
How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing

1
School of Molecular Biosciences, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow G12 8QQ, UK
2
Department of Molecular Biology and Biochemical Engineering, Universidad Pablo de Olavide, 41013 Seville, Spain
*
Author to whom correspondence should be addressed.
Antioxidants 2023, 12(4), 831; https://doi.org/10.3390/antiox12040831
Submission received: 28 February 2023
/
Revised: 16 March 2023
/
Accepted: 25 March 2023
/
Published: 29 March 2023
(This article belongs to the Special Issue The Ubiquitous and Multifaceted Coenzyme Q)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:In the past, mitochondrial reactive oxygen species (mtROS) were considered a byproduct of cellular metabolism. Due to the capacity of mtROS to cause oxidative damage, they were proposed as the main drivers of ageing and age-related diseases. Today, we know that mtROS are cellular messengers instrumental in maintaining cellular homeostasis. As cellular messengers, they are produced in specific places at specific times, and the intensity and duration of the ROS signal determine the downstream effects of mitochondrial redox signalling. We do not know yet all the processes for which mtROS are important, but we have learnt that they are essential in decisions that affect cellular differentiation, proliferation and survival. On top of causing damage due to their capacity to oxidize cellular components, mtROS contribute to the onset of degenerative diseases when redox signalling becomes dysregulated. Here, we review the best-characterized signalling pathways in which mtROS participate and those pathological processes in which they are involved. We focus on how mtROS signalling is altered during ageing and discuss whether the accumulation of damaged mitochondria without signalling capacity is a cause or a consequence of ageing.
1. Introduction
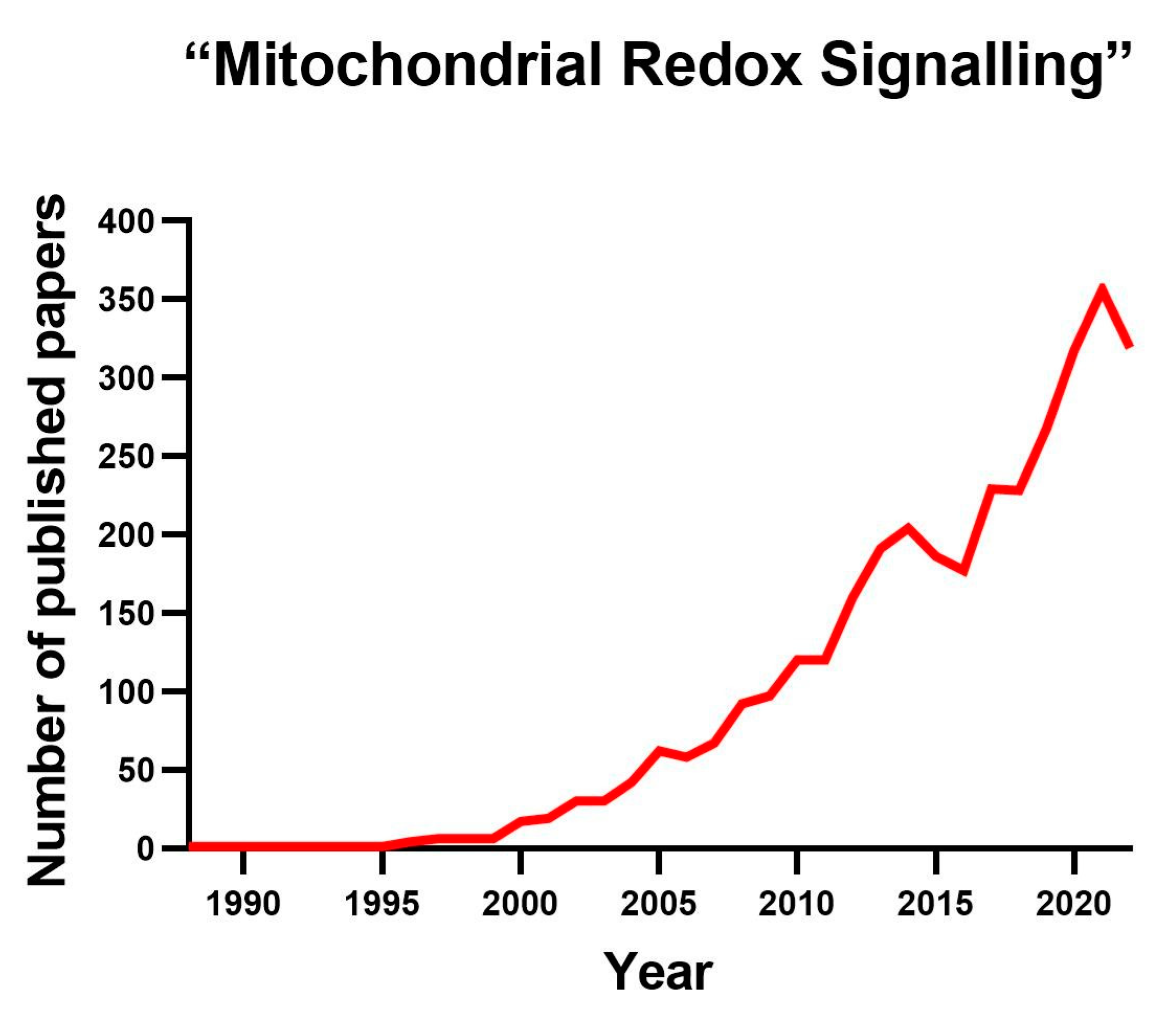
Reactive oxygen species (ROS) is an umbrella concept that includes multiple molecules resulting from the incomplete reduction of oxygen to water. The former occurs when oxygen is reduced with three, two or one electron [1]. ROS can attack other molecules, modifying their structure; by doing so, they change the way the modified molecule behaves [1]. While some ROS, such as superoxide, are free radicals, others, such as hydrogen peroxide, are not. Initially, ROS were considered toxic agents responsible for ageing and age-related diseases. This concept originated within the free radical theory of ageing framework, which presents ROS as simple byproducts of cellular metabolism [2,3]. However, we know that ROS are also messengers controlling critical cellular functions, and the interest in studying them has exponentially increased in recent years (Figure 1). ROS can be produced both externally and internally. Ultraviolet light, ionizing radiation and smoking are external sources of ROS, whereas internally, ROS are generated during inflammation, protein folding, fatty acid oxidation or mitochondrial respiration. Although ROS can be generated in various locations within a cell, this review will concentrate solely on ROS produced within the mitochondria, known as mitochondrial ROS (mtROS). We refer the reader to other excellent papers in which extra-mitochondrially produced ROS are exhaustively discussed [4,5,6,7].
