1, NF-κB S536 Phosphorylation and Mitochondrial Dynamics in the Rat Hippocampus following Status Epilepticus
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Chemicals
2.2. Surgical Procedures and SE Induction
2.3. Western Blot
2.4. Immunohistochemistry and Mitochondrial Morphometry
2.5. Fluoro-Jade B (FJB) Staining
2.6. Data Analysis
3. Results
3.1. EGCG Attenuates SE-Induced CA1 Neuronal Death
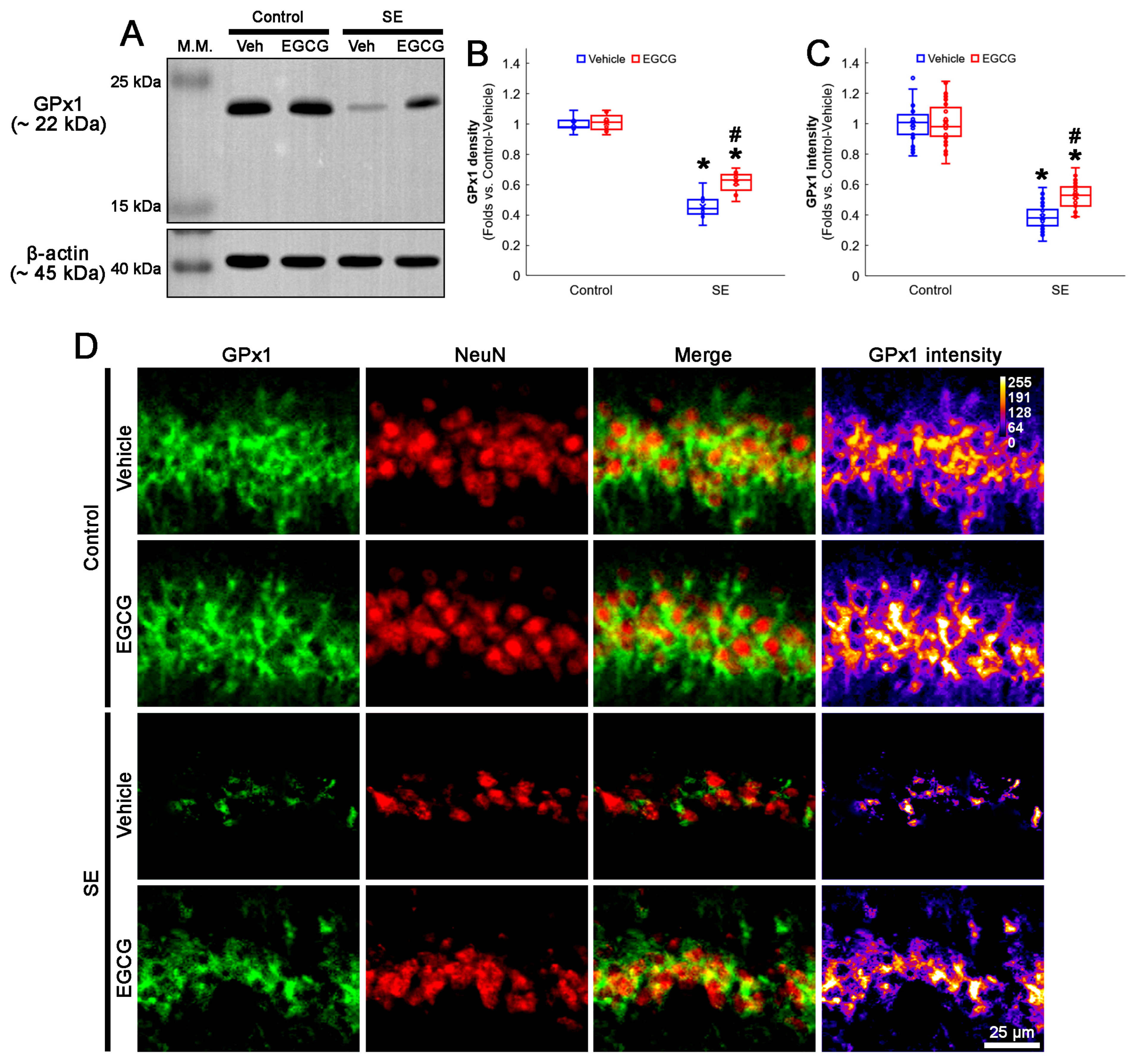
3.2. EGCG Diminishes SE-Induced GPx1 Downregulation in CA1 Neurons
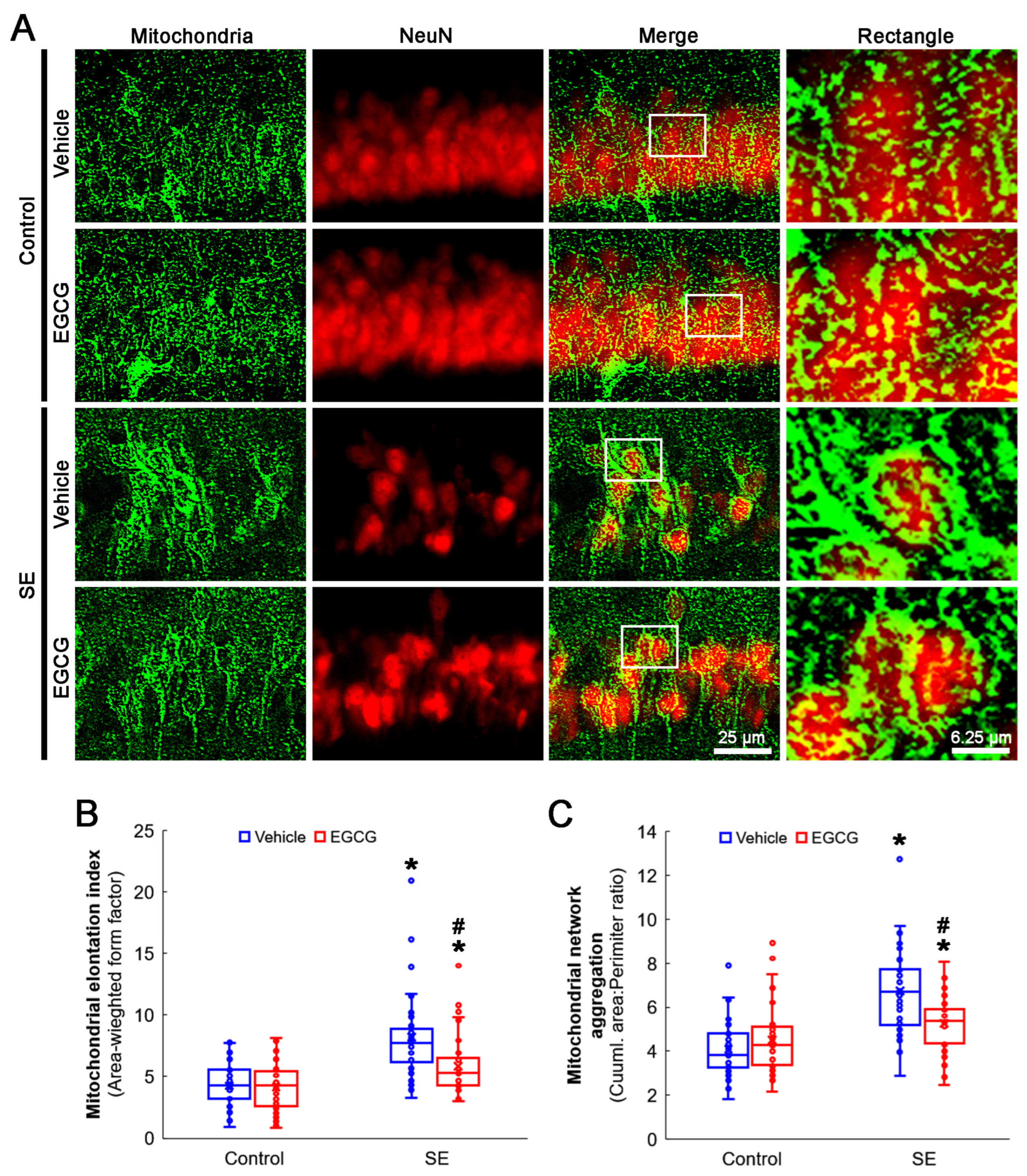
3.3. EGCG Ameliorates SE-Induced Aberrant Mitochondrial Elongation in CA1 Neurons
3.4. EGCG Attenuates a Decrease in DRP1 Expression and Its S616 Phosphorylation in CA1 Neurons
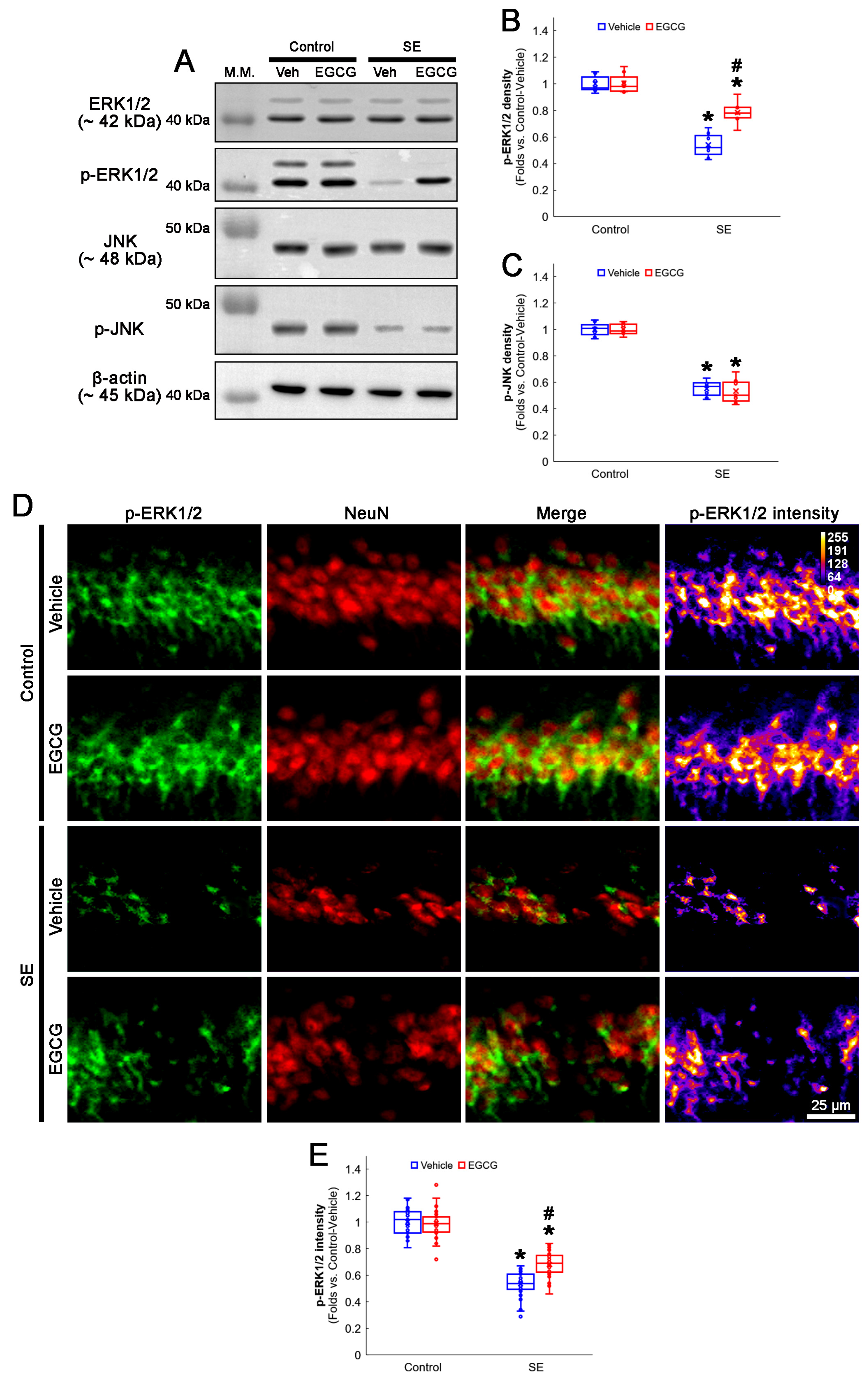
3.5. EGCG Enhances ERK1/2 but Not JNK Phosphorylation in CA1 Neurons following SE
3.6. EGCG Ameliorates NF-κB S536 Phosphorylation in CA1 Neurons following SE
3.7. ERK1/2 Inhibition Abrogates the Effect of EGCG on SE-Induced CA1 Neuronal Degeneration
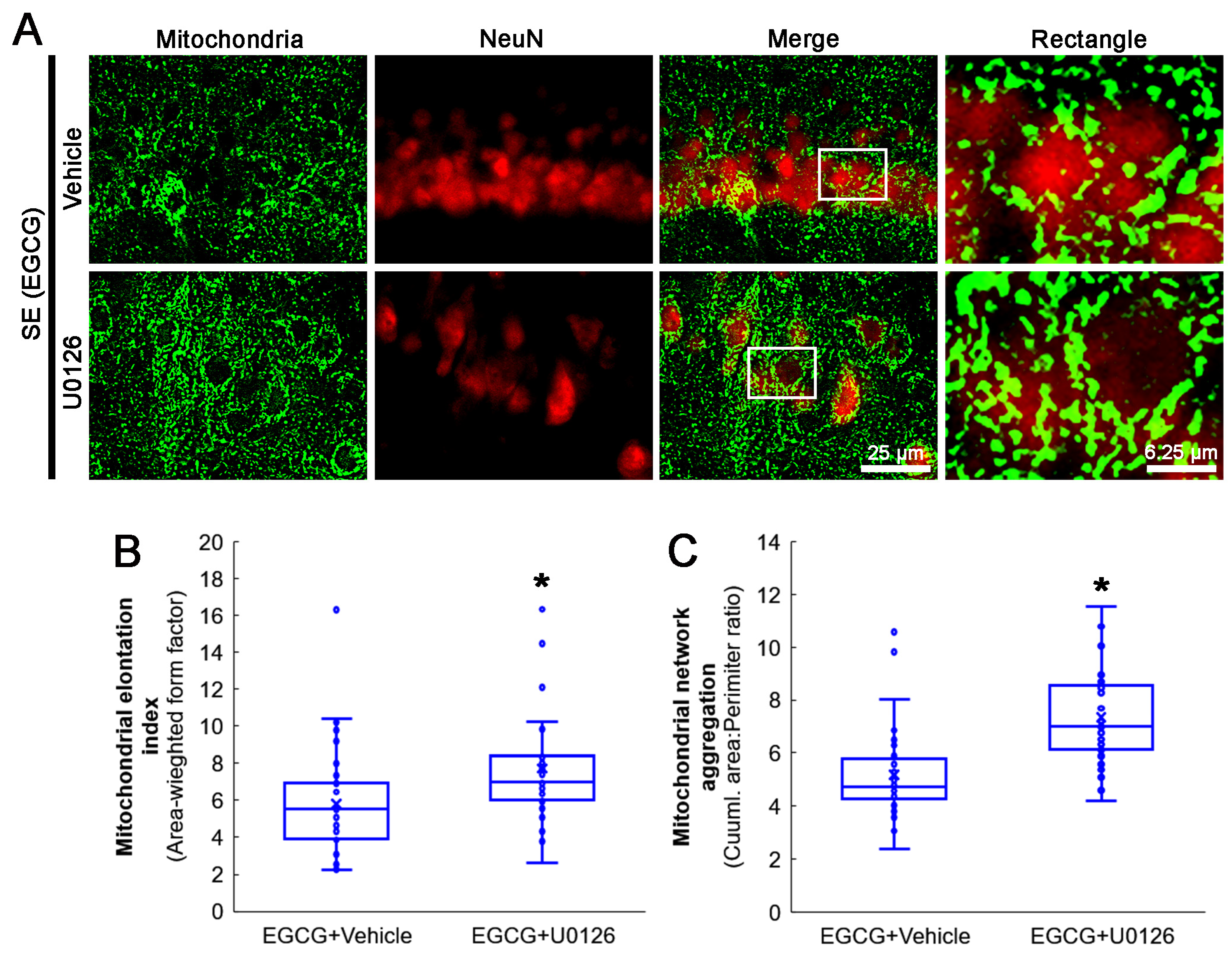
3.8. ERK1/2 Inhibition Abrogates the Effect of EGCG on Mitochondrial Hyperfusion
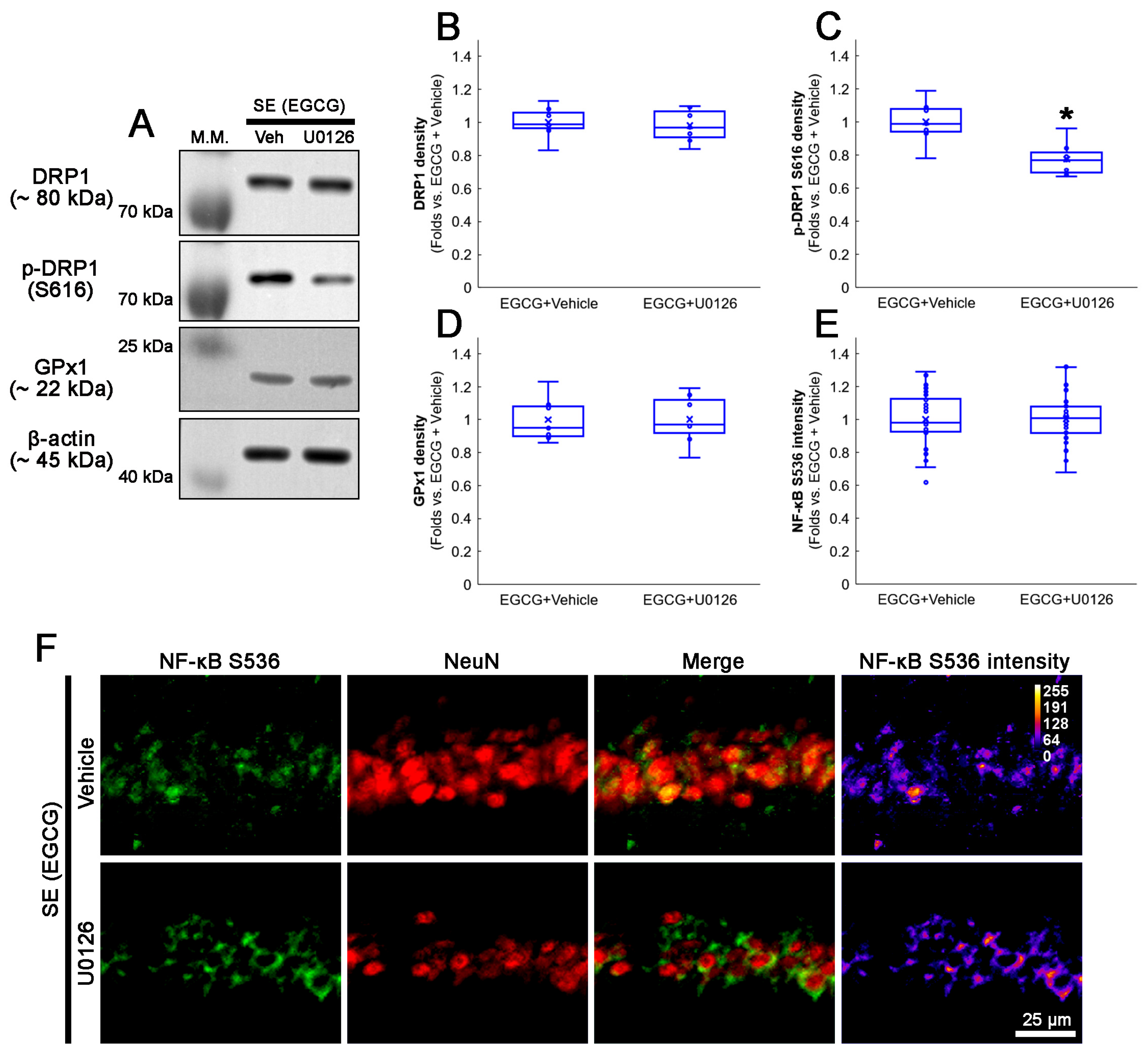
3.9. U0126 Co-Treatment Inhibits EGCG-Induced DRP1 S616 Phosphorylation without Affecting GPx1 Induction and NF-κB S536 Phosphorylation following SE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus--Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Neligan, A.; Noyce, A.J.; Gosavi, T.D.; Shorvon, S.D.; Köhler, S.; Walker, M.C. Change in Mortality of Generalized Convulsive Status Epilepticus in High-Income Countries Over Time: A Systematic Review and Meta-analysis. JAMA Neurol. 2019, 76, 897–905. [Google Scholar] [CrossRef]
- Kim, J.E.; Ryu, H.J.; Kim, M.J.; Kang, T.C. LIM kinase-2 induces programmed necrotic neuronal death via dysfunction of DRP1-mediated mitochondrial fission. Cell Death Differ. 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, D.G. Programmed mechanisms of status epilepticus-induced neuronal necrosis. Epilepsia Open 2022. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef]
- Campello, S.; Scorrano, L. Mitochondrial shape changes: Orchestrating cell pathophysiology. EMBO Rep. 2010, 11, 678–684. [Google Scholar] [CrossRef]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef]
- Kang, T.C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Fahrner, J.A.; Liu, R.; Perry, M.S.; Klein, J.; Chan, D.C. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet. A 2016, 170, 2002–2011. [Google Scholar] [CrossRef]
