
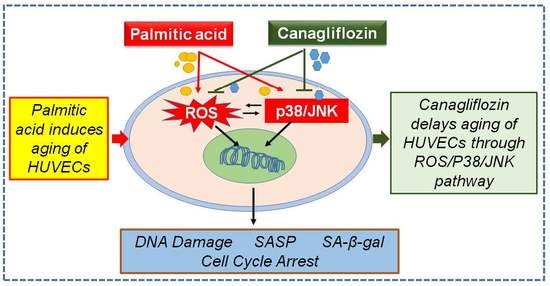
Canagliflozin Delays Aging of HUVECs Induced by Palmitic Acid via the ROS/p38/JNK Pathway
 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
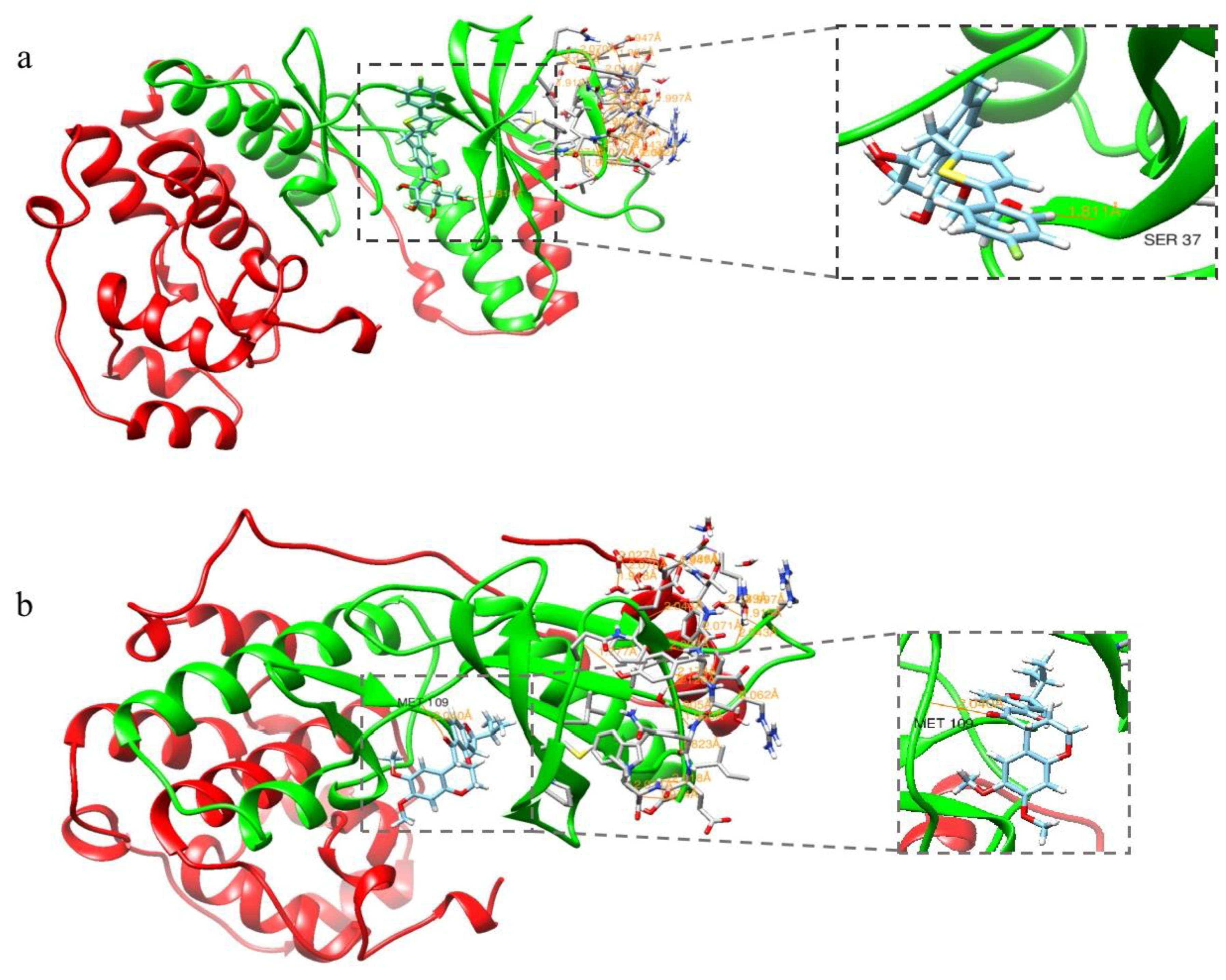
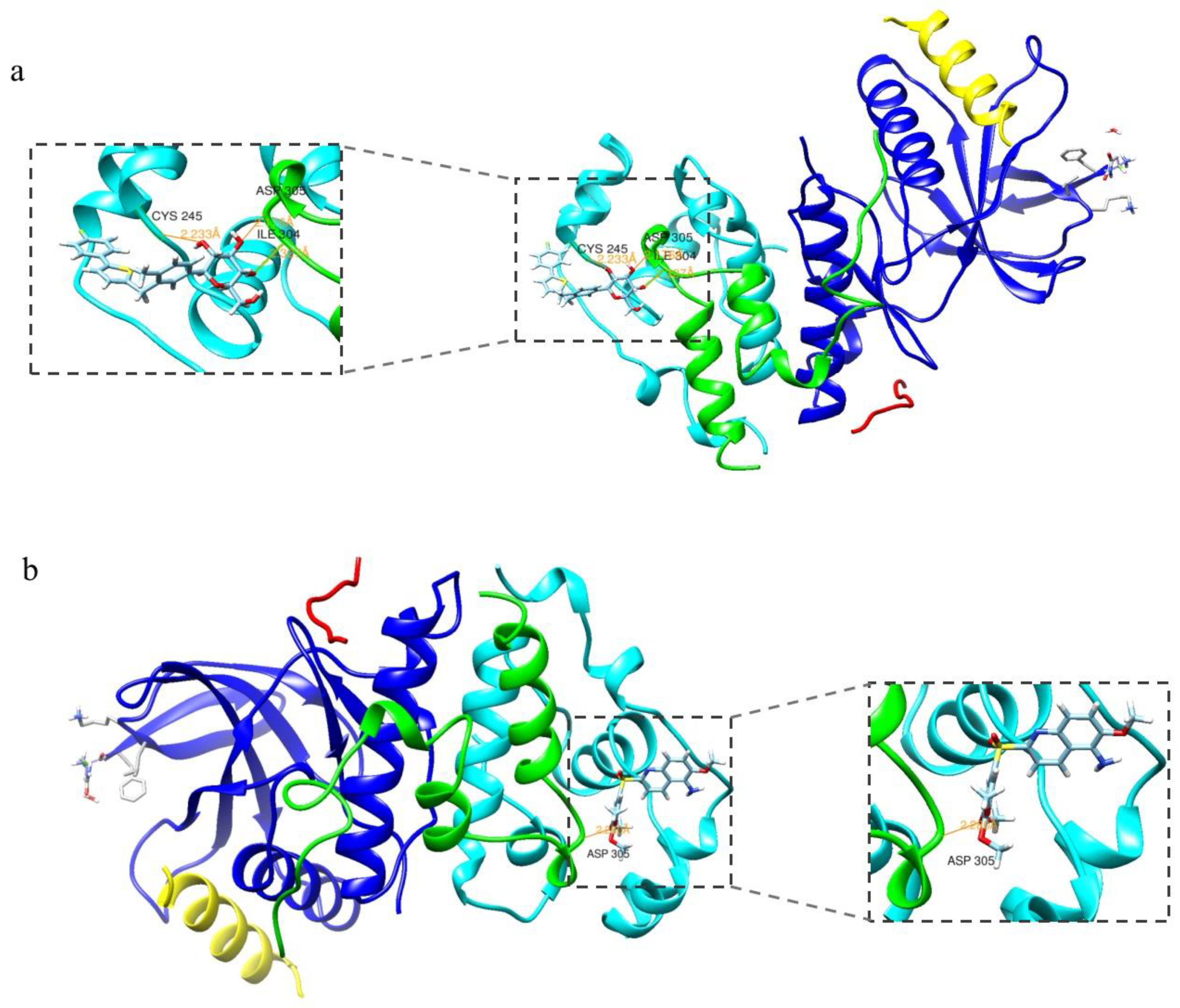
2.1. Molecular Docking Assay
2.2. Cell Culture and Treatment
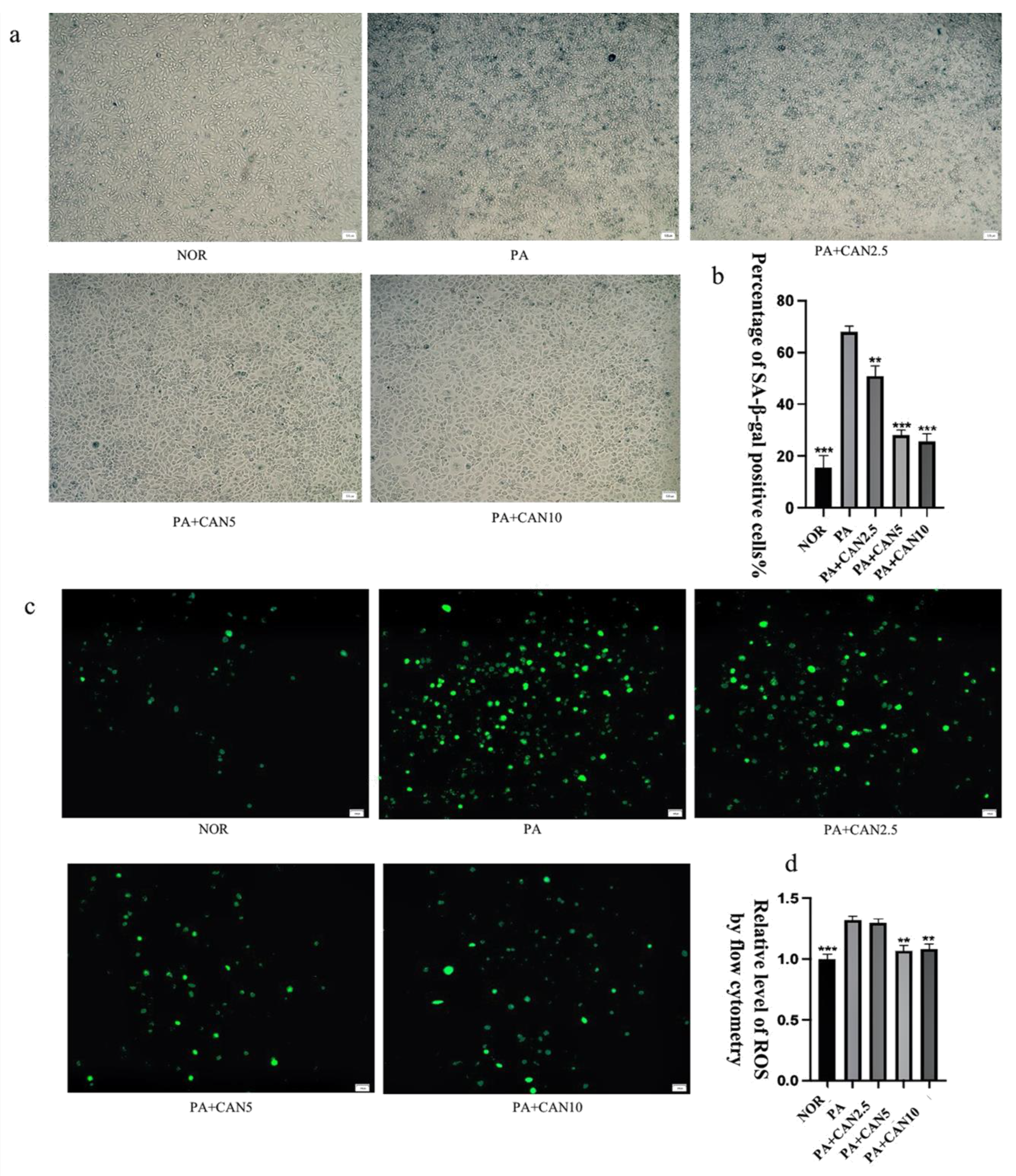
2.3. Senescence-Associated β-Galactosidase Staining Assay
2.4. Cellular ROS Assay—DCFH-DA Probe
2.5. Cell Viability—MTT Assay
2.6. Western Blot
2.7. Cell Cycle
2.8. Statistical Analysis
3. Results and Discussion
3.1. Molecular Docking Assay
3.2. CAN Delays PA-Induced HUVEC Cell Senescence
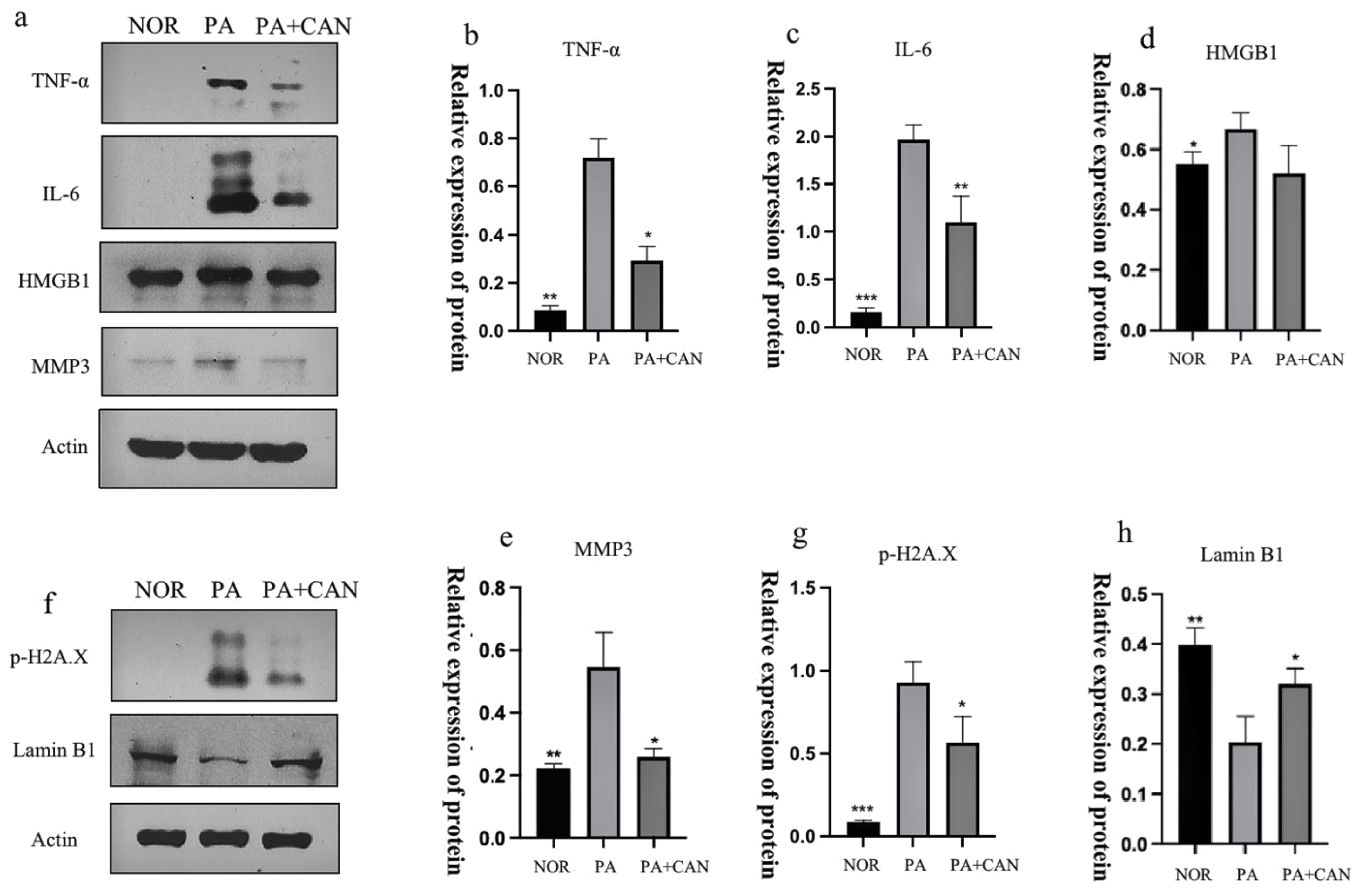
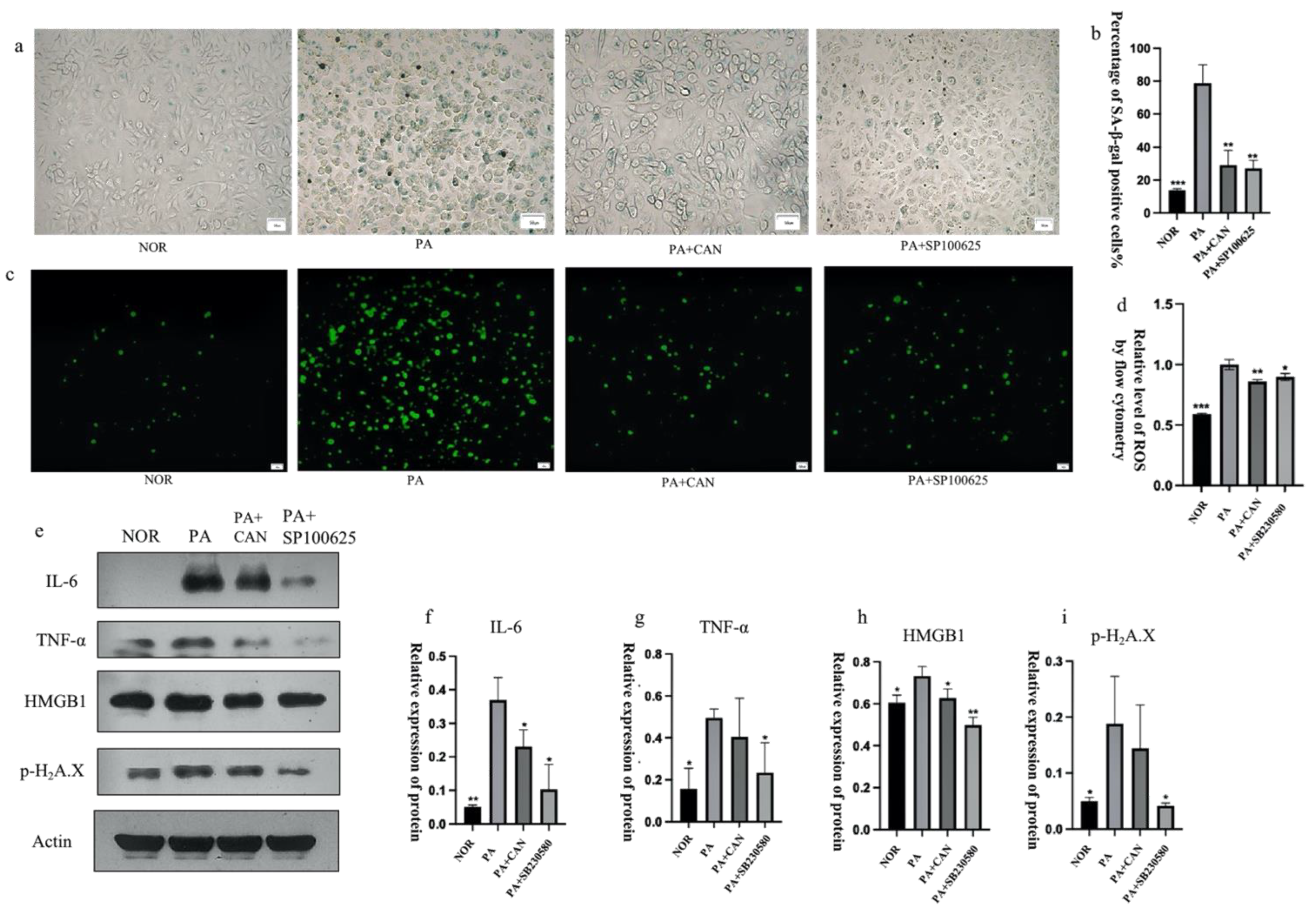
3.3. CAN Regulates SASP and Senescence Signature Protein Content
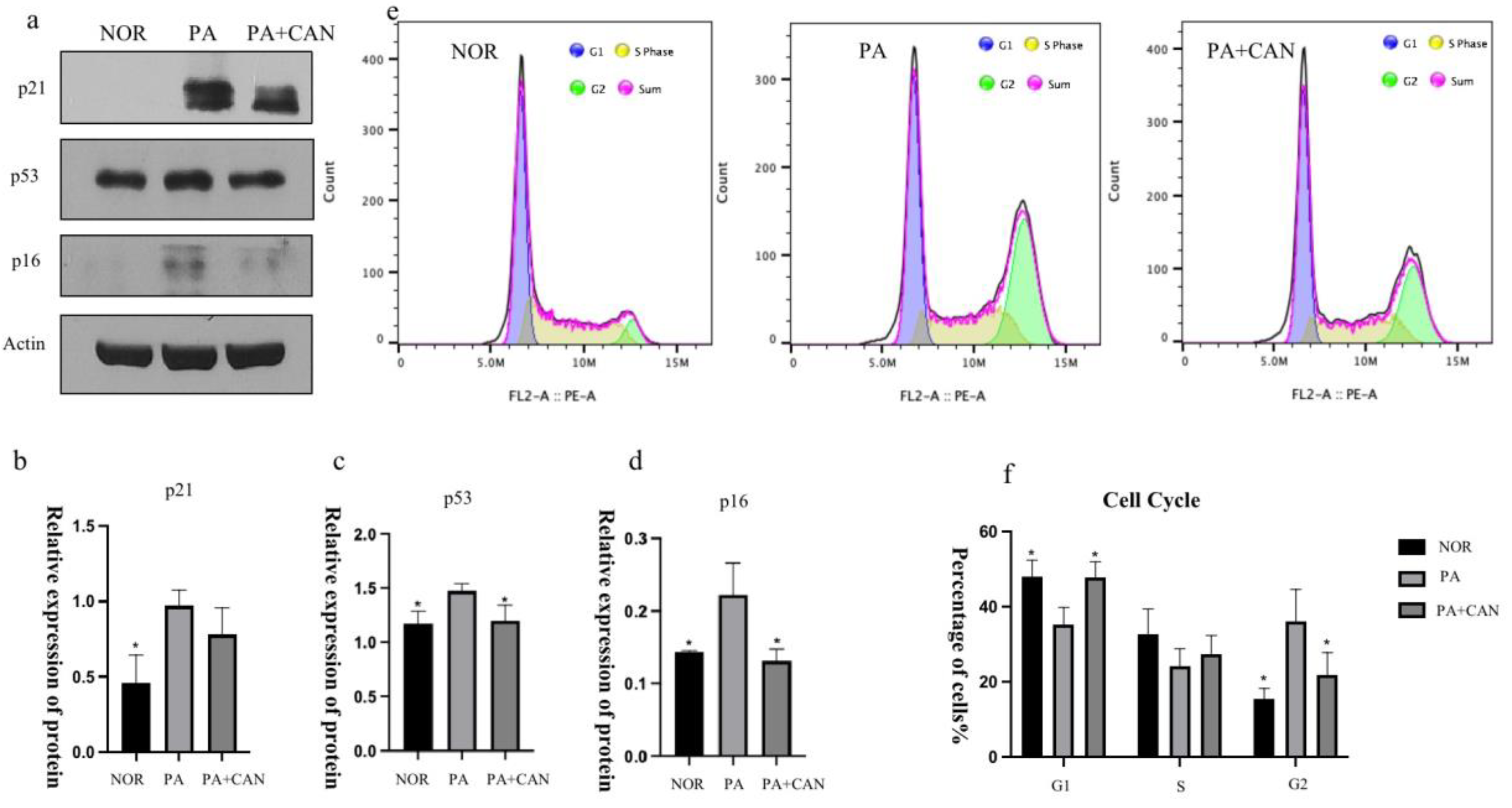
3.4. CAN Regulates Cell Cycle Arrest Due to Aging
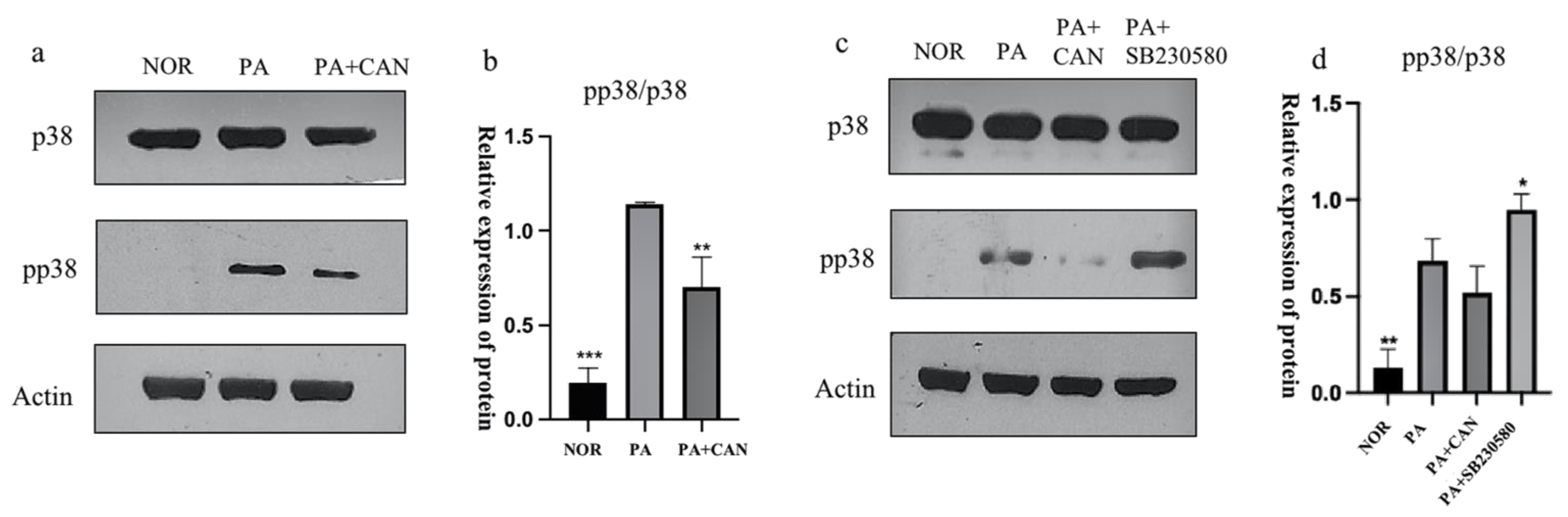
3.5. CAN Alleviates PA-Induced HUVEC Replicative Senescence by Inhibiting p38 Activation
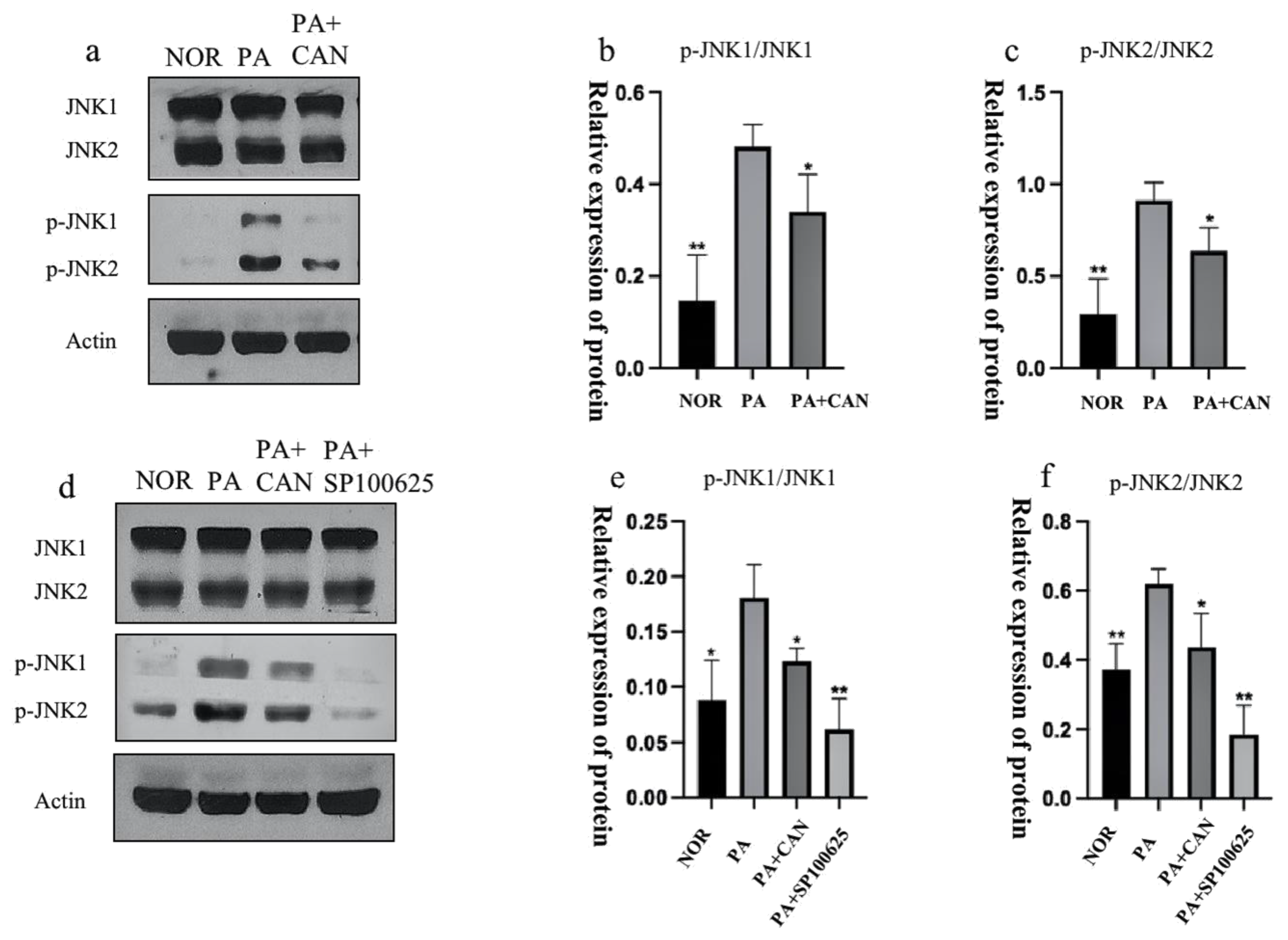
3.6. CAN Alleviates PA-Induced HUVEC Replicative Senescence by Inhibiting JNK Activation
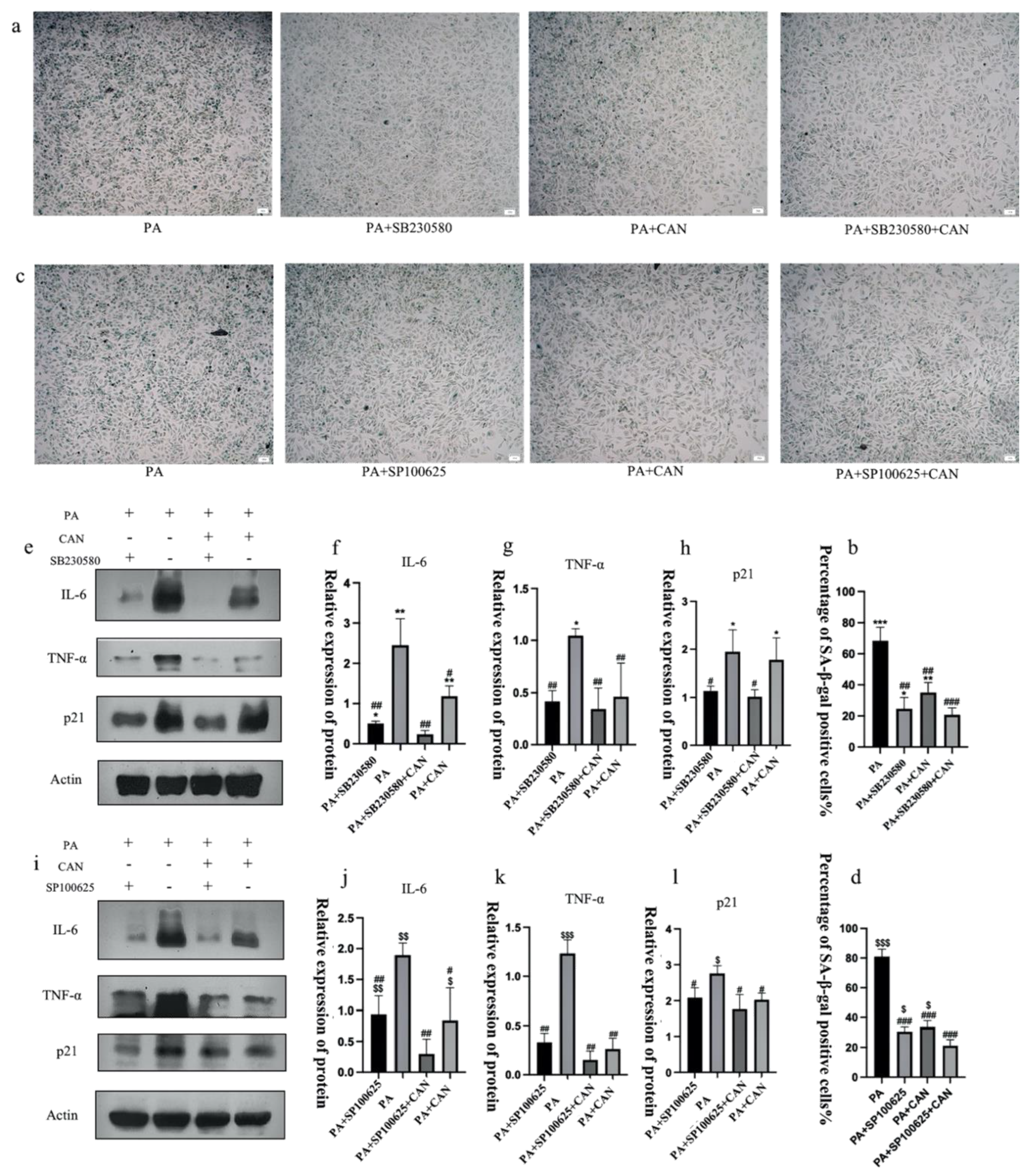
3.7. CAN Delays PA-Induced Senescence in HUVEC Cells through p38/JNK Dual Targeting
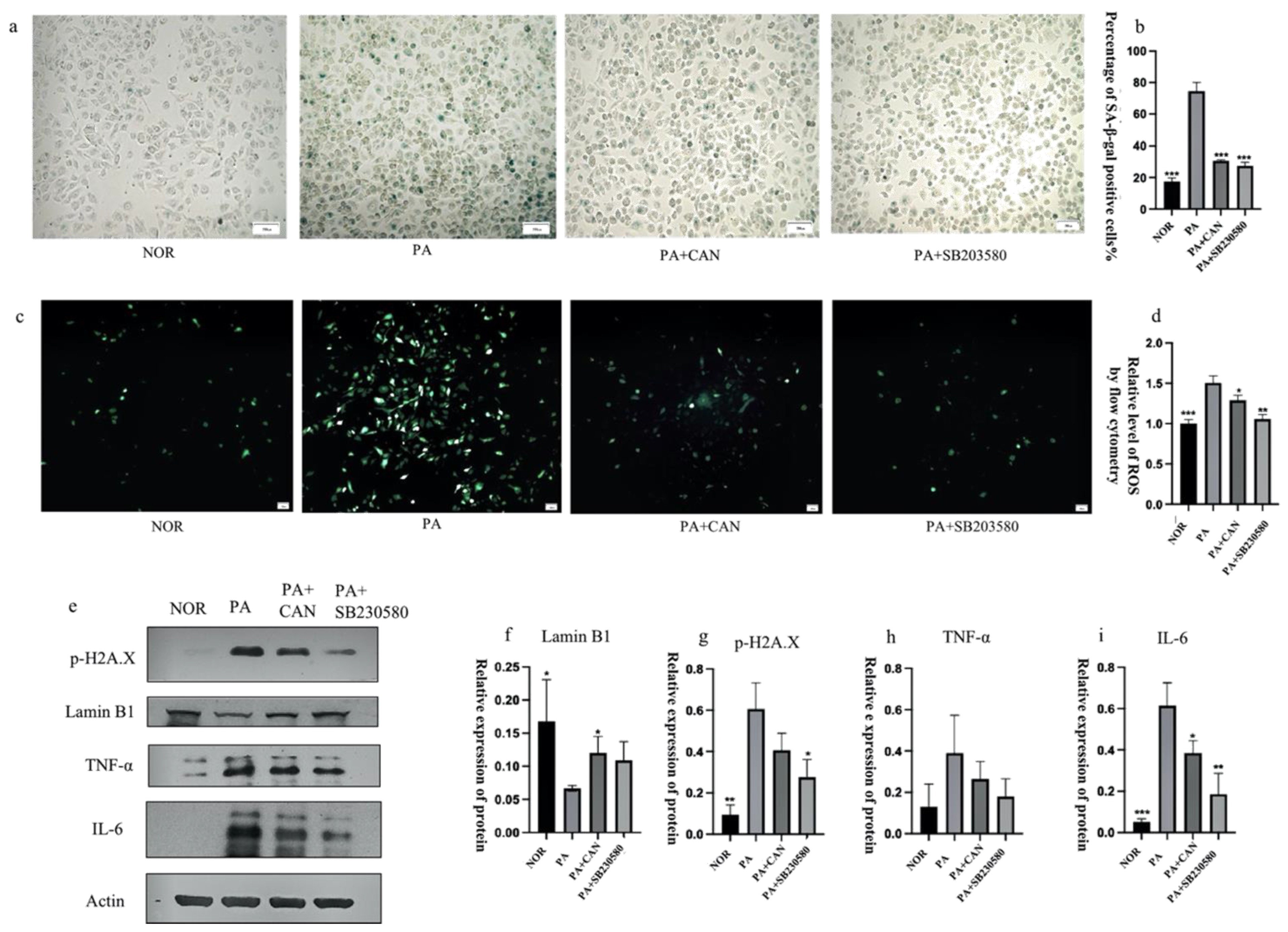
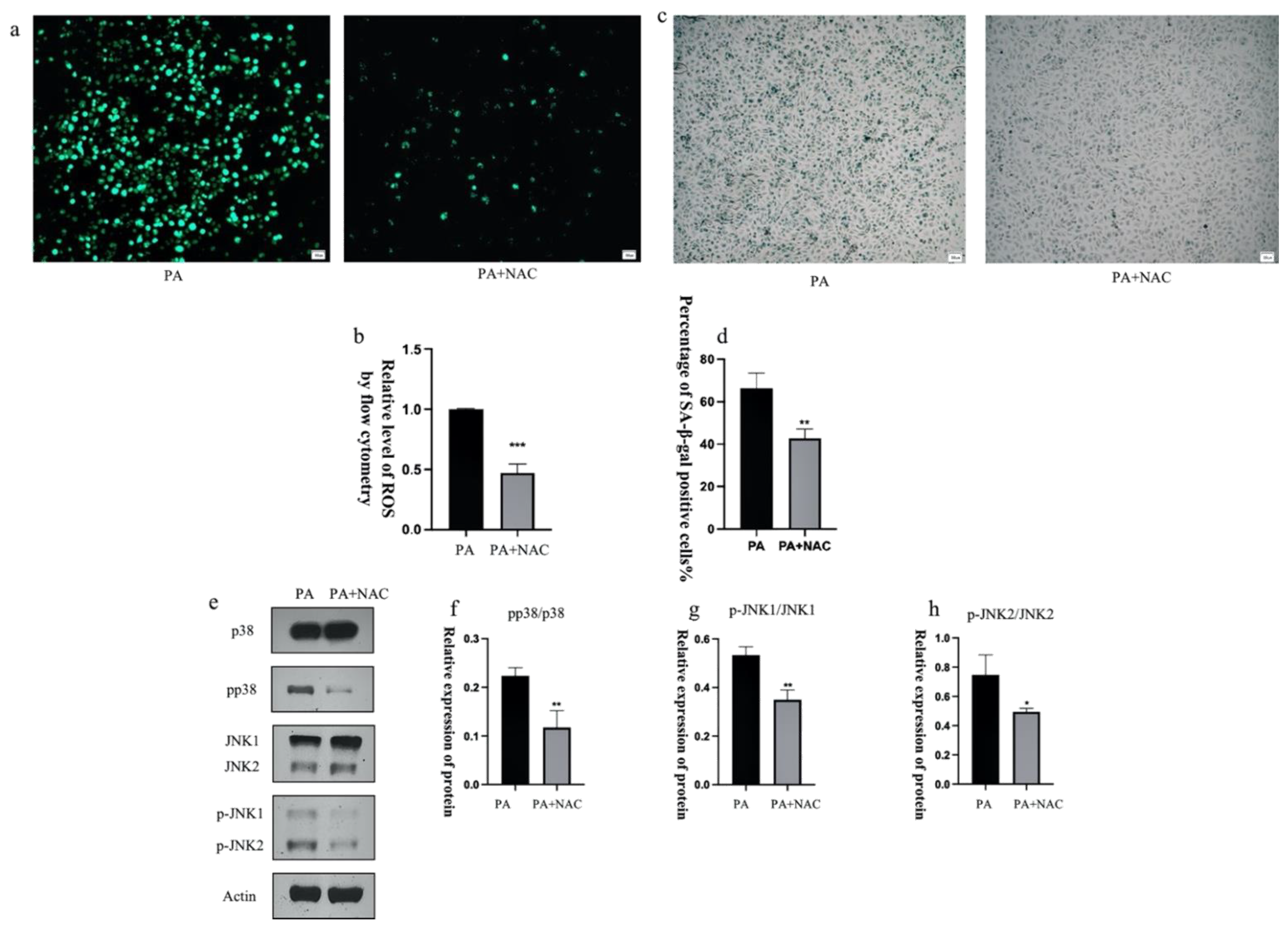