Mitochondria are double-membrane organelles with their own DNA [8]. Within mitochondria, the generation of ROS is mainly associated with oxidative phosphorylation (OXPHOS). The mitochondrial electron transport chain (ETC) is canonically composed of respiratory complexes I-IV. As electrons move through the ETC, energy is released to move protons into the intermembrane space, creating an electrochemical gradient. Complex V then uses this gradient to convert ADP to ATP through a process called oxidative phosphorylation (OXPHOS) [9]. Mitochondrial ATP generation is essential for cell survival; however, ATP production is just one of the many functions performed by mitochondria. Most mitochondrial functions require a functional ETC to be completed. Calcium homeostasis and synthesis of iron–sulphur clusters and pyrimidines are just a couple of examples of the essential tasks performed by mitochondria coupled with OXPHOS [10]. For the ETC to work correctly, it is necessary to transfer electrons to the final electron acceptor, oxygen. When oxygen receives four electrons, it is completely reduced to water; however, when it receives one, two or three electrons, ROS are formed [1]. Superoxide results from the reduction of oxygen with one electron, the reduction of hydrogen peroxide with two electrons and the reduction of a hydroxyl radical with three electrons. In addition, the former ROS can further react with other molecules to produce more damaging chemicals, such as peroxynitrite [11].
Molecules that neutralize ROS are called antioxidants. However, the existence of oxidized proteins, lipids and DNA shows that antioxidants are not 100% efficient [12]. Because ROS are not only byproducts of metabolism, antioxidants are not only detoxifiers; they are also information transducers, allowing, for example, for the transformation of a superoxide signal into a hydrogen peroxide signal [13]. There are both enzymatic and non-enzymatic antioxidants [14]. Enzymatic antioxidants are specific against superoxide (superoxide dismutases) or peroxides (catalases, peroxiredoxins or glutathione peroxidases). In contrast, non-enzymatic antioxidants such as vitamin C or E are non-specific and intercept more damaging ROS such as hydroxyl radicals. The discovery of the enzyme superoxide dismutase confirmed the generation of ROS within the cell [15]. Superoxide dismutases dismutate superoxide to hydrogen peroxide. There are three different enzymatic systems dedicated to detoxifying hydrogen peroxide: (1) glutathione peroxidases, (2) peroxiredoxins and (3) catalases. Catalases decompose hydrogen peroxide to oxygen and water. Peroxide oxidizes glutathione peroxidases and peroxiredoxins, which are then reduced by thioredoxin and glutathione. Finally, NADPH, which is produced mainly by the pentose phosphate pathway, is the final electron donor for thioredoxin and glutathione regeneration as the last electron donor. The former shows that maintaining redox homeostasis requires constant metabolic adjustments [16].
One of the most important questions in redox biology is how ROS signals are transmitted. The most popular answer is that redox information is transmitted through the reversible oxidation of specific cysteine residues [17]. However, oxidative modifications of other amino acids, such as methionine, are also important [18]. Peroxiredoxins intercept most of the hydrogen peroxide produced by cells [19]. When catalase detoxifies hydrogen peroxide, the information is lost. However, oxidized peroxiredoxins can convey the information carried by hydrogen peroxide. Four models have been proposed to explain how ROS act as cellular messengers, as reviewed in [20]. In two of these models, peroxiredoxins are instrumental. The first model proposes direct interaction between ROS and the target protein, avoiding antioxidant interception. The second suggests binding the target protein