- Schmid, S.J.; Wagner, M.; Goetz, C.; Makowski, C.; Freisinger, P.; Berweck, S.; Mall, V.; Burdach, S.; Juenger, H. A De Novo Dominant Negative Mutation in DNM1L Causes Sudden Onset Status Epilepticus with Subsequent Epileptic Encephalopathy. Neuropediatrics 2019, 50, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Vanstone, J.R.; Smith, A.M.; McBride, S.; Naas, T.; Holcik, M.; Antoun, G.; Harper, M.E.; Michaud, J.; Sell, E.; Chakraborty, P.; et al. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur. J. Hum. Genet. 2016, 24, 1084–1088. [Google Scholar] [CrossRef]
- Shahni, R.; Cale, C.M.; Anderson, G.; Osellame, L.D.; Hambleton, S.; Jacques, T.S.; Wedatilake, Y.; Taanman, J.W.; Chan, E.; Qasim, W.; et al. Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission. Brain 2015, 138, 2834–2846. [Google Scholar] [CrossRef] [PubMed]
- Córdova-Dávalos, L.; Carrera-Calvo, D.; Solís-Navarrete, J.; Mercado-Gómez, O.F.; Arriaga-Ávila, V.; Agredano-Moreno, L.T.; Jiménez-García, L.F.; Guevara-Guzmán, R. Status epilepticus triggers early mitochondrial fusion in the rat hippocampus in a lithium-pilocarpine model. Epilepsy Res. 2016, 123, 11–19. [Google Scholar] [CrossRef]
- Lee, D.S.; Kim, J.E. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. 2018, 9, 869. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. CDDO-Me Selectively Attenuates CA1 Neuronal Death Induced by Status Epilepticus via Facilitating Mitochondrial Fission Independent of LONP1. Cells 2019, 8, 833. [Google Scholar] [CrossRef]
- Kim, J.E.; Kang, T.C. p47Phox/CDK5/DRP1-Mediated Mitochondrial Fission Evokes PV Cell Degeneration in the Rat Dentate Gyrus Following Status Epilepticus. Front. Cell. Neurosci. 2017, 11, 267. [Google Scholar] [CrossRef]
- Gundimeda, U.; McNeill, T.H.; Fan, T.K.; Deng, R.; Rayudu, D.; Chen, Z.; Cadenas, E.; Gopalakrishna, R. Green tea catechins potentiate the neuritogenic action of brain-derived neurotrophic factor: Role of 67-kDa laminin receptor and hydrogen peroxide. Biochem. Biophys. Res. Commun. 2014, 445, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, H.; Jeong, M.J.; Kang, T.C. Epigallocatechin-3-Gallate and PEDF 335 Peptide, 67LR Activators, Attenuate Vasogenic Edema, and Astroglial Degeneration Following Status Epilepticus. Antioxidants 2020, 9, 854. [Google Scholar] [CrossRef] [PubMed]
- Nanjo, F.; Mori, M.; Goto, K.; Hara, Y. Radical scavenging activity of tea catechins and their related compounds. Biosci. Biotechnol. Biochem. 1999, 63, 1621–1623. [Google Scholar] [CrossRef]
- Sarkar, J.; Chakraborti, T.; Chowdhury, A.; Bhuyan, R.; Chakraborti, S. Protective role of epigallocatechin-3-gallate in NADPH oxidase-MMP2-Spm-Cer-S1P signalling axis mediated ET-1 induced pulmonary artery smooth muscle cell proliferation. J. Cell Commun. Signal. 2019, 13, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Cos, P.; Ying, L.; Calomme, M.; Hu, J.P.; Cimanga, K.; van Poel, B.; Pieters, L.; Vlietinck, A.J.; Vanden Berghe, D. Structure-activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J. Nat. Prod. 1998, 61, 71–76. [Google Scholar] [CrossRef]