3.8. ROS Inhibitors Attenuate the Activation of p38 and JNK Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, Y.; Xu, A.; Wang, Y. SIRT1 in Endothelial Cells as a Novel Target for the Prevention of Early Vascular Aging. J. Cardiovasc. Pharmacol. 2016, 67, 465–473. [Google Scholar] [PubMed]
- Tian, X.L.; Li, Y. Endothelial cell senescence and age-related vascular diseases. J. Genet. Genom. 2014, 41, 485–495. [Google Scholar] [PubMed]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [PubMed] [Green Version]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar]
- Pearson, K.J.; Baur, J.A.; Lewis, K.N.; Peshkin, L.; Price, N.L.; Labinskyy, N.; Swindell, W.R.; Kamara, D.; Minor, R.K.; Perez, E.; et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008, 8, 157–168. [Google Scholar]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Rossman, M.J.; Kaplon, R.E.; Hill, S.D.; McNamara, M.N.; Santos-Parker, J.R.; Pierce, G.L.; Seals, D.R.; Donato, A.J. Endothelial cell senescence with aging in healthy humans: Prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H890–H895. [Google Scholar]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar]
- Benincasa, G.; Coscioni, E.; Napoli, C. Cardiovascular risk factors and molecular routes underlying endothelial dysfunction: Novel opportunities for primary prevention. Biochem. Pharmacol. 2022, 202, 115108. [Google Scholar] [PubMed]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 2020, 126, 298–314. [Google Scholar] [PubMed]