to the generator of ROS. The third model, called the floodgate model, proposes the hyper-oxidation of peroxiredoxins as the mechanism required for transmission of the redox signal. Finally, the relay model states that (oxidized) peroxiredoxins directly oxidize the target protein, modifying its function. The last two models are the most supported by experimental evidence [21,22]. An example of the relay model occurs in Caenorhabditis elegans in response to metformin feeding [23]. Inhibition of CI by metformin triggers ROS production, causing the oxidation of PRDX-2 and subsequent activation (by the latest) of an MAPK cascade that prolongs the worms’ lifespan.
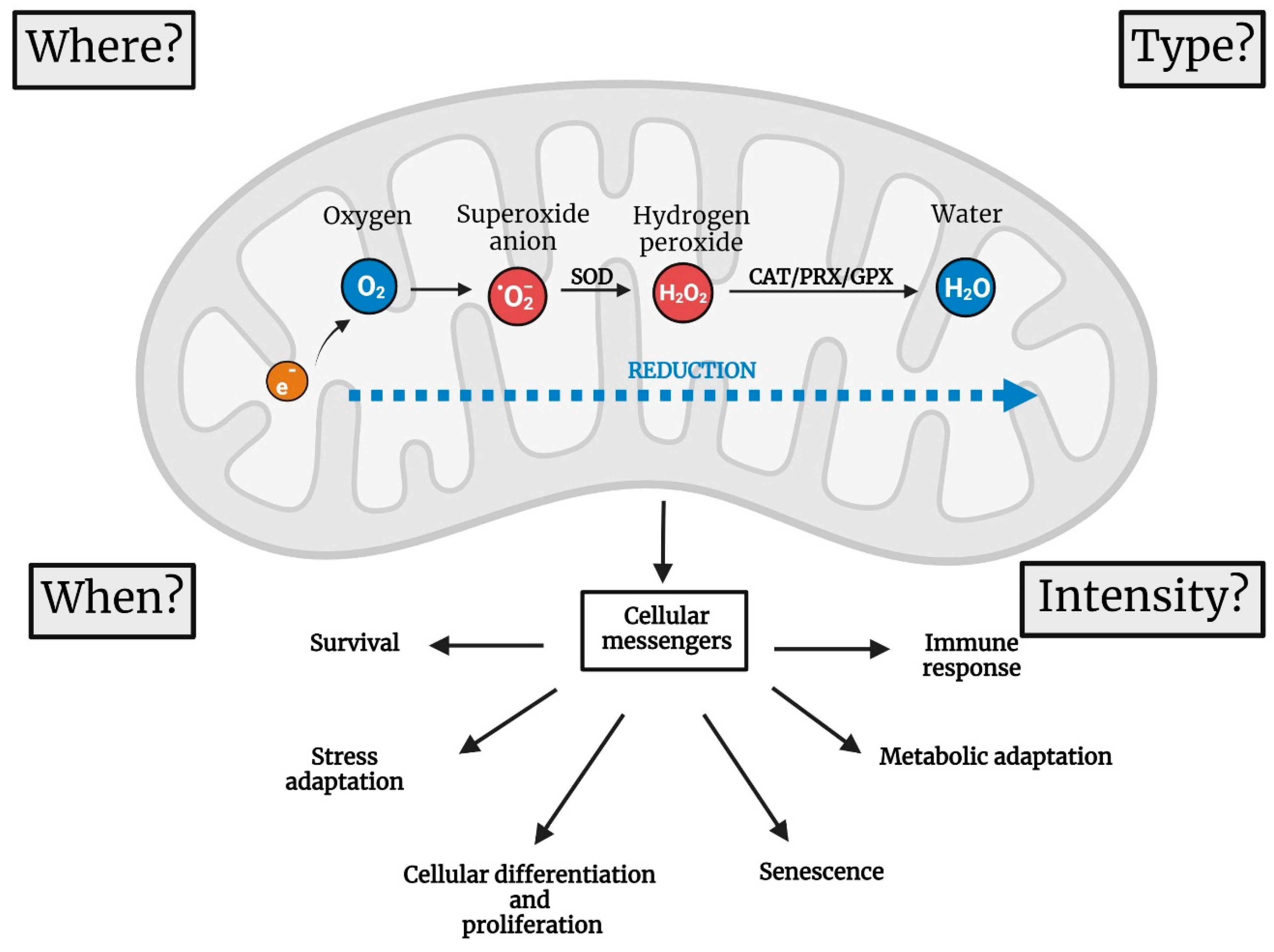
2. Four Parameters to Describe the Functions of mtROS
To understand the physiological role of mtROS, it is imperative to recognize them as signalling molecules that are generated to transmit information and exert an effect. Therefore, they can cause damage by triggering oxidative stress or defects in signalling processes. Since ROS can function as cellular messengers, it is important to consider four aspects when studying their roles (Figure 2): (1) where ROS are produced, (2) when ROS are generated, (3) the type of ROS and (4) the amount of ROS or intensity of the ROS signal. Studying ROS as signalling agents requires an appropriate experimental setup. Here, we do not pretend to revise the different ways of measuring mtROS or provide advice on performing such measurements. Therefore, we refer the reader to other excellent reviews and technical papers [1,24]. Nevertheless, we will briefly discuss how ROS are measured to provide the appropriate context for the rest of this review. For historical reasons, most information about how mitochondria work and produce ROS comes from studies using isolated mitochondria. Isolated mitochondria measurements provide a high level of resolution, allowing us to identify which respiratory complex generates the ROS. However, they lack physiological relevance (Sanz 2016). These experiments are also performed in the context of supraphysiological oxygen levels, causing overoxidation of mitochondrial components. High resolution is achieved using isolated mitochondria via the combination of specific substrates and inhibitors of different ETC complexes [25]. Working with intact cells increases the physiological relevance of mtROS experiments. However, the problem with supraphysiological oxygen levels remains the same unless it is adequately addressed within a normoxia chamber [26]. Furthermore, in vitro cell measurements do not provide sufficient resolution to identify the generator of ROS per se. Resolution is increased by using inhibitors similar to those employed in isolated mitochondria or a genetic approach to silence the ROS generator. For obvious reasons, the most informative measurements are those performed in vivo using animal models such as worms [27], flies [28] or mice [29]. Finally, mtROS measurements must be validated using different methods. Validation with antioxidants serves a dual purpose. First, it confirms the type of ROS being assessed. For example, superoxide dismutase overexpression reduces superoxide, while catalase diminishes hydrogen peroxide levels [30]. The physiological importance of the signal is confirmed if the analysed phenotype is modified accordingly due to the change in antioxidant levels [31].
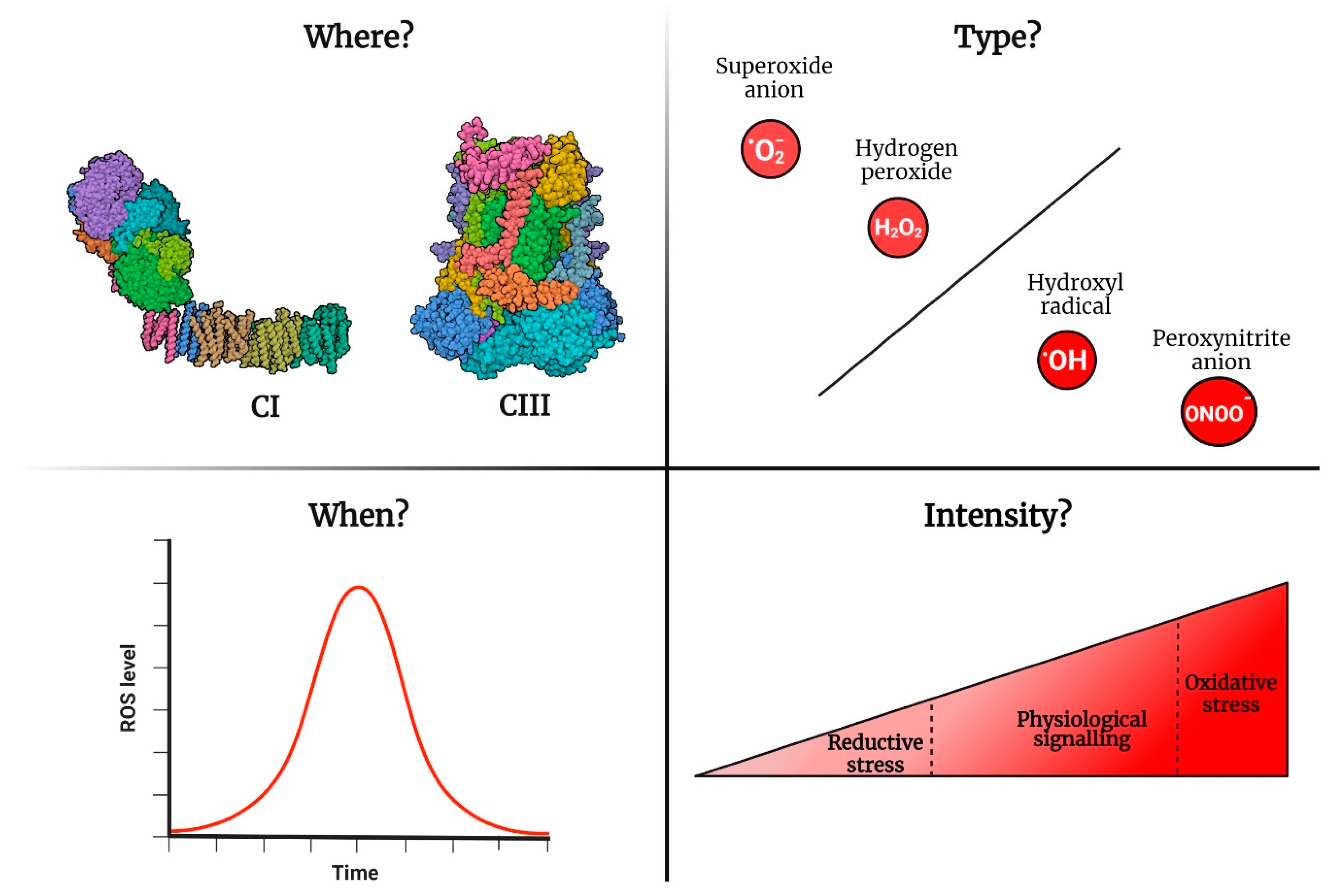
As mentioned before, the characterization of ROS signalling involves determining the intensity of the signal, in addition to when and where the signal is generated and the type of signal (superoxide, hydrogen peroxide, etc.). In the following sections, we will discuss examples of the importance of specific spatiotemporal ROS signalling in physiological and pathological situations and argue why ROS signalling becomes problematic during ageing (Figure 3).
