

Oxidative Stress and Neuroinflammation in Parkinson’s Disease: The Role of Dopamine Oxidation Products

{kind=link}
{kind=link}
Abstract
:1. Introduction
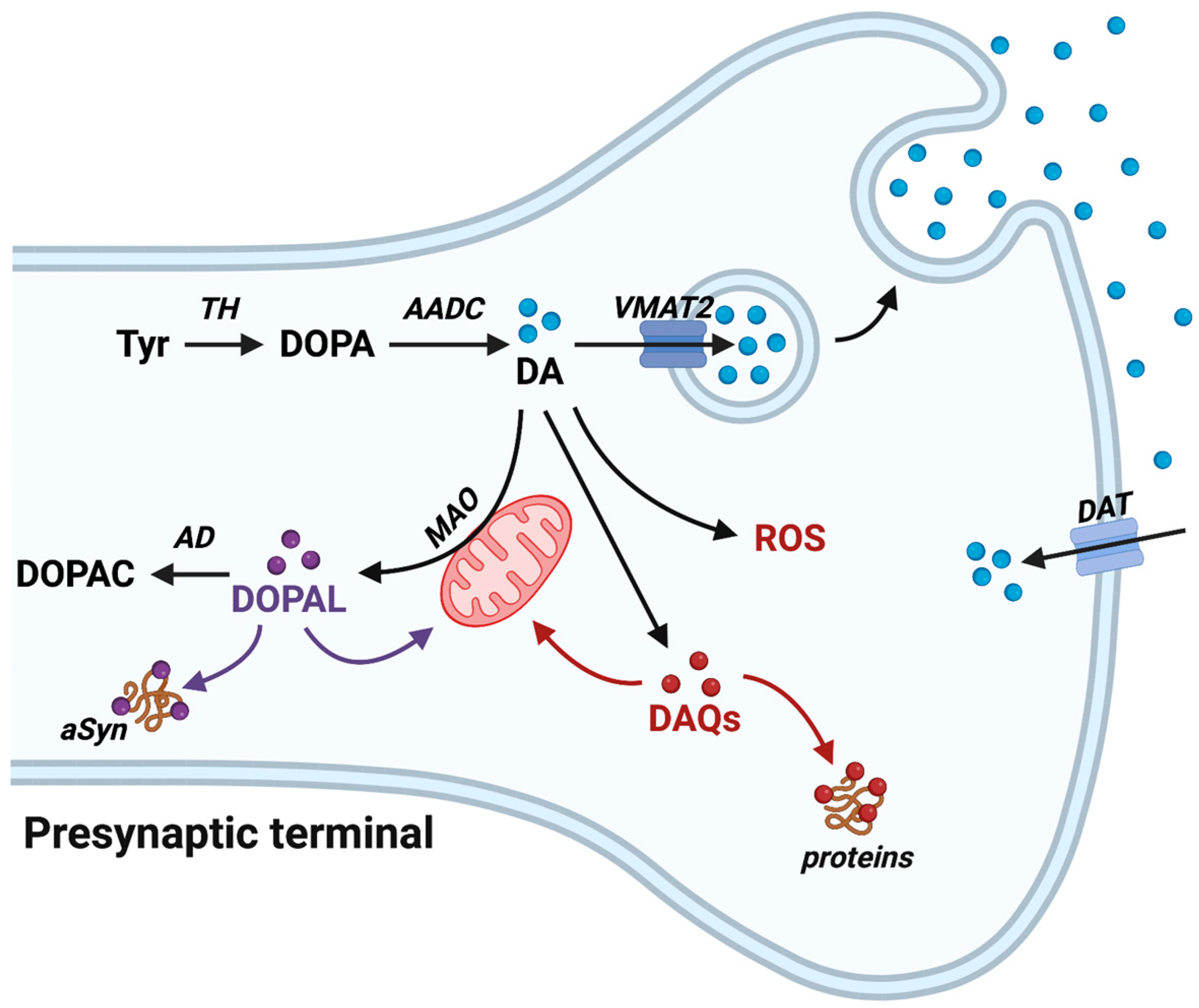
2. DA Metabolism and Oxidative Chemistry
3. DA-Related Toxicity
4. Protein Targets of DAQs
5. DAQs’ Reactivity toward Mitochondria
6. DOPAL Toxicity: The Catecholaldehyde Hypothesis
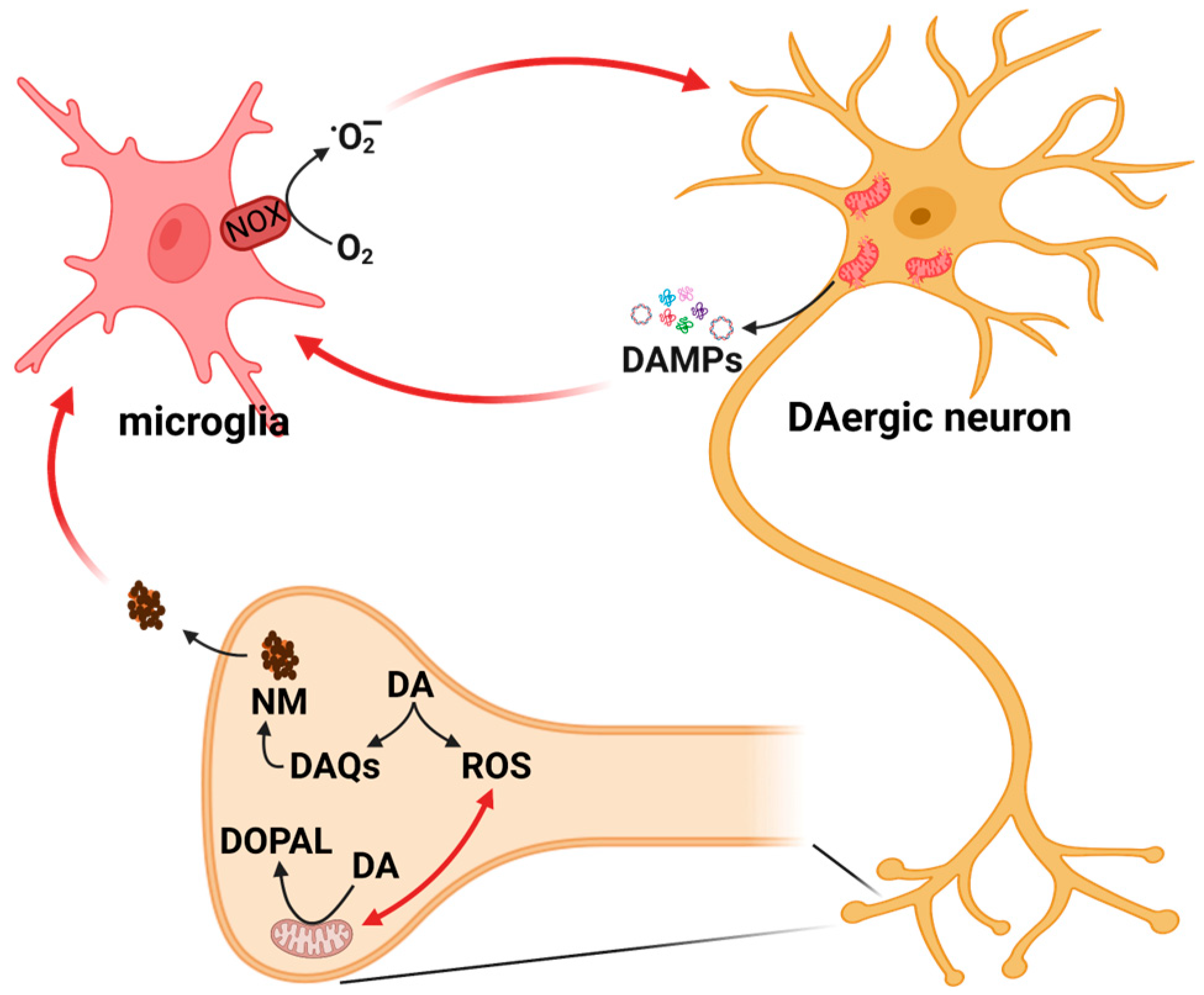
7. DA and Neuroinflammation: The Role of Neuromelanin
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fieblinger, T. Striatal Control of Movement: A Role for New Neuronal (Sub-) Populations? Front. Hum. Neurosci. 2021, 15, 697284. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Ross, O.A.; Puschmann, A.; Dickson, D.W.; Wszolek, Z.K. Autosomal Dominant Parkinson’s Disease Caused by SNCA Duplications. Park. Relat Disord 2016, 22 (Suppl. S1), S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Ogaki, K.; Tacik, P.M.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K. Update on Novel Familial Forms of Parkinson’s Disease and Multiple System Atrophy. Park. Relat. Disord. 2014, 20 (Suppl. S1), S29–S34. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Greenamyre, J.T. Gene-Environment Interactions in Parkinson’s Disease: Specific Evidence in Humans and Mammalian Models. Neurobiol. Dis. 2013, 57, 38–46. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- De Lazzari, F.; Bubacco, L.; Whitworth, A.J.; Bisaglia, M. Superoxide Radical Dismutation as New Therapeutic Strategy in Parkinson’s Disease. Aging Dis. 2018, 9, 716–728. [Google Scholar] [CrossRef]
- De Lazzari, F.; Sandrelli, F.; Whitworth, A.J.; Bisaglia, M. Antioxidant Therapy in Parkinson’s Disease: Insights from Drosophila Melanogaster. Antioxidants 2020, 9, 52. [Google Scholar] [CrossRef]
- Chang, K.H.; Chen, C.M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Zhuang, Q.Q.; Zhu, L.B.; Zhu, H.; Li, T.; Li, R.; Chen, S.F.; Huang, C.P.; Zhang, X.; Zhu, J.H. Meta-Analysis of Brain Iron Levels of Parkinson’s Disease Patients Determined by Postmortem and MRI Measurements. Sci. Rep. 2016, 6, 36669. [Google Scholar] [CrossRef]
- Pyatigorskaya, N.; Sanz-Morère, C.B.; Gaurav, R.; Biondetti, E.; Valabregue, R.; Santin, M.; Yahia-Cherif, L.; Lehéricy, S. Iron Imaging as a Diagnostic Tool for Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 00366. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D.; Nakamura, K. Understanding the Susceptibility of Dopamine Neurons to Mitochondrial Stressors in Parkinson’s Disease. FEBS Lett. 2015, 589, 3702–3713. [Google Scholar] [CrossRef]
- He, J.; Zhu, G.; Wang, G.; Zhang, F. Oxidative Stress and Neuroinflammation Potentiate Each Other to Promote Progression of Dopamine Neurodegeneration. Oxid. Med. Cell. Longev. 2020, 2020, 6137521. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Pinton, P. Mitochondria in the Line of Fire. Cell Death Differ. 2022, 29, 1301–1303. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Cristóvão, A.C.; Guhathakurta, S.; Lee, J.; Joh, T.H.; Beal, M.F.; Kim, Y.S. NADPH Oxidase 1-Mediated Oxidative Stress Leads to Dopamine Neuron Death in Parkinson’s Disease. Antioxid. Redox Signal. 2012, 16, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Teismann, P.; Tieu, K.; Vila, M.; Jackson-Lewis, V.; Ischiropoulos, H.; Przedborski, S. NADPH Oxidase Mediates Oxidative Stress in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Model of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 6145–6150. [Google Scholar] [CrossRef]