- Yousefian, M.; Shakour, N.; Hosseinzadeh, H.; Hayes, A.W.; Hadizadeh, F.; Karimi, G. The natural phenolic compounds as modulators of NADPH oxidases in hypertension. Phytomedicine 2019, 55, 200–213. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.; Sun, X.; Shi, X.; Wang, L.; Huang, L.; Zhou, W. Evaluation of the neuroprotective effect of EGCG: A potential mechanism of mitochondrial dysfunction and mitochondrial dynamics after subarachnoid hemorrhage. Food Funct. 2018, 9, 6349–6359. [Google Scholar] [CrossRef]
- Qu, Z.; Jia, L.; Xie, T.; Zhen, J.; Si, P.; Cui, Z.; Xue, Y.; Sun, C.; Wang, W. (-)-Epigallocatechin-3-Gallate Protects Against Lithium-Pilocarpine-Induced Epilepsy by Inhibiting the Toll-Like Receptor 4 (TLR4)/Nuclear Factor-κB (NF-κB) Signaling Pathway. Med. Sci. Monit. 2019, 25, 1749–1758. [Google Scholar] [CrossRef]
- Machova Urdzikova, L.; Ruzicka, J.; Karova, K.; Kloudova, A.; Svobodova, B.; Amin, A.; Dubisova, J.; Schmidt, M.; Kubinova, S.; Jhanwar-Uniyal, M.; et al. A green tea polyphenol epigallocatechin-3-gallate enhances neuroregeneration after spinal cord injury by altering levels of inflammatory cytokines. Neuropharmacology 2017, 126, 213–223. [Google Scholar] [CrossRef]
- Gundimeda, U.; McNeill, T.H.; Elhiani, A.A.; Schiffman, J.E.; Hinton, D.R.; Gopalakrishna, R. Green tea polyphenols precondition against cell death induced by oxygen-glucose deprivation via stimulation of laminin receptor, generation of reactive oxygen species, and activation of protein kinase Cε. J. Biol. Chem. 2012, 287, 34694–34708. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Antunes, F.; Pinto, R.E. Role of glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase in the reduction of lysophospholipid hydroperoxides. Free Radic. Biol. Med. 1997, 22, 871–883. [Google Scholar] [CrossRef]
- de Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O’Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J. Biol. Chem. 1998, 273, 22528–22536. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Lee, D.S.; Kim, T.H.; Kang, T.C. Glutathione Regulates GPx1 Expression during CA1 Neuronal Death and Clasmatodendrosis in the Rat Hippocampus following Status Epilepticus. Antioxidants 2022, 11, 756. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Park, J.Y.; Lambert, J.D. Differential prooxidative effects of the green tea polyphenol, (-)-epigallocatechin-3-gallate, in normal and oral cancer cells are related to differences in sirtuin 3 signaling. Mol. Nutr. Food Res. 2015, 59, 203–211. [Google Scholar] [CrossRef]