- Niu, Y.; Zhang, Y.; Chen, Y.; Zhang, W.; Hao, W.; Lu, J.; Zhou, J.; Wang, L.; Xie, W. Canagliflozin Ameliorates NLRP3 Inflammasome-Mediated Inflammation Through Inhibiting NF-κB Signaling and Upregulating Bif-1. Front Pharmacol. 2022, 13, 820541. [Google Scholar] [PubMed]
- Zhong, J.; Sun, P.; Xu, N.; Liao, M.; Xu, C.; Ding, Y.; Cai, J.; Zhang, Y.; Xie, W. Canagliflozin inhibits p-gp function and early autophagy and improves the sensitivity to the antitumor effect of doxorubicin. Biochem. Pharmacol. 2020, 175, 113856. [Google Scholar]
- Xu, C.; Wang, W.; Zhong, J.; Lei, F.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem. Pharmacol. 2018, 152, 45–59. [Google Scholar]
- Yang, X.; Liu, Q.; Li, Y.; Tang, Q.; Wu, T.; Chen, L.; Pu, S.; Zhao, Y.; Zhang, G.; Huang, C.; et al. The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK-Sirt1-Pgc-1α signalling pathway. Adipocyte 2020, 9, 484–494. [Google Scholar]
- Papadopoli, D.; Uchenunu, O.; Palia, R.; Chekkal, N.; Hulea, L.; Topisirovic, I.; Pollak, M.; St-Pierre, J. Perturbations of cancer cell metabolism by the antidiabetic drug canagliflozin. Neoplasia 2021, 23, 391–399. [Google Scholar]
- Sun, P.; Wang, Y.; Ding, Y.; Luo, J.; Zhong, J.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin attenuates lipotoxicity in cardiomyocytes and protects diabetic mouse hearts by inhibiting the mTOR/HIF-1α pathway. iScience 2021, 24, 102521. [Google Scholar]