- Bisaglia, M. Mediterranean Diet and Parkinson’s Disease. Int. J. Mol. Sci. 2022, 24, 42. [Google Scholar] [CrossRef]
- Silva, D.F.; Empadinhas, N.; Cardoso, S.M.; Esteves, A.R. Neurodegenerative Microbially-Shaped Diseases: Oxidative Stress Meets Neuroinflammation. Antioxidants 2022, 11, 2141. [Google Scholar] [CrossRef]
- Bisaglia, M.; Greggio, E.; Beltramini, M.; Bubacco, L. Dysfunction of Dopamine Homeostasis: Clues in the Hunt for Novel Parkinson’s Disease Therapies. FASEB J. 2013, 27, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.H.; Sa, M.; Ju, Y.H.; Park, M.G.; Lee, C.J. Revisiting the Role of Astrocytic MAOB in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 4453. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Korkina, L.G.; Greenfield, S.A. Autoxidation of Dopamine: A Comparison of Luminescent and Spectrophotometric Detection in Basic Solutions. Free Radic. Biol. Med. 1995, 18, 215–222. [Google Scholar] [CrossRef]
- Wise, R.M.; Wagener, A.; Fietzek, U.M.; Klopstock, T.; Mosharov, E.V.; Zucca, F.A.; Sulzer, D.; Zecca, L.; Burbulla, L.F. Interactions of Dopamine, Iron, and Alpha-Synuclein Linked to Dopaminergic Neuron Vulnerability in Parkinson’s Disease and Neurodegeneration with Brain Iron Accumulation Disorders. Neurobiol. Dis. 2022, 175, 105920. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Capucciati, A.; Bellei, C.; Sarna, M.; Sarna, T.; Monzani, E.; Casella, L.; Zecca, L. Neuromelanins in Brain Aging and Parkinson’s Disease: Synthesis, Structure, Neuroinflammatory, and Neurodegenerative Role. IUBMB Life 2023, 75, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin Biosynthesis Is Driven by Excess Cytosolic Catecholamines Not Accumulated by Synaptic Vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef]
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of Oxidation in the Neurotoxic Effects of Intrastriatal Dopamine Injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961. [Google Scholar] [CrossRef]
- Bucher, M.L.; Barrett, C.W.; Moon, C.J.; Mortimer, A.D.; Burton, E.A.; Timothy Greenamyre, J.; Hastings, T.G. Acquired Dysregulation of Dopamine Homeostasis Reproduces Features of Parkinson’s Disease. NPJ Park. Dis. 2020, 6, 34. [Google Scholar] [CrossRef]
- Vecchio, L.M.; Sullivan, P.; Dunn, A.R.; Bermejo, M.K.; Fu, R.; Masoud, S.T.; Gregersen, E.; Urs, N.M.; Nazari, R.; Jensen, P.H.; et al. Enhanced Tyrosine Hydroxylase Activity Induces Oxidative Stress, Causes Accumulation of Autotoxic Catecholamine Metabolites, and Augments Amphetamine Effects in Vivo. J. Neurochem. 2021, 158, 960–979. [Google Scholar] [CrossRef]
- Spencer, J.P.E.; Jenner, P.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Halliwell, B. Conjugates of Catecholamines with Cysteine and GSH in Parkinson’s Disease: Possible Mechanisms of Formation Involving Reactive Oxygen Species. J. Neurochem. 1998, 71, 2112–2122. [Google Scholar] [CrossRef]