2.1. Where Are mtROS Produced?
For many years, mitochondria were considered the main generator of ROS [32]. The former is supported by mitochondria consuming most of the oxygen cells use. However, a few recent studies have questioned the former [5,33,34]. According to these studies, mitochondria account for about 50% of the hydrogen peroxide that is released from a cell. The remaining 50% is produced by enzymes called NADPH oxidases (NOX). However, since the detection system was located outside the cell, the former experiments did not account for the hydrogen peroxide (or superoxide) intercepted by intracellular antioxidants. Accordingly, studies in yeast using genetically encoded hydrogen peroxide reporters (that detect hydrogen peroxide generated within the cell) showed higher levels of hydrogen peroxide in the mitochondria than in the cytosol [35,36]. Regrettably, these reports did not quantify the levels of hydrogen peroxide in other significant generators of ROS, such as peroxisomes, the endoplasmic reticulum and lysosomes. Therefore, it is not possible to conclude that mitochondria are the main ROS generator. In the future, we will have to quantify ROS generated simultaneously at different locations to determine their relative importance depending on the physiological conditions.
Even if mitochondria are not the main generators of ROS within the cell, mtROS play a significant role in redox signalling [37]. Up to 11 ROS generators have been found in isolated mitochondria [34]. However, most do not generate significant amounts of ROS in vivo. Under physiological conditions, complex I (CI) and complex III (CIII) are the main physiological ROS producers [31]. Complex II (CII) produces ROS only in pathological situations such as cancer or neurodegenerative diseases [38]. CI generates ROS in both the forward and reverse directions [39]. In the forward direction, ROS production depends on the redox state of the NAD(H) pool, whereas in the reverse direction, it depends on the redox state of the coenzyme Q (CoQ) pool and the proton motive force (pmf) [31,40]. The generation of CIII ROS relies on the entry of electrons through different mitochondrial dehydrogenases (e.g., CI and CII), the redox state of CoQ and the pmf [41,42]. Supporting a leading role of CI/CIII in ROS generation, specific suppressors of superoxide production in CI (S1QELs) and CIII (S3QELs) have been recently developed [43,44]. S1QELs and S3QELs do not alter electron flow or oxygen consumption but prevent the reduction of oxygen to superoxide by leaked electrons from the ETC. S1/3QELs represent convenient research tools with potential as therapeutic agents. Accordingly, they have demonstrated efficacy in protecting against excessive ROS levels in the heart, liver and gut [43,45].
When we discuss redox signalling, an additional layer of complexity is that mitochondria produce mtROS in different ways depending on the cell type. For example, mitochondria from glial cells generate more mtROS than neuronal mitochondria [46]. The former is caused by higher levels of free CI in glial cells. Furthermore, different types of mitochondria within a cell also generate ROS differently. For example, in rat muscle, subsarcolemmal mitochondria generate more hydrogen peroxide than interfibrillar mitochondria [47]. The variety in terms of how different cells and mitochondria produce ROS needs further study, since specific physiological and pathological processes are linked explicitly with mtROS produced by CI or CIII [31]. Furthermore, cellular differentiation is a process in which mtROS play a leading role. Accordingly, mtROS are essential to correctly differentiate the haematopoietic cells of flies [48]. Interestingly, some cell types require CI or CIII ROS to differentiate appropriately. CI ROS are specifically needed to transform myoblasts into myotubes [49]. During the differentiation process, myoblasts start to produce ROS via reverse electron transport (RET), and suppression of the former by rotenone stops the differentiation process. Adipocytes also use an mtROS signal to become fully differentiated [50]. However, in the case of adipocytes, the signal is produced by CIII, not CI. Accordingly, the knockdown of the CIII Rieske Fe-S subunit prevents ROS generation and stops the specialization of adipocytes.
Different aspects of the immune response seem to be controlled explicitly by ROS generated in CI or CIII [51]. For example, proinflammatory reprogramming of macrophages in response to infection requires CI ROS [52], while activation of T cells is reported to be mediated by CIII ROS [53]. As experiments implicating CI or CIII in ROS generation are typically conducted by different research teams using various experimental setups, we cannot rule out the possibility that ROS produced by other complexes may also be necessary (or sufficient). To determine the veracity of these findings, it is advisable to repeat these experiments using multiple approaches to inhibit ROS production by both CI and CIII under identical experimental conditions.
2.2. When Are ROS Produced?
As with all signals, ROS signals must be turned on and off. Due to practical constraints, ROS measurements are generally presented as static, single-time-point values rather than dynamic processes that evolve over time. Nonetheless, the literature offers instances of mtROS signalling whereby an mtROS signal is triggered by specific stimuli and is concluded when the stimulus subsides. The most conspicuous example is the adaptive response to cellular hypoxia. In response to hypoxia, CIII produces ROS instrumental in stabilizing HIF1a [54]. Chemical (with myxothiazol) or genetic suppression (using cytochrome b mutants or RNA interference against the Rieske protein) of mtROS prevents the adaptation response to hypoxia [55,56]. Once the initial hypoxic response (i.e., stabilization of HIF1a) is completed, the CIII ROS signal is turned off [57,58]. Another example of mediation by CI ROS is how the specialized cells of the carotid body detect and react to low levels of oxygen in the blood [59,60]. These chemoreceptor cells have a specific mitochondrial metabolism characterized by high succinate levels. The former favours ROS generation via RET in response to low oxygen concentration. ROS-RET activation leads to elevated respiration, which helps to restore normal oxygen levels in the bloodstream. Once this has been achieved, the signal ceases. Similarly, macrophage activation in response to infection uses a similar ROS-RET mechanism that is also terminated when the damage signals disappear from the tissue [52].
By no means does the signal’s duration determine the importance of the downstream processes triggered by it. For example, fly brain mitochondria produce an ROS-RET signal in response to thermal stress [61]. Although the signal only persists for a few hours, it is vital for stress adaptation. If the signal is disrupted, fly survival under stress is significantly reduced. Similarly, ROS levels during the early stages of worm development determine the duration of the adult lifespan [62]. Worms with higher levels of ROS during L2 are long-lived, and experimental manipulations that elevate ROS in this period extend their lifespan. This phenomenon is conserved in fruit flies, where exposure to oxidants during development increases longevity [63]. On the other hand, extending ROS signalling for too long has harmful consequences. An excellent example of the former is tissue repair, which is controlled by TGF-beta signalling. The former stimulates CIII ROS production, which is instrumental in completing the healing process in the lungs [64]. Preventing the generation of ROS impairs the repair activity. However, excessive or sustained CIII ROS signalling can result in fibrosis, highlighting the need to regulate ROS signalling with appropriate intensity and duration. Another example of the former is pulmonary vasoconstriction. Low oxygen levels trigger CIII ROS signalling in the lungs [65,66]. ROS signalling plays a crucial role in triggering the vasoconstriction response, which preserves the integrity of the lungs. Therefore, insufficient or excessive ROS signalling can compromise lung integrity and endanger the survival of the individual [67].