- Zhang, W.; Lu, J.; Wang, Y.; Sun, P.; Gao, T.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin Attenuates Lipotoxicity in Cardiomyocytes by Inhibiting Inflammation and Ferroptosis through Activating AMPK Pathway. Int. J. Mol. Sci. 2023, 24, 858. [Google Scholar]
- Miller, R.A.; Harrison, D.E.; Allison, D.B.; Bogue, M.; Debarba, L.; Diaz, V.; Fernandez, E.; Galecki, A.; Garvey, W.T.; Jayarathne, H.; et al. Canagliflozin extends life span in genetically heterogeneous male but not female mice. JCI Insight 2020, 5, e140019. [Google Scholar]
- Anerillas, C.; Abdelmohsen, K.; Gorospe, M. Regulation of senescence traits by MAPKs. Geroscience 2020, 42, 397–408. [Google Scholar] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [PubMed] [Green Version]
- Hsin, K.Y.; Ghosh, S.; Kitano, H. Combining machine learning systems and multiple docking simulation packages to improve docking prediction reliability for network pharmacology. PLoS ONE 2013, 8, e83922. [Google Scholar]
- Itahana, K.; Campisi, J.; Dimri, G.P. Methods to detect biomarkers of cellular senescence: The senescence-associated beta-galactosidase assay. Methods Mol. Biol. 2007, 371, 21–31. [Google Scholar] [PubMed]
- Papaconstantinou, J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells 2019, 8, 1383. [Google Scholar]