- Ganguly, U.; Ganguly, A.; Sen, O.; Ganguly, G.; Cappai, R.; Sahoo, A.; Chakrabarti, S. Dopamine Cytotoxicity on SH-SY5Y Cells: Involvement of α-Synuclein and Relevance in the Neurodegeneration of Sporadic Parkinson’s Disease. Neurotox. Res. 2019, 35, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Arduini, I.; Soriano, M.E.; Giorgio, V.; Bernardi, P.; Bisaglia, M.; Bubacco, L. Dopamine Oxidation Products as Mitochondrial Endotoxins, a Potential Molecular Mechanism for Preferential Neurodegeneration in Parkinson’s Disease. ACS Chem. Neurosci. 2018, 9, 2849–2858. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Munshi, S.; Sen, O.; Pramanik, V.; Roy Mukherjee, T.; Chakrabarti, S. Dopamine Cytotoxicity Involves Both Oxidative and Nonoxidative Pathways in SH-SY5Y Cells: Potential Role of Alpha-Synuclein Overexpression and Proteasomal Inhibition in the Etiopathogenesis of Parkinson’s Disease. Park. Dis. 2014, 2014, 878935. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Sinha, M.; Chanda, D.; Roy, T.; Banerjee, K.; Munshi, S.; Patro, B.S.; Chakrabarti, S. Mitochondrial Dysfunction Mediated by Quinone Oxidation Products of Dopamine: Implications in Dopamine Cytotoxicity and Pathogenesis of Parkinson’s Disease. Biochim. Biophys. Acta 2011, 1812, 663–673. [Google Scholar] [CrossRef]
- Bisaglia, M.; Greggio, E.; Maric, D.; Miller, D.W.; Cookson, M.R.; Bubacco, L. Alpha-Synuclein Overexpression Increases Dopamine Toxicity in BE2-M17 Cells. BMC Neurosci. 2010, 11, 41. [Google Scholar] [CrossRef]
- Giménez-Xavier, P.; Gómez-Santos, C.; Castaño, E.; Francisco, R.; Boada, J.; Unzeta, M.; Sanz, E.; Ambrosio, S. The Decrease of NAD(P)H Has a Prominent Role in Dopamine Toxicity. Biochim. Biophys. Acta 2006, 1762, 564–574. [Google Scholar] [CrossRef]
- Pedrosa, R.; Soares-da-Silva, P. Oxidative and Non-Oxidative Mechanisms of Neuronal Cell Death and Apoptosis by L-3,4-Dihydroxyphenylalanine (L-DOPA) and Dopamine. Br. J. Pharmacol. 2002, 137, 1305–1313. [Google Scholar] [CrossRef]
- Lai, C.T.; Yu, P.H. Dopamine- and L-β-3,4-Dihydroxyphenylalanine Hydrochloride (L-DOPA)-Induced Cytotoxicity towards Catecholaminergic Neuroblastoma SH-SY5Y Cells. Effects of Oxidative Stress and Antioxidative Factors. Biochem. Pharmacol. 1997, 53, 363–372. [Google Scholar] [CrossRef]
- Offen, D.; Ziv, I.; Sternin, H.; Melamed, E.; Hochman, A. Prevention of Dopamine-Induced Cell Death by Thiol Antioxidants: Possible Implications for Treatment of Parkinson’s Disease. Exp. Neurol. 1996, 141, 32–39. [Google Scholar] [CrossRef]
- Biosa, A.; De Lazzari, F.; Masato, A.; Filograna, R.; Plotegher, N.; Beltramini, M.; Bubacco, L.; Bisaglia, M. Superoxide Dismutases SOD1 and SOD2 Rescue the Toxic Effect of Dopamine-Derived Products in Human SH-SY5Y Neuroblastoma Cells. Neurotox. Res. 2019, 36, 746–755. [Google Scholar] [CrossRef]