- Ayyalasomayajula, N.; Bandaru, L.J.M.; Chetty, C.S.; Dixit, P.K.; Challa, S. Mitochondria-Mediated Moderation of Apoptosis by EGCG in Cytotoxic Neuronal Cells Induced by Lead (Pb) and Amyloid Peptides. Biol. Trace Elem. Res. 2022, 200, 3582–3593. [Google Scholar] [CrossRef]
- Aras-López, R.; Almeida, L.; Andreu-Fernández, V.; Tovar, J.; Martínez, L. Anti-oxidants correct disturbance of redox enzymes in the hearts of rat fetuses with congenital diaphragmatic hernia. Pediatr. Surg. Int. 2018, 34, 307–313. [Google Scholar] [CrossRef]
- Akhtar, M.W.; Sunico, C.R.; Nakamura, T.; Lipton, S.A. Redox Regulation of Protein Function via Cysteine S-Nitrosylation and Its Relevance to Neurodegenerative Diseases. Int. J. Cell Biol. 2012, 2012, 463756. [Google Scholar] [CrossRef]
- Haun, F.; Nakamura, T.; Lipton, S.A. Dysfunctional Mitochondrial Dynamics in the Pathophysiology of Neurodegenerative Diseases. J. Cell Death 2013, 6, 27–35. [Google Scholar] [CrossRef]
- Wu, M.; Gu, X.; Ma, Z. Mitochondrial Quality Control in Cerebral Ischemia-Reperfusion Injury. Mol. Neurobiol. 2021, 58, 5253–5271. [Google Scholar] [CrossRef]
- Yan, J.; Feng, Z.; Liu, J.; Shen, W.; Wang, Y.; Wertz, K.; Weber, P.; Long, J.; Liu, J. Enhanced autophagy plays a cardinal role in mitochondrial dysfunction in type 2 diabetic Goto-Kakizaki (GK) rats: Ameliorating effects of (-)-epigallocatechin-3-gallate. J. Nutr. Biochem. 2012, 23, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Casanova, E.; Baselga-Escudero, L.; Ribas-Latre, A.; Arola-Arnal, A.; Bladé, C.; Arola, L.; Salvadó, M.J. Epigallocatechin gallate counteracts oxidative stress in docosahexaenoxic acid-treated myocytes. Biochim. Biophys. Acta 2014, 1837, 783–791. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 3rd ed.; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Weise, J.; Engelhorn, T.; Dörfler, A.; Aker, S.; Bähr, M.; Hufnagel, A. Expression time course and spatial distribution of activated caspase-3 after experimental status epilepticus: Contribution of delayed neuronal cell death to seizure-induced neuronal injury. Neurobiol. Dis. 2005, 18, 582–590. [Google Scholar] [CrossRef]
- Scholl, E.A.; Dudek, F.E.; Ekstrand, J.J. Neuronal degeneration is observed in multiple regions outside the hippocampus after lithium pilocarpine-induced status epilepticus in the immature rat. Neuroscience 2013, 252, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, J.J.; Pouliot, W.; Scheerlinck, P.; Dudek, F.E. Lithium pilocarpine-induced status epilepticus in postnatal day 20 rats results in greater neuronal injury in ventral versus dorsal hippocampus. Neuroscience 2011, 192, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol. 2009, 457, 231–253. [Google Scholar]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef]