Finally, how is the ROS signal turned on and off? Although this is a question of crucial importance, it is one of the most understudied aspects of redox signalling. ROS-RET signalling is started by increasing the entry of electrons to the ETC. This is achieved by enhancing access via single or multiple entry points. An example of single-point entry is ischemia–reperfusion. During reperfusion, CII oxidizes the succinate accumulated during ischemia, triggering ROS-RET [68]. However, ROS-RET is activated by electrons introduced via CI, CII and other mitochondrial dehydrogenases (not part of the canonical ETC) in response to thermal stress in fly brain mitochondria [61]. Recently, it was reported that oocytes have negligible amounts of CI [69]. This was interpreted as a mechanism to prevent the accumulation of oxidative damage in cells that are extremely important for reproduction. However, preventing CI assembly could be a way to keep signalling inactive until it is required, i.e., after fertilization. Accordingly, the assembly of CI upon fertilization triggers mtROS signalling, which is instrumental in correctly initiating the developmental program [70].
An interesting new mechanism has been described to explain how CIII ROS signalling initiates the hypoxia response [58,71]. Hypoxia deactivates CI, leading to acidification of the mitochondrial matrix and the exchange of mitochondrial Ca2+ with cytosolic Na+. The increased Na+ levels within the mitochondrial matrix limit the mobility of CoQ, resulting in a longer semiquinone lifetime within CIII and the subsequent generation of superoxide [58]. Investigating whether this mechanism is replicated in other situations in which CIII-derived ROS play a crucial role is imperative. There are several ways to turn off mtROS signalling. Unfortunately, there are not many studies addressing this vital issue. One possibility is to reduce the entry of electrons into the ETC. This has been shown to be effective in cancelling ROS-RET and CIII ROS signalling [61,68,72]. Another alternative is to increase the levels of antioxidants, which eliminates the benefits conferred by increased mtROS levels during exercise in humans [73].
3. Pathological Role of mtROS
3.1. The Two Ways by Which ROS Cause Damage
There are two ways ROS cause cellular distress: dysregulation in redox signalling and oxidative damage. Aberrant redox signalling occurs when signals are not produced or are generated at the incorrect time/place or with the wrong intensity [74]. We presented a few examples of the former above. Damage is provoked by too much signalling or a lack thereof. Ablation of mitochondrial redox signalling in mouse astrocytes causes metabolic and behavioural alterations [29]. On the other hand, overexpression of the ATPase inhibitory factor 1 (IF1) in mouse neurons increases mtROS generation, boosting the learning skills of mice [75]. Conversely, knocking out IF1 impairs memorization and performance in different cognitive tests. The other way ROS cause harm is through oxidative damage. High concentrations of ROS trigger oxidative stress [76]. This happens when the concentration of oxidants exceeds that of antioxidants, leading to oxidative damage [11]. For instance, animals fed oxidants such as hydrogen peroxide or paraquat exemplify the former scenario and die as a result [77]. Likewise, in mice, SOD2 knockout is lethal during embryonic development [78]. Additionally, ischemia–reperfusion provides a more physiological example of oxidative stress culminating in cell death [79]. As we have seen, ROS-RET triggered by succinate accumulation during ischemia causes oxidative damage, subsequently triggering tissue damage [68]. Accordingly, preventing RET attenuates oxidative stress and preserves the function of the affected tissue [43,68,80,81]. Some cancers are caused by mutations in genes encoding CII subunits that increase mtROS levels [82,83]. Recent studies have revealed that ROS-RET is upregulated in cancer stem cells with aberrant NOTCH signalling, a critical factor in the growth of tumour cells in mouse and fly brain models. [84]. Tumour necrosis factor (TNF) is critical to protect cells against mycobacteria [85]. However, excessive TNF signalling triggers ROS-RET, which causes necrosis in macrophages infected by mycobacteria [86]. Accordingly, metformin inhibits both ROS-RET and macrophage death. These findings open the possibility of using metformin in combination with antituberculosis drugs to prevent the harmful effects of the latter.
Several diseases are associated with excessive CIII ROS. As previously noted, unchecked CIII ROS generation can lead to lung fibrosis [64]. Similarly, pollution causes thrombosis in the lungs by triggering CIII ROS [72,87]. Like CI ROS, CIII ROS play a crucial role in the proliferation and metastasis of specific tumour types, such as those regulated by KRAS signalling [88,89]. Again, it remains unclear whether CI/CIII ROS are responsible for the proliferation and metastasis of specific tumour cells or if the divergent outcomes are attributable to methodological differences in the execution of the experiments. Finally, some unidentified in vivo ROS generators may be crucial for other afflictions, since at least 11 sites produce superoxide in isolated mitochondria [90].
3.2. The Role of mtROS in Ageing
Damage caused by endogenously produced mitochondrial free radicals was initially proposed as the cause of ageing by Denham Harman [2,3]. In his famous “Mitochondrial Free Radical Theory of Ageing” (MFRTA), Harman proposed that mtROS produced as byproducts of metabolism cause the accumulation of oxidative damage responsible for ageing and age-related diseases. Over the years, the MFRTA has been supported by two types of evidence [91]: (1) observational studies describing the age-related accumulation of oxidative damage [92,93] and (2) correlative studies showing that mitochondria from long-lived animals produce fewer ROS [94,95]. While the theory was highly prevalent at the turn of the 21st century, it has since experienced a significant decline in popularity and is now considerably less popular than it once was [96,97]. The demise of MFRTA began when its forecasts were subjected to experimental verification. The first prediction states that increasing levels of antioxidants will extend lifespan and that, conversely, decreasing their concentration will shorten survival. Supplementing non-enzymatic antioxidants fails to increase longevity in most animal models [94]. Similarly, overexpression of enzymatic antioxidants reduces oxidative stress but does not extend the lifespan of mice [98,99]. An important exception is the targeted expression of catalase to mitochondria, significantly improving the median and maximum survival of mice [100]. However, the same strategy in fruit flies shortens lifespan [101] and causes deleterious alterations in mouse behaviour [29]. Unexpectedly, genetic depletion of most enzymatic antioxidants elevates oxidative damage without impacting mouse survival [102,103,104]. Nevertheless, there are some exceptions, such as knocking out SOD1 in mice or knocking down peroxiredoxins 3 or 5 in fruit flies [105,106]. However, these three enzymes are instrumental in redox signalling, and therefore, it is impossible to conclude whether the animals die because of the accumulation of oxidative damage, interruption in redox signalling or both.