- Whitehead, R.E.; Ferrer, J.V.; Javitch, J.A.; Justice, J.B. Reaction of Oxidized Dopamine with Endogenous Cysteine Residues in the Human Dopamine Transporter. J. Neurochem. 2001, 76, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.M.; Arthur, R.E.; Thomas, D.M.; Elferink, L.A. Tyrosine Hydroxylase Is Inactivated by Catechol-Quinones and Converted to a Redox-Cycling Quinoprotein: Possible Relevance to Parkinson’s Disease. J. Neurochem. 1999, 73, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-Induced Neurotoxicity: The Role of Dopamine Quinone Formation and Tyrosinase in a Model of Parkinson’s Disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Mishizen, A.J.; Cascio, M.; Hastings, T.G. Proteomic Identification of Dopamine-Conjugated Proteins from Isolated Rat Brain Mitochondria and SH-SY5Y Cells. Neurobiol. Dis. 2009, 34, 487–500. [Google Scholar] [CrossRef]
- Girotto, S.; Sturlese, M.; Bellanda, M.; Tessari, I.; Cappellini, R.; Bisaglia, M.; Bubacco, L.; Mammi, S. Dopamine-Derived Quinones Affect the Structure of the Redox Sensor DJ-1 through Modifications at Cys-106 and Cys-53. J. Biol. Chem. 2012, 287, 18738–18749. [Google Scholar] [CrossRef] [PubMed]
- LaVoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine Covalently Modifies and Functionally Inactivates Parkin. Nat. Med. 2005, 11, 1214–1221. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Lim, T.M. Dopamine (DA) Induced Irreversible Proteasome Inhibition via DA Derived Quinones. Free Radic. Res. 2009, 43, 417–430. [Google Scholar] [CrossRef]
- Zafar, K.S.; Siegel, D.; Ross, D. A Potential Role for Cyclized Quinones Derived from Dopamine, DOPA, and 3,4-Dihydroxyphenylacetic Acid in Proteasomal Inhibition. Mol. Pharmacol. 2006, 70, 1079–1086. [Google Scholar] [CrossRef]
- Volles, M.J.; Lansbury, P.T. Zeroing in on the Pathogenic Form of Alpha-Synuclein and Its Mechanism of Neurotoxicity in Parkinson’s Disease. Biochemistry 2003, 42, 7871–7878. [Google Scholar] [CrossRef]
- Conway, K.A.; Rochet, J.C.; Bieganski, R.M.; Lansbury, J. Kinetic Stabilization of the Alpha-Synuclein Protofibril by a Dopamine-Alpha-Synuclein Adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Rekas, A.; Knott, R.B.; Sokolova, A.; Barnham, K.J.; Perez, K.A.; Masters, C.L.; Drew, S.C.; Cappai, R.; Curtain, C.C.; Pham, C.L.L. The Structure of Dopamine Induced Alpha-Synuclein Oligomers. Eur. Biophys. J. 2010, 39, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Tosatto, L.; Munari, F.; Tessari, I.; de Laureto, P.P.; Mammi, S.; Bubacco, L. Dopamine Quinones Interact with Alpha-Synuclein to Form Unstructured Adducts. Biochem. Biophys. Res. Commun. 2010, 394, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Cappai, R.; Leck, S.; Tew, D.J.; Williamson, N.A.; Smith, D.P.; Galatis, D.; Sharpies, R.A.; Curtain, C.C.; Ali, F.E.; Cherny, R.A.; et al. Dopamine Promotes Alpha-Synuclein Aggregation into SDS-Resistant Soluble Oligomers via a Distinct Folding Pathway. FASEB J. 2005, 19, 1377–1379. [Google Scholar] [CrossRef]
- Li, H.T.; Lin, D.H.; Luo, X.Y.; Zhang, F.; Ji, L.N.; Du, H.N.; Song, G.Q.; Hu, J.; Zhou, J.W.; Hu, H.Y. Inhibition of Alpha-Synuclein Fibrillization by Dopamine Analogs via Reaction with the Amino Groups of Alpha-Synuclein. Implication for Dopaminergic Neurodegeneration. FEBS J. 2005, 272, 3661–3672. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Armakola, M.; Dumoulin, M.; Parastatidis, I.; Ischiropoulos, H. Cellular Oligomerization of Alpha-Synuclein Is Determined by the Interaction of Oxidized Catechols with a C-Terminal Sequence. J. Biol. Chem. 2007, 282, 31621–31630. [Google Scholar] [CrossRef] [PubMed]