- Schroeder, E.K.; Kelsey, N.A.; Doyle, J.; Breed, E.; Bouchard, R.J.; Loucks, F.A.; Harbison, R.A.; Linseman, D.A. Green tea epigallocatechin 3-gallate accumulates in mitochondria and displays a selective antiapoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antioxid. Redox Signal. 2009, 11, 469–480. [Google Scholar] [CrossRef]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-García, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, X.; Ni, C.; Dai, Y.; Guo, Y. Zearalenone regulates endometrial stromal cell apoptosis and migration via the promotion of mitochondrial fission by activation of the JNK/Drp1 pathway. Mol. Med. Rep. 2018, 17, 7797–7806. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kim, M.J.; Jo, S.M.; Kang, T.C. ReLA/P65-serine 536 nuclear factor-kappa B phosphorylation is related to vulnerability to status epilepticus in the rat hippocampus. Neuroscience 2011, 187, 93–102. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, D.S.; Ryu, H.J.; Kim, W.I.; Kim, M.J.; Kim, D.W.; Choi, S.Y.; Kang, T.C. The effect of P2X7 receptor activation on nuclear factor-κB phosphorylation induced by status epilepticus in the rat hippocampus. Hippocampus 2013, 23, 500–514. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, W.; Shao, Q.; Yang, Y.; Xu, Z.; Chen, J.; Zhang, X.; Ge, X. Drp1, a potential therapeutic target for Parkinson’s disease, is involved in olfactory bulb pathological alteration in the Rotenone-induced rat model. Toxicol. Lett. 2020, 325, 1–13. [Google Scholar] [CrossRef]
- Battaglia, C.R.; Cursano, S.; Calzia, E.; Catanese, A.; Boeckers, T.M. Corticotropin-releasing hormone (CRH) alters mitochondrial morphology and function by activating the NF-kB-DRP1 axis in hippocampal neurons. Cell Death Dis. 2020, 11, 1004. [Google Scholar] [CrossRef]
- Crack, P.J.; Taylor, J.M.; Ali, U.; Mansell, A.; Hertzog, P.J. Potential contribution of NF-kappaB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke 2006, 37, 1533–1538. [Google Scholar] [CrossRef]
- Wang, X.; Han, Y.; Chen, F.; Wang, M.; Xiao, Y.; Wang, H.; Xu, L.; Liu, W. Glutathione Peroxidase 1 Protects Against Peroxynitrite-Induced Spiral Ganglion Neuron Damage Through Attenuating NF-κB Pathway Activation. Front. Cell. Neurosci. 2022, 16, 841731. [Google Scholar] [CrossRef]