The second prediction of MFRTA argues that decreasing mtROS levels must extend lifespan and that increasing mtROS generation must shorten survival. However, both in worms and flies, we find examples of experiments disproving both predictions. Ectopic expression of the alternative oxidase (AOX) from Ciona intestinalis in Drosophila mitochondria reduces mtROS but does not extend lifespan [107,108]. In Caenorhabditis elegans, it is possible to increase mtROS production by using inhibitors of the ETC or knocking down subunits of the respiratory complexes. Both types of interruptions in electron flow increase longevity [109]. To the best of our knowledge, ETC inhibitors have not been reported to extend the lifespan of fruit flies, although inhibition of CV does (see below). Surprisingly, feeding rotenone to killifish extends longevity by 15% [110]. In flies, the knockdown of CI, CIII, CIV and CV extends survival [111]. The most consistent results are obtained by knocking down CI subunits, where lifespan is prolonged by an mtROS-dependant mechanism [112]. However, a recent report showed a reduction in mtROS associated with CI depletion in Drosophila flight muscle and extended lifespan [113]. Extending the lifespan of Drosophila is achieved by expressing the internal alternative NADH dehydrogenase 1 (NDI1) from Saccharomyces cerevisiae [114,115]. NDI1 promotes longevity, stimulating the generation of mtROS via RET. Similarly, several reports show that chemical or genetic inhibition of ATP synthesis by blocking CV increases mtROS levels and the survival of fruit flies and worms [116,117,118,119]. In summary, all the former results do not support the role of mtROS limiting lifespan per se. Conversely, they indicate that adequate mitochondrial redox signalling is essential for a healthy lifespan. As we will see next, mitochondrial redox signalling becomes a problem for aged individuals.
3.3. Accumulation of Damaged Mitochondria during Ageing Causes an Interruption in Mitochondrial Redox Signalling
We have seen that mitochondrial redox signalling is a sophisticated communication system that becomes a source of damage during ageing. Controlled and regulated occurrence of ROS-RET and CIII ROS production requires specific conditions [40]. For example, ROS-RET requires the appropriate levels of reduction of the CoQ pool and a sufficiently high pmf to provide the necessary energy for RET [120]. In the ageing process, a significant concern arises due to the difficulty experience by old mitochondria in maintaining a high mitochondrial membrane potential [121]. Thus, the age-related decline in the NAD+-to-NADH ratio can potentially modify how CI generates ROS [122]. Furthermore, the depletion of NAD+ has the potential to impact the activity of SOD2, as it necessitates deacetylation by SIRT3 for activation, and SIRT3, in turn, is reliant on NAD+ [123]. This may also impede the transformation of superoxide signals into more stable hydrogen peroxide signals, thereby modifying the cellular signalling process. Initiating the CIII signalling process requires an influx of electrons, while a proper efflux is necessary to conclude the process [31]. Therefore, any process that alters the entry or exit of electrons, such as those associated with ageing, as described below, also modifies CIII redox signalling. In conclusion, it is unsurprising that alterations in the mitochondrial electron flow caused by the disruption in the OXPHOS machinery profoundly affect redox signalling [48,124]. Accordingly, one of the hallmarks of ageing is the accumulation of dysfunctional mitochondria that generate fewer ATP and higher levels of mtROS [125].
Reduced mitochondrial oxygen consumption is a common characteristic of ageing found in multiple animal species [28,126,127,128]. The referred decrease is related to problems with both the entry (due to problems with CI) and the exit of electrons (caused by issues with CIV) [28,129,130,131,132]. Age-related reduction in the levels of CI has been reported in the human cortex [133], naked mole rat skeletal muscle [134]) and fruit fly mitochondria [135]. However, other studies inform of a preferential decrease in CIV activity correlated with high levels of mtROS in flies and mouse adipocytes [131,136]. Reducing the entry of electrons can impede the generation of ROS signals [30], while blocking their exit results in the cessation of ROS signalling due to the ongoing production of uncontrolled ROS [61]. Interestingly, the opposite also occurs, i.e., increased mitochondrial oxygen consumption that disrupts mitochondrial redox signalling during ageing. The former happens during cellular senescence when an expansion in mitochondrial mass increases both cellular oxygen consumption and levels of ROS [137]. In senescence cells, mtROS reprogram the cell, causing DNA damage and releasing proinflammatory cytokines such as IL-6. Accordingly, strategies that reduce either the number of mitochondria or the amount of ROS decrease DNA damage and the senescence-associated secretory phenotype [138]. All the examples presented above indicate that age-related alterations in mitochondrial respiration disrupt physiological redox signalling. Notably, modifications in redox signalling can arise through different mechanisms, which can entail increments and decrements in oxygen consumption. A significant challenge for future research is to conduct tissue-specific analysis of mitochondrial function, investigate how these alterations affect redox signalling and explore the potential consequences for the cellular processes regulated by mtROS.
Numerous examples in the literature demonstrate age-related changes in mitochondrial redox signalling, leading to severe disruptions in cellular homeostasis. In the fly brain and muscle, there is a significant increase in the levels of mtROS during ageing [28,61,113,126]. However, old and young brain mitochondria produce ROS in different ways. Young brain mitochondria have low levels of mtROS that are increased in response to specific types of stress. In contrast, old mitochondria continuously produce high levels of ROS and are less responsive to external stimuli [61,62]. In young flies, blocking the exit of electrons by knocking down subunits of CIV increases mtROS and causes interruptions in redox signalling. However, interventions targeting the entry of electrons (by knocking down CI subunits) prevent the generation of mtROS signals without increasing the amount of ROS [61,113]. A separate study reported an increase in mtROS in the flight muscle of Drosophila, which is crucial for extending lifespan resulting from CI depletion [112]. Along the same line, the knockout of the CIV assembly factor COX15 activates an ROS-dependent adaptation program in mouse muscle [124]. Interruption of this program by expressing AOX damages the antistress response and shortens survival. Similarly, in Drosophila flies, blocking mitochondrial redox signalling by expressing a mitochondrially targeted catalase damages the antistress response [30]. Conversely, boosting it by overexpressing SOD2 extends survival under stress and non-stress conditions [30,139]. Similarly, ectopic catalase expression in the mitochondria of astrocytes interrupts normal redox signalling, disrupting brain activity [29]. The former data indicate that young mitochondria produce low basal ROS levels and only increase ROS production in response to specific stimuli. Once the stress situation is over, mtROS levels return to normal. However, old mitochondria continually produce ROS and are unresponsive to stress (Figure 4). A recent paper showed that old fly mitochondria generate more ROS, triggering RET in CI [113]. Accordingly, restricting the inhibition of ROS-RET to flies older than 20 days using a novel, specific CI blocker (6-chloro-3-(2,4-dichloro-5-methoxyphenyl)-2-mecapto-7-methoxyquinazolin-4(3H)-one) leads to an extension of longevity. However, other manuscripts reported that ROS-RET extends fly lifespan [28], is instrumental for stress adaptation [30] and does not occur in the brain of flies older than 25 days [61]. Therefore, although different data support the loss of redox signalling during ageing, more studies are required to clarify how the former occurs mechanistically. An essential aspect of elucidating this mechanism is whether identical mechanisms generate ROS in both young and old mitochondria and whether there is a shift in the distribution of low- and highly reactive oxygen species in older individuals. Increased concentrations of hydroxyl radicals, peroxynitrite and other highly reactive free radicals may account for the accumulation of oxidative damage observed in aged individuals. Such an increase in the levels of aggressive ROS could be attributed to the liberation of iron and other metals from the catalytic centres of mitochondrial enzymes due to the amplified intensity of ROS signalling [140].