- de Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Man, C.D.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: Potential biochemical mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar]
- Baranov, V.S.; Baranova, E.V. Aging and Ambiguous ROS. System Genetics Analysis. Curr. Aging Sci. 2017, 10, 6–11. [Google Scholar] [CrossRef]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar]
- Buj, R.; Leon, K.E.; Anguelov, M.A.; Aird, K.M. Suppression of p16 alleviates the senescence-associated secretory phenotype. Aging 2021, 13, 3290–3312. [Google Scholar] [CrossRef] [PubMed]
- Radspieler, M.M.; Schindeldecker, M.; Stenzel, P.; Försch, S.; Tagscherer, K.E.; Herpel, E.; Hohenfellner, M.; Hatiboglu, G.; Roth, W.; Macher-Goeppinger, S. Lamin-B1 is a senescence-associated biomarker in clear-cell renal cell carcinoma. Oncol. Lett. 2019, 18, 2654–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. A common signature of cellular senescence; does it exist? Ageing Res. Rev. 2021, 71, 101458. [Google Scholar] [PubMed]
- Mao, Z.; Ke, Z.; Gorbunova, V.; Seluanov, A. Replicatively senescent cells are arrested in G1 and G2 phases. Aging 2012, 4, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Bu, X.; Liu, X.; Tankersley, C.G.; Wang, C.; Huang, K. Age-induced augmentation of p38 MAPK phosphorylation in mouse lung. Exp. Gerontol. 2011, 46, 694–702. [Google Scholar] [CrossRef]