- Norris, E.H.; Giasson, B.I.; Hodara, R.; Xu, S.; Trojanowski, J.Q.; Ischiropoulos, H.; Lee, V.M.Y. Reversible Inhibition of Alpha-Synuclein Fibrillization by Dopaminochrome-Mediated Conformational Alterations. J. Biol. Chem. 2005, 280, 21212–21219. [Google Scholar] [CrossRef]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired Dopamine Metabolism in Parkinson’s Disease Pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef]
- Goldstein, D.S. The Catecholaldehyde Hypothesis for the Pathogenesis of Catecholaminergic Neurodegeneration: What We Know and What We Do Not Know. Int. J. Mol. Sci. 2021, 22, 5999. [Google Scholar] [CrossRef]
- Hauser, D.N.; Dukes, A.A.; Mortimer, A.D.; Hastings, T.G. Dopamine Quinone Modifies and Decreases the Abundance of the Mitochondrial Selenoprotein Glutathione Peroxidase 4. Free Radic. Biol. Med. 2013, 65, 419–427. [Google Scholar] [CrossRef]
- Wang, D.; Peng, Y.; Xie, Y.; Zhou, B.; Sun, X.; Kang, R.; Tang, D. Antiferroptotic Activity of Non-Oxidative Dopamine. Biochem. Biophys. Res. Commun. 2016, 480, 602–607. [Google Scholar] [CrossRef]
- Dukes, A.A.; Van Laar, V.S.; Cascio, M.; Hastings, T.G. Changes in Endoplasmic Reticulum Stress Proteins and Aldolase A in Cells Exposed to Dopamine. J. Neurochem. 2008, 106, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Dukes, A.A.; Cascio, M.; Hastings, T.G. Proteomic Analysis of Rat Brain Mitochondria Following Exposure to Dopamine Quinone: Implications for Parkinson Disease. Neurobiol. Dis. 2008, 29, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Hurben, A.K.; Erber, L.N.; Tretyakova, N.Y.; Doran, T.M. Proteome-Wide Profiling of Cellular Targets Modified by Dopamine Metabolites Using a Bio-Orthogonally Functionalized Catecholamine. ACS Chem. Biol. 2021, 16, 2581–2594. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-Modified Alpha-Synuclein Blocks Chaperone-Mediated Autophagy. J. Clin. Investig. 2008, 118, 777–778. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stafanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant Alpha-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine Oxidation Mediates Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef]
- Berman, S.B.; Hastings, T.G. Dopamine Oxidation Alters Mitochondrial Respiration and Induces Permeability Transition in Brain Mitochondria: Implications for Parkinson’s Disease. J. Neurochem. 1999, 73, 1127–1137. [Google Scholar] [CrossRef]
- Bisaglia, M.; Soriano, M.E.; Arduini, I.; Mammi, S.; Bubacco, L. Molecular Characterization of Dopamine-Derived Quinones Reactivity toward NADH and Glutathione: Implications for Mitochondrial Dysfunction in Parkinson Disease. Biochim. Biophys. Acta 2010, 1802, 699–706. [Google Scholar] [CrossRef]
- Gluck, M.R.; Zeevalk, G.D. Inhibition of Brain Mitochondrial Respiration by Dopamine and Its Metabolites: Implications for Parkinson’s Disease and Catecholamine-Associated Diseases. J. Neurochem. 2004, 91, 788–795. [Google Scholar] [CrossRef]