- Levites, Y.; Amit, T.; Youdim, M.B.; Mandel, S. Involvement of protein kinase C activation and cell survival/ cell cycle genes in green tea polyphenol (-)-epigallocatechin 3-gallate neuroprotective action. J. Biol. Chem. 2002, 277, 30574–30580. [Google Scholar] [CrossRef]
- Liu, M.; Chen, F.; Sha, L.; Wang, S.; Tao, L.; Yao, L.; He, M.; Yao, Z.; Liu, H.; Zhu, Z.; et al. (-)-Epigallocatechin-3-gallate ameliorates learning and memory deficits by adjusting the balance of TrkA/p75NTR signaling in APP/PS1 transgenic mice. Mol. Neurobiol. 2014, 49, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, F.; Jin, H.; Li, R.; Wang, Y.; Zhang, W.; Wang, H.; Chen, W. Involvement of PKCα and ERK1/2 signaling pathways in EGCG’s protection against stress-induced neural injuries in Wistar rats. Neuroscience 2017, 346, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, R.; Jin, H.; Jin, H.; Wang, Y.; Zhang, W.; Wang, H.; Chen, W. Epigallocatechin-3-gallate confers protection against corticosterone-induced neuron injuries via restoring extracellular signal-regulated kinase 1/2 and phosphatidylinositol-3 kinase/protein kinase B signaling pathways. PLoS ONE 2018, 13, e0192083. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.V.; Shin, E.J.; Nguyen, L.T.T.; Lee, Y.; Kim, D.J.; Jeong, J.H.; Jang, C.G.; Nah, S.Y.; Toriumi, K.; Nabeshima, T.; et al. Protein Kinase Cδ Gene Depletion Protects Against Methamphetamine-Induced Impairments in Recognition Memory and ERK1/2 Signaling via Upregulation of Glutathione Peroxidase-1 Gene. Mol. Neurobiol. 2018, 55, 4136–4159. [Google Scholar] [CrossRef] [PubMed]
- Mai, H.N.; Pham, D.T.; Chung, Y.H.; Sharma, N.; Cheong, J.H.; Yun, J.; Nah, S.Y.; Jeong, J.H.; Gen Lei, X.; Shin, E.J.; et al. Glutathione peroxidase-1 knockout potentiates behavioral sensitization induced by cocaine in mice via σ-1 receptor-mediated ERK signaling: A comparison with the case of glutathione peroxidase-1 overexpressing transgenic mice. Brain Res. Bull. 2020, 164, 107–120. [Google Scholar] [CrossRef]
- Shin, E.J.; Chung, Y.H.; Sharma, N.; Nguyen, B.T.; Lee, S.H.; Kang, S.W.; Nah, S.Y.; Wie, M.B.; Nabeshima, T.; Jeong, J.H.; et al. Glutathione Peroxidase-1 Knockout Facilitates Memory Impairment Induced by β-Amyloid (1-42) in Mice via Inhibition of PKC βII-Mediated ERK Signaling; Application with Glutathione Peroxidase-1 Gene-Encoded Adenovirus Vector. Neurochem. Res. 2020, 45, 2991–3002. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, B.; Chen, H.; Qin, Y.; Cheng, J.; He, B.; Wan, Y.; Zhu, D.; Gao, F. Epigallocatechin-3-Gallate Ameliorated Iron Accumulation and Apoptosis and Promoted Neuronal Regeneration and Memory/Cognitive Functions in the Hippocampus Induced by Exposure to a Chronic High-Altitude Hypoxia Environment. Neurochem. Res. 2022, 47, 2254–2262. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Smith, A.; Lin, X.; Yuan, F.; Copes, N.; Delic, V.; Tan, J.; Cao, C.; Shytle, R.D.; Bradshaw, P.C. Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 2011, 26, 507–521. [Google Scholar] [CrossRef]