- Kumar, S.; Jiang, M.S.; Adams, J.L.; Lee, J.C. Pyridinylimidazole Compound SB 203580 Inhibits the Activity but Not the Activation of p38 Mitogen-Activated Protein Kinase. Biochem. Biophys. Res. Commun. 1999, 263, 825–831. [Google Scholar] [CrossRef]
- Vallerie, S.N.; Hotamisligil, G.S. The Role of JNK Proteins in Metabolism. Sci. Transl. Med. 2010, 2, 60rv65. [Google Scholar] [CrossRef]
- Müller, K.; Honcharova-Biletska, H.; Koppe, C.; Egger, M.; Chan, L.K.; Schneider, A.T.; Küsgens, L.; Böhm, F.; Boege, Y.; Healy, M.E.; et al. JNK signaling prevents biliary cyst formation through a CASPASE-8-dependent function of RIPK1 during aging. Proc. Natl. Acad. Sci. USA 2021, 118, e2007194118. [Google Scholar] [CrossRef]
- Yin, F.; Jiang, T.; Cadenas, E. Metabolic triad in brain aging: Mitochondria, insulin/IGF-1 signalling and JNK signalling. Biochem. Soc. Trans. 2013, 41, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Sun, P.; Zhang, X.; Lin, G.; Xin, Q.; Niu, Y.; Chen, Y.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin Modulates Hypoxia-Induced Metastasis, Angiogenesis and Glycolysis by Decreasing HIF-1α Protein Synthesis via AKT/mTOR Pathway. Int. J. Mol. Sci. 2021, 22, 13336. [Google Scholar] [CrossRef]
- Samsonov, M.V.; Podkuychenko, N.V.; Khapchaev, A.Y.; Efremov, E.E.; Yanushevskaya, E.V.; Vlasik, T.N.; Lankin, V.Z.; Stafeev, I.S.; Skulachev, M.V.; Shestakova, M.V.; et al. AICAR Protects Vascular Endothelial Cells from Oxidative Injury Induced by the Long-Term Palmitate Excess. Int. J. Mol. Sci. 2021, 23, 211. [Google Scholar] [CrossRef]