- Gluck, M.; Ehrhart, J.; Jayatilleke, E.; Zeevalk, G.D. Inhibition of Brain Mitochondrial Respiration by Dopamine: Involvement of H(2)O(2) and Hydroxyl Radicals but Not Glutathione-Protein-Mixed Disulfides. J. Neurochem. 2002, 82, 66–74. [Google Scholar] [CrossRef]
- Jana, S.; Maiti, A.K.; Bagh, M.B.; Banerjee, K.; Das, A.; Roy, A.; Chakrabarti, S. Dopamine but Not 3,4-Dihydroxy Phenylacetic Acid (DOPAC) Inhibits Brain Respiratory Chain Activity by Autoxidation and Mitochondria Catalyzed Oxidation to Quinone Products: Implications in Parkinson’s Disease. Brain Res. 2007, 1139, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.H.; Sen, T.; Maiti, A.K.; Jana, S.; Chatterjee, U.; Chakrabarti, S. Inhibition of Rat Brain Mitochondrial Electron Transport Chain Activity by Dopamine Oxidation Products during Extended in Vitro Incubation: Implications for Parkinson’s Disease. Biochim. Biophys. Acta 2005, 1741, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S. The Catecholaldehyde Hypothesis: Where MAO Fits In. J. Neural Transm. 2020, 127, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Panneton, W.M.; Kumar, V.B.; Gan, Q.; Burke, W.J.; Galvin, J.E. The Neurotoxicity of DOPAL: Behavioral and Stereological Evidence for Its Role in Parkinson Disease Pathogenesis. PLoS ONE 2010, 5, e15251. [Google Scholar] [CrossRef]
- Burke, W.J.; Li, S.W.; Williams, E.A.; Nonneman, R.; Zahm, D.S. 3,4-Dihydroxyphenylacetaldehyde Is the Toxic Dopamine Metabolite in Vivo: Implications for Parkinson’s Disease Pathogenesis. Brain Res. 2003, 989, 205–213. [Google Scholar] [CrossRef]
- Ritz, B.R.; Paul, K.C.; Bronstein, J.M. Of Pesticides and Men: A California Story of Genes and Environment in Parkinson’s Disease. Curr. Environ. Health Rep. 2016, 3, 40–52. [Google Scholar] [CrossRef]
- Fitzmaurice, A.G.; Rhodes, S.L.; Cockburn, M.; Ritz, B.; Bronstein, J.M. Aldehyde Dehydrogenase Variation Enhances Effect of Pesticides Associated with Parkinson Disease. Neurology 2014, 82, 419–426. [Google Scholar] [CrossRef]
- Fitzmaurice, A.G.; Rhodes, S.L.; Lulla, A.; Murphy, N.P.; Lam, H.A.; O’Donnell, K.C.; Barnhill, L.; Casida, J.E.; Cockburn, M.; Sagasti, A.; et al. Aldehyde Dehydrogenase Inhibition as a Pathogenic Mechanism in Parkinson Disease. Proc. Natl. Acad. Sci. USA 2013, 110, 636–641. [Google Scholar] [CrossRef]
- Grünblatt, E.; Ruder, J.; Monoranu, C.M.; Riederer, P.; Youdim, M.B.; Mandel, S.A. Differential Alterations in Metabolism and Proteolysis-Related Proteins in Human Parkinson’s Disease Substantia Nigra. Neurotox. Res. 2018, 33, 560–568. [Google Scholar] [CrossRef]
- Mandel, S.; Grunblatt, E.; Riederer, P.; Amariglio, N.; Hirsch, J.J.; Rechavi, G.; Youdim, M.B.H. Gene Expression Profiling of Sporadic Parkinson’s Disease Substantia Nigra Pars Compacta Reveals Impairment of Ubiquitin-Proteasome Subunits, SKP1A, Aldehyde Dehydrogenase, and Chaperone HSC-70. Ann. N. Y. Acad. Sci. 2005, 1053, 356–375. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Sullivan, P.; Holmes, C.; Miller, G.W.; Alter, S.; Strong, R.; Mash, D.C.; Kopin, I.J.; Sharabi, Y. Determinants of Buildup of the Toxic Dopamine Metabolite DOPAL in Parkinson’s Disease. J. Neurochem. 