The proposed model presented in the previous figure (Figure 4) has not been experimentally verified, and there is indirect supporting and opposing evidence. For example, disruption of redox signalling explains why boosting antioxidants fails to positively impact lifespan in animal models [32] or human health in clinical trials [141]. Antioxidants neutralize oxidative damage but cannot restore mitochondrial redox signalling. The transcriptome of old flies presents many similarities with the transcriptome of flies exposed to hyperoxia [142], indicating an increase in the oxidation state of fly cells during ageing. This observation supports a model in which the interruption of redox signalling induces oxidative stress. However, the redoxome of neither flies nor mice consistently shows an increase in the number of oxidized cysteines in aged individuals. [77,143]. The former is surprising, considering the age-related decrease in reduced glutathione in insects and rodents [144,145]. On the one hand, the lack of consistent age-related modifications in the redoxome does not support a generalized interruption of redox signalling caused by mitochondrial dysfunction. On the other hand, specific tissues in mice exhibit age-related modifications in highly reactive cysteines in proteins associated with age-related diseases [143]. These specific modifications could account for the changes observed in the transcriptome of Drosophila without significant alterations in the redoxome. The consequences of losing redox signalling at the onset of ageing and age-related diseases deserve further investigation. Future studies must consider ROS as signals, not as simple metabolic byproducts. The majority of ROS-regulated processes discussed earlier have not been investigated in the context of ageing, a process during which mitochondrial function is notably impaired. This is not surprising, given that redox signalling remains a developing area of research, with limited studies dedicated to investigating its role in ageing, particularly in mammals. For example, an appropriate generation of mtROS is required to trigger sleep in fruit flies [146]. Accordingly, overexpression of catalase or AOX reduces sleep in transgenic flies, likely due to interruptions in redox signalling. However, it remains to be seen whether the age-related increase in mtROS generation is involved in disrupting sleep patterns in old animals [147]. To employ manipulations in redox signalling as a therapeutic strategy, it is crucial to first determine how the interruption of redox signalling affects ROS-regulated processes in both young and aged individuals. Second, it is essential to understand the underlying mechanisms driving changes in redox signalling and whether restoring redox signalling could potentially promote healthy ageing in humans.
Author Contributions
Writing—original draft preparation, A.S.; writing—review and editing, B.C.-V., M.D.C. & A.S. All authors have read and agreed to the published version of the manuscript.
Funding
A. Sanz is a Wellcome Trust Senior Research Fellow (212241_A_18_Z), and the research conducted in Sanz lab that was partly covered in this review is supported by BBSRC grants (BB/W006774/1, BB/W002892/1, BB/R008167/2). The APC was funded by Wellcome/BBSRC.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No original data was used in this review.
Acknowledgments
The figures presented in this review were generated with the assistance of Biorender.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AOX | Alternative oxidase |
| CoQ | Coenzyme Q |
| CI | Complex I |
| CII | Complex II |
| CIII | Complex III |
| CIV | Complex IV |
| CV | Complex V |
| ETC | Electron transport chain |
| HIF1a | Hypoxia-inducible factor 1-alpha |
| IF1 | ATPase inhibitory factor 1 |
| IL-6 | Interleukin 6 |
| MAPK | Mitogen-activated protein kinase |
| MFRTA | The mitochondrial free radical theory of ageing |
| mROS | Mitochondrial reactive oxygen species |
| NDI1 | NADH dehydrogenase internal 1 |
| NOTCH | Neurogenic locus notch homolog protein |
| NOX | NADPH oxidase |
| OXPHOS | Oxidative phosphorylation |
| PRDX2 | Peroxiredoxin-2 |
| pmf | Proton motive force |
| RET | Reverse electron transport |
| S1QELs | Suppressors of superoxide production in CI |
| S3QELs | Suppressors of superoxide production in CIII |
| SOD1 | Superoxide dismutase-1 |
| SOD2 | Superoxide dismutase-2 |
| TGF-beta | Transforming growth factor beta |
| TNF | Tumour necrosis factor |
References
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, Y.; Gerencser, A.A.; Brand, M.D. Effects of sugars, fatty acids and amino acids on cytosolic and mitochondrial hydrogen peroxide release from liver cells. Free Radic. Biol. Med. 2022, 188, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [Green Version]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Gorlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [Green Version]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Luzikov, V.N. Principles of control over formation of structures responsible for respiratory functions of mitochondria. Biochemistry 2009, 74, 1443–1456. [Google Scholar] [CrossRef]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flora, S.J. Structural, chemical and biological aspects of antioxidants for strategies against metal and metalloid exposure. Oxid. Med. Cell. Longev. 2009, 2, 191–206. [Google Scholar] [CrossRef] [Green Version]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.J.; Dong, W.; Stephanopoulos, G.N.; Sikes, H.D. Oxidative pentose phosphate pathway and glucose anaplerosis support maintenance of mitochondrial NADPH pool under mitochondrial oxidative stress. Bioeng. Transl. Med. 2020, 5, e10184. [Google Scholar] [CrossRef]
- Lennicke, C.; Cocheme, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Kaya, A.; Lee, B.C.; Gladyshev, V.N. Regulation of protein function by reversible methionine oxidation and the role of selenoprotein MsrB1. Antioxid. Redox Signal. 2015, 23, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Nordzieke, D.E.; Medrano-Fernandez, I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants 2018, 7, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2017, 14, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Vivancos, A.P.; Castillo, E.A.; Biteau, B.; Nicot, C.; Ayte, J.; Toledano, M.B.; Hidalgo, E. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 8875–8880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Sanz, A.; Stefanatos, R.; McIlroy, G. Production of reactive oxygen species by the mitochondrial electron transport chain in Drosophila melanogaster. J. Bioenerg. Biomembr. 2010, 42, 135–142. [Google Scholar] [CrossRef]
- Keeley, T.P.; Mann, G.E. Defining Physiological Normoxia for Improved Translation of Cell Physiology to Animal Models and Humans. Physiol. Rev. 2019, 99, 161–234. [Google Scholar] [CrossRef] [Green Version]