- Varela, R.; Rauschert, I.; Romanelli, G.; Alberro, A.; Benech, J.C. Hyperglycemia and hyperlipidemia can induce morphophysiological changes in rat cardiac cell line. Biochem. Biophys. Rep. 2021, 26, 100983. [Google Scholar] [CrossRef]
- Jayarathne, H.S.M.; Debarba, L.K.; Jaboro, J.J.; Ginsburg, B.C.; Miller, R.A.; Sadagurski, M. Neuroprotective effects of Canagliflozin: Lessons from aged genetically diverse UM-HET3 mice. Aging Cell 2022, 21, e13653. [Google Scholar] [CrossRef]
- Snyder, J.M.; Casey, K.M.; Galecki, A.; Harrison, D.E.; Jayarathne, H.; Kumar, N.; Macchiarini, F.; Rosenthal, N.; Sadagurski, M.; Salmon, A.B.; et al. Canagliflozin retards age-related lesions in heart, kidney, liver, and adrenal gland in genetically heterogenous male mice. Geroscience 2023, 45, 385–397. [Google Scholar] [CrossRef]
- Biteau, B.; Karpac, J.; Hwangbo, D.; Jasper, H. Regulation of Drosophila lifespan by JNK signaling. Exp. Gerontol. 2011, 46, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Kanatsu-Shinohara, M.; Yamamoto, T.; Toh, H.; Kazuki, Y.; Kazuki, K.; Imoto, J.; Ikeo, K.; Oshima, M.; Shirahige, K.; Iwama, A.; et al. Aging of spermatogonial stem cells by Jnk-mediated glycolysis activation. Proc. Natl. Acad. Sci. USA 2019, 116, 16404–16409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ding, Y.; Sun, P.; Zhang, W.; Xin, Q.; Wang, N.; Niu, Y.; Chen, Y.; Luo, J.; Lu, J.; et