2013, 126, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Sullivan, P.; Holmes, C.; Kopin, I.J.; Basile, M.J.; Mash, D.C. Catechols in Post-Mortem Brain of Patients with Parkinson Disease. Eur. J. Neurol. 2011, 18, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Jinsmaa, Y.; Sharabi, Y.; Sullivan, P.; Isonaka, R.; Goldstein, D.S. 3,4-Dihydroxyphenylacetaldehyde-Induced Protein Modifications and Their Mitigation by N-Acetylcysteine. J. Pharmacol. Exp. Ther. 2018, 366, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Kristal, B.S.; Conway, A.D.; Brown, A.M.; Jain, J.C.; Ulluci, P.A.; Li, S.W.; Burke, W.J. Selective Dopaminergic Vulnerability: 3,4-Dihydroxyphenylacetaldehyde Targets Mitochondria. Free Radic. Biol. Med. 2001, 30, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Bubacco, L. Lysines, Achilles’ Heel in Alpha-Synuclein Conversion to a Deadly Neuronal Endotoxin. Ageing Res. Rev. 2016, 26, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Greggio, E.; Bisaglia, M.; Bubacco, L. Biophysical Groundwork as a Hinge to Unravel the Biology of α-Synuclein Aggregation and Toxicity. Q Rev. Biophys. 2014, 47, 1–48. [Google Scholar] [CrossRef]
- Burke, W.J.; Kumar, V.B.; Pandey, N.; Panneton, W.M.; Gan, Q.; Franko, M.W.; O’Dell, M.; Li, S.W.; Pan, Y.; Chung, H.D.; et al. Aggregation of Alpha-Synuclein by DOPAL, the Monoamine Oxidase Metabolite of Dopamine. Acta Neuropathol. 2008, 115, 193–203. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Sullivan, P.; Gross, D.; Cooney, A.; Sharabi, Y.; Goldstein, D.S. Divalent Metal Ions Enhance DOPAL-Induced Oligomerization of Alpha-Synuclein. Neurosci. Lett. 2014, 569, 27–32. [Google Scholar] [CrossRef]
- Anderson, D.G.; Florang, V.R.; Schamp, J.H.; Buettner, G.R.; Doorn, J.A. Antioxidant-Mediated Modulation of Protein Reactivity for 3,4-Dihydroxyphenylacetaldehyde, a Toxic Dopamine Metabolite. Chem. Res. Toxicol. 2016, 29, 1098–1107. [Google Scholar] [CrossRef]
- Follmer, C.; Coelho-Cerqueira, E.; Yatabe-Franco, D.Y.; Araujo, G.D.T.; Pinheiro, A.S.; Domont, G.B.; Eliezer, D. Oligomerization and Membrane-Binding Properties of Covalent Adducts Formed by the Interaction of α-Synuclein with the Toxic Dopamine Metabolite 3,4-Dihydroxyphenylacetaldehyde (DOPAL). J. Biol. Chem. 2015, 290, 27660–27679. [Google Scholar] [CrossRef]
- Plotegher, N.; Berti, G.; Ferrari, E.; Tessari, I.; Zanetti, M.; Lunelli, L.; Greggio, E.; Bisaglia, M.; Veronesi, M.; Girotto, S.; et al. DOPAL Derived Alpha-Synuclein Oligomers Impair Synaptic Vesicles Physiological Function. Sci. Rep. 2017, 7, 40699. [Google Scholar] [CrossRef] [PubMed]
- Sarafian, T.A.; Yacoub, A.; Kunz, A.; Aranki, B.; Serobyan, G.; Cohn, W.; Whitelegge, J.P.; Watson, J.B. Enhanced Mitochondrial Inhibition by 3,4-Dihydroxyphenyl-Acetaldehyde (DOPAL)-Oligomerized α-Synuclein. J. Neurosci. Res. 2019, 97, 1689–1705. [Google Scholar] [CrossRef] [PubMed]
- Masato, A.; Plotegher, N.; Terrin, F.; Sandre, M.; Faustini, G.; Thor, A.; Adams, S.; Berti, G.; Cogo, S.; De Lazzari, F.; et al. DOPAL Initiates ASynuclein-Dependent Impaired Proteostasis and Degeneration of Neuronal Projections in Parkinson’s Disease. NPJ Park. Dis. 2023, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Vanna, R.; Cupaioli, F.A.; Bellei, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Grassi, S.; Prinetti, A.; Casella, L.; et al. Neuromelanin Organelles Are Specialized Autolysosomes That Accumulate Undegraded Proteins and Lipids in Aging Human Brain and Are Likely Involved in Parkinson’s Disease. NPJ Park. Dis. 2018, 4, 17. [Google Scholar] [CrossRef]