- Lee, J.H.; Moon, J.H.; Kim, S.W.; Jeong, J.K.; Nazim, U.M.; Lee, Y.J.; Seol, J.W.; Park, S.Y. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget 2015, 6, 9701–9717. [Google Scholar] [CrossRef]
- Castellano-González, G.; Pichaud, N.; Ballard, J.W.; Bessede, A.; Marcal, H.; Guillemin, G.J. Epigallocatechin-3-gallate induces oxidative phosphorylation by activating cytochrome c oxidase in human cultured neurons and astrocytes. Oncotarget 2016, 7, 7426–7440. [Google Scholar] [CrossRef]
- Kamalden, T.A.; Ji, D.; Osborne, N.N. Rotenone-induced death of RGC-5 cells is caspase independent, involves the JNK and p38 pathways and is attenuated by specific green tea flavonoids. Neurochem. Res. 2012, 37, 1091–1101. [Google Scholar] [CrossRef]
- He, Q.; Bao, L.; Zimering, J.; Zan, K.; Zhang, Z.; Shi, H.; Zu, J.; Yang, X.; Hua, F.; Ye, X.; et al. The protective role of (-)-epigallocatechin-3-gallate in thrombin-induced neuronal cell apoptosis and JNK-MAPK activation. Neuroreport 2015, 26, 416–423. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Madrid, L.V.; Mayo, M.W.; Reuther, J.Y.; Baldwin, A.S., Jr. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 2001, 276, 18934–18940. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.Y.; Neeman, I.; Resnick, N. A novel mechanism for the inhibition of NF-kappaB activation in vascular endothelial cells by natural antioxidants. FASEB J. 2002, 16, 1931–1933. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kang, T.C. The Regional Specific Alterations in BBB Permeability are Relevant to the Differential Responses of 67-kDa LR Expression in Endothelial Cells and Astrocytes Following Status Epilepticus. Int. J. Mol. Sci. 2019, 20, 6025. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, C.; Vinet, J.; Meletti, S.; Biagini, G. Ischemic-hypoxic mechanisms leading to hippocampal dysfunction as a consequence of status epilepticus. Epilepsy Behav. 2015, 49, 47–54. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Raghunayakula, S.; Kumar, R. Neuronal hypoxia disrupts mitochondrial fusion. Neuroscience 2015, 301, 71–78. [Google Scholar] [CrossRef]
- Do Val-da Silva, R.A.; Peixoto-Santos, J.E.; Scandiuzzi, R.C.; Balista, P.A.; Bassi, M.; Glass, M.L.; Romcy-Pereira, R.N.; Galvis-Alonso, O.Y.; Leite, J.P. Decreased neuron loss and memory dysfunction in pilocarpine-treated rats pre-exposed to hypoxia. Neuroscience 2016, 332, 88–100. [Google Scholar] [CrossRef]