al. Empagliflozin-Enhanced Antioxidant Defense Attenuates Lipotoxicity and Protects Hepatocytes by Promoting FoxO3a- and Nrf2-Mediated Nuclear Translocation via the CAMKK2/AMPK Pathway. Antioxidants 2022, 11, 799. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Papaconstantinou, J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. 2006, 20, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishitoh, H.; Saitoh, M.; Mochida, Y.; Takeda, K.; Nakano, H.; Rothe, M.; Miyazono, K.; Ichijo, H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 1998, 2, 389–395. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, W.; Shan, W.; Wan, F.; Luo, J.; Niu, Y.; Zhou, J.; Zhang, Y.; Xu, N.; Xie, W. Canagliflozin Delays Aging of HUVECs Induced by Palmitic Acid via the ROS/p38/JNK Pathway. Antioxidants 2023, 12, 838. https://doi.org/10.3390/antiox12040838
Hao W, Shan W, Wan F, Luo J, Niu Y, Zhou J, Zhang Y, Xu N, Xie W. Canagliflozin Delays Aging of HUVECs Induced by Palmitic Acid via the ROS/p38/JNK Pathway. Antioxidants. 2023; 12(4):838. https://doi.org/10.3390/antiox12040838
Chicago/Turabian StyleHao, Wenhui, Wenjie Shan, Fang Wan, Jingyi Luo, Yaoyun Niu, Jin Zhou, Yaou Zhang, Naihan Xu, and Weidong Xie. 2023. "Canagliflozin Delays Aging of HUVECs Induced by Palmitic Acid via the ROS/p38/JNK Pathway" Antioxidants 12, no. 4: 838. https://doi.org/10.3390/antiox12040838