- Bisaglia, M.; Bubacco, L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules 2020, 10, 195. [Google Scholar] [CrossRef]
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, Oxidative Stress and Protein-Quinone Modifications in Parkinson’s and Other Neurodegenerative Diseases. Angew Chem. Int. Ed. Engl. 2019, 58, 6512–6527. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of Major Histocompatibility Complex Class II-Positive Microglia and Cytokine Profile of Parkinson’s Disease Brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Ferrari, E.; Capucciati, A.; Prada, I.; Zucca, F.A.; D’Arrigo, G.; Pontiroli, D.; Bridelli, M.G.; Sturini, M.; Bubacco, L.; Monzani, E.; et al. Synthesis, Structure Characterization, and Evaluation in Microglia Cultures of Neuromelanin Analogues Suitable for Modeling Parkinson’s Disease. ACS Chem. Neurosci. 2017, 8, 501–512. [Google Scholar] [CrossRef]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin Activates Microglia and Induces Degeneration of Dopaminergic Neurons: Implications for Progression of Parkinson’s Disease. Neurotox. Res. 2011, 19, 63–72. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective Neuronal Vulnerability in Parkinson Disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Bubacco, L.; Chakrabarti, S.; Bisaglia, M. α-Synuclein Toxicity in Drosophila Melanogaster Is Enhanced by the Presence of Iron: Implications for Parkinson’s Disease. Antioxidants 2023, 12, 261. [Google Scholar] [CrossRef] [PubMed]
- Oun, A.; Soliman, A.; Trombetta-Lima, M.; Tzepapadaki, A.; Tsagkari, D.; Kortholt, A.; Dolga, A.M. LRRK2 Protects Immune Cells against Erastin-Induced Ferroptosis. Neurobiol. Dis. 2022, 175, 105917. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.J.; Chen, S.D.; Lin, K.L.; Liou, C.W.; Lan, M.Y.; Chuang, Y.C.; Wang, P.W.; Lee, J.J.; Wang, F.S.; Lin, H.Y.; et al. Iron Brain Menace: The Involvement of Ferroptosis in Parkinson Disease. Cells 2022, 11, 3829. [Google Scholar] [CrossRef]
- Tan, Y.Y.; Jenner, P.; Chen, S. Di Monoamine Oxidase-B Inhibitors for the Treatment of Parkinson’s Disease: Past, Present, and Future. J. Park. Dis. 2022, 12, 477–493. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakrabarti, S.; Bisaglia, M. Oxidative Stress and Neuroinflammation in Parkinson’s Disease: The Role of Dopamine Oxidation Products. Antioxidants 2023, 12, 955. https://doi.org/10.3390/antiox12040955
Chakrabarti S, Bisaglia M. Oxidative Stress and Neuroinflammation in Parkinson’s Disease: The Role of Dopamine Oxidation Products. Antioxidants. 2023; 12(4):955. https://doi.org/10.3390/antiox12040955
Chicago/Turabian StyleChakrabarti, Sasanka, and Marco Bisaglia. 2023. "Oxidative Stress and Neuroinflammation in Parkinson’s Disease: The Role of Dopamine Oxidation Products" Antioxidants 12, no. 4: 955. https://doi.org/10.3390/antiox12040955