- Wei, B.B.; Liu, M.Y.; Zhong, X.; Yao, W.F.; Wei, M.J. Increased BBB permeability contributes to EGCG-caused cognitive function improvement in natural aging rats: Pharmacokinetic and distribution analyses. Acta Pharmacol. Sin. 2019, 40, 1490–1500. [Google Scholar] [CrossRef]
- Unno, K.; Pervin, M.; Nakagawa, A.; Iguchi, K.; Hara, A.; Takagaki, A.; Nanjo, F.; Minami, A.; Nakamura, Y. Blood-Brain Barrier Permeability of Green Tea Catechin Metabolites and their Neuritogenic Activity in Human Neuroblastoma SH-SY5Y Cells. Mol. Nutr. Food Res. 2017, 61, 1700294. [Google Scholar] [CrossRef]
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F.; et al. Blood brain barrier permeability of (-)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem. Biophys. Rep. 2017, 9, 180–186. [Google Scholar] [CrossRef]
- Harland, M.; Torres, S.; Liu, J.; Wang, X. Neuronal Mitochondria Modulation of LPS-Induced Neuroinflammation. J. Neurosci. 2020, 40, 1756–1765. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Hose | Manufacturer (Catalog Number) | Dilution |
|---|---|---|---|
| DRP1 | Rabbit | Thermo (PA1-16987) | 1:500 (IH) 1:1000 (WB) |
| ERK1/2 | Rabbit | Biorbyt (Orb160960) | 1:2000 (WB) |
| GPx1 | Sheep | Biosensis (S-072-100) | 1:2000 (IH) 1:10,000 (WB) |
| JNK | Rabbit | Proteintech (10023-1-AP) | 1:1000 (WB) |
| Mitochondrial marker (Mitochondrial complex IV subunit 1, MTCO1) | Mouse | Abcam (#ab14705) | 1:500 (IH) |
| NeuN | Guinea pig | Millipore (#ABN90P) | 1:1000 (IH) |
| NF-κB S536 | Rabbit | Abcam (#ab28856) | 1:100 (IH) |
| p-DRP1 S616 | Rabbit | Cell Signaling (#4494) | 1:1000 (WB) |
| p-ERK1/2 | Rabbit | Millipore (#05-797R) | 1:100 (IH) 1:1000 (WB) |
| p-JNK | Rabbit | Millipore (#07-175) | 1:1000 (WB) |
| β-actin | Mouse | Sigma (A5316) | 1:5000 (WB) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-E.; Kim, T.-H.; Kang, T.-C. EGCG Attenuates CA1 Neuronal Death by Regulating GPx1, NF-κB S536 Phosphorylation and Mitochondrial Dynamics in the Rat Hippocampus following Status Epilepticus. Antioxidants 2023, 12, 966. https://doi.org/10.3390/antiox12040966
Kim J-E, Kim T-H, Kang T-C. EGCG Attenuates CA1 Neuronal Death by Regulating GPx1, NF-κB S536 Phosphorylation and Mitochondrial Dynamics in the Rat Hippocampus following Status Epilepticus. Antioxidants. 2023; 12(4):966. https://doi.org/10.3390/antiox12040966
Chicago/Turabian StyleKim, Ji-Eun, Tae-Hyun Kim, and Tae-Cheon Kang. 2023. "EGCG Attenuates CA1 Neuronal Death by Regulating GPx1, NF-κB S536 Phosphorylation and Mitochondrial Dynamics in the Rat Hippocampus following Status Epilepticus" Antioxidants 12, no. 4: 966. https://doi.org/10.3390/antiox12040966