- Maremonti, E.; Eide, D.M.; Rossbach, L.M.; Lind, O.C.; Salbu, B.; Brede, D.A. In vivo assessment of reactive oxygen species production and oxidative stress effects induced by chronic exposure to gamma radiation in Caenorhabditis elegans. Free. Radic. Biol. Med. 2020, 152, 583–596. [Google Scholar] [CrossRef]
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef]
- Scialo, F.; Sriram, A.; Stefanatos, R.; Spriggs, R.V.; Loh, S.H.Y.; Martins, L.M.; Sanz, A. Mitochondrial complex I derived ROS regulate stress adaptation in Drosophila melanogaster. Redox Biol. 2020, 32, 101450. [Google Scholar] [CrossRef]
- Scialo, F.; Sanz, A. Coenzyme Q redox signalling and longevity. Free Radic. Biol. Med. 2021, 164, 187–205. [Google Scholar] [CrossRef]
- Sanz, A.; Pamplona, R.; Barja, G. Is the mitochondrial free radical theory of aging intact? Antioxid. Redox Signal. 2006, 8, 582–599. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wong, H.S.; Brand, M.D. Production of superoxide and hydrogen peroxide in the mitochondrial matrix is dominated by site IQ of complex I in diverse cell lines. Redox Biol. 2020, 37, 101722. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.S.; Watson, M.A.; Wong, H.S.; Orr, A.L.; Brand, M.D. The use of site-specific suppressors to measure the relative contributions of different mitochondrial sites to skeletal muscle superoxide and hydrogen peroxide production. Redox Biol. 2020, 28, 101341. [Google Scholar] [CrossRef]
- de Cubas, L.; Pak, V.V.; Belousov, V.V.; Ayte, J.; Hidalgo, E. The Mitochondria-to-Cytosol H2O2 Gradient Is Caused by Peroxiredoxin-Dependent Cytosolic Scavenging. Antioxidants 2021, 10, 731. [Google Scholar] [CrossRef] [PubMed]
- Carmona, M.; de Cubas, L.; Bautista, E.; Moral-Blanch, M.; Medrano-Fernandez, I.; Sitia, R.; Boronat, S.; Ayte, J.; Hidalgo, E. Monitoring cytosolic H2O2 fluctuations arising from altered plasma membrane gradients or from mitochondrial activity. Nat. Commun. 2019, 10, 4526. [Google Scholar] [CrossRef] [Green Version]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2014, 33C, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. Commun. Free Radic. Res. 2020, 25, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Treberg, J.R.; Perevoshchikova, I.V.; Orr, A.L.; Brand, M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic. Biol. Med. 2012, 53, 1807–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Mezera, V.; Dighe, P.; Melov, S.; Gerencser, A.A.; Sweis, R.F.; Pliushchev, M.; Wang, Z.; Esbenshade, T.; McKibben, B.; et al. Superoxide produced by mitochondrial site IQ inactivates cardiac succinate dehydrogenase and induces hepatic steatosis in Sod2 knockout mice. Free Radic. Biol. Med. 2021, 164, 223–232. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolanos, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Judge, S.; Jang, Y.M.; Smith, A.; Selman, C.; Phillips, T.; Speakman, J.R.; Hagen, T.; Leeuwenburgh, C. Exercise by lifelong voluntary wheel running reduces subsarcolemmal and interfibrillar mitochondrial hydrogen peroxide production in the heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1564–R1572. [Google Scholar] [CrossRef] [Green Version]
- Owusu-Ansah, E.; Yavari, A.; Mandal, S.; Banerjee, U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 2008, 40, 356–361. [Google Scholar] [CrossRef]
- Lee, S.; Tak, E.; Lee, J.; Rashid, M.A.; Murphy, M.P.; Ha, J.; Kim, S.S. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 2011, 21, 817–834. [Google Scholar] [CrossRef] [Green Version]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernansanz-Agustin, P.; Ramos, E.; Navarro, E.; Parada, E.; Sanchez-Lopez, N.; Pelaez-Aguado, L.; Cabrera-Garcia, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Pina, T.; Moreno, L.; Izquierdo-Alvarez, A.; Cabrera-Garcia, J.D.; Cortes, A.; et al. Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291. [Google Scholar] [CrossRef]
- Fernandez-Aguera, M.C.; Gao, L.; Gonzalez-Rodriguez, P.; Pintado, C.O.; Arias-Mayenco, I.; Garcia-Flores, P.; Garcia-Perganeda, A.; Pascual, A.; Ortega-Saenz, P.; Lopez-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Arias-Mayenco, I.; Gonzalez-Rodriguez, P.; Torres-Torrelo, H.; Gao, L.; Fernandez-Aguera, M.C.; Bonilla-Henao, V.; Ortega-Saenz, P.; Lopez-Barneo, J. Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab. 2018, 28, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Graham, C.; Stefanatos, R.; Yek, A.E.H.; Spriggs, R.V.; Loh, S.H.Y.; Uribe, A.H.; Zhang, T.; Martins, L.M.; Maddocks, O.D.K.; Scialo, F.; et al. Mitochondrial ROS signalling requires uninterrupted electron flow and is lost during ageing in flies. Geroscience 2022, 44, 1961–1974. [Google Scholar] [CrossRef] [PubMed]
- Bazopoulou, D.; Knoefler, D.; Zheng, Y.; Ulrich, K.; Oleson, B.J.; Xie, L.; Kim, M.; Kaufmann, A.; Lee, Y.T.; Dou, Y.; et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 2019, 576, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Obata, F.; Fons, C.O.; Gould, A.P. Early-life exposure to low-dose oxidants can increase longevity via microbiome remodelling in Drosophila. Nat. Commun. 2018, 9, 975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Marks, J.D.; Guzy, R.D.; Mungai, P.T.; Schriewer, J.M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 2013, 187, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Sommer, N.; Alebrahimdehkordi, N.; Pak, O.; Knoepp, F.; Strielkov, I.; Scheibe, S.; Dufour, E.; Andjelkovic, A.; Sydykov, A.; Saraji, A.; et al. Bypassing mitochondrial complex III using alternative oxidase inhibits acute pulmonary oxygen sensing. Sci. Adv. 2020, 6, eaba0694. [Google Scholar] [CrossRef] [Green Version]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Nuevo, A.; Torres-Sanchez, A.; Duran, J.M.; De Guirior, C.; Martinez-Zamora, M.A.; Boke, E. Oocytes maintain ROS-free mitochondrial metabolism by suppressing complex I. Nature 2022, 607, 756–761. [Google Scholar] [CrossRef]
- Han, Y.; Ishibashi, S.; Iglesias-Gonzalez, J.; Chen, Y.; Love, N.R.; Amaya, E. Ca2+-Induced Mitochondrial ROS Regulate the Early Embryonic Cell Cycle. Cell Rep. 2018, 22, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Hernansanz-Agustin, P.; Enriquez, J.A. Sodium in Mitochondrial Redox Signaling. Antioxid. Redox Signal. 2022, 37, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Soberanes, S.; Misharin, A.V.; Jairaman, A.; Morales-Nebreda, L.; McQuattie-Pimentel, A.C.; Cho, T.; Hamanaka, R.B.; Meliton, A.Y.; Reyfman, P.A.; Walter, J.M.; et al. Metformin Targets Mitochondrial Electron Transport to Reduce Air-Pollution-Induced Thrombosis. Cell Metab. 2019, 29, 335–347.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Esparza-Molto, P.B.; Romero-Carraminana, I.; Nunez de Arenas, C.; Pereira, M.P.; Blanco, N.; Pardo, B.; Bates, G.R.; Sanchez-Castillo, C.; Artuch, R.; Murphy, M.P.; et al. Generation of mitochondrial reactive oxygen species is controlled by ATPase inhibitory factor 1 and regulates cognition. PLoS Biol. 2021, 19, e3001252. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [Green Version]
- Menger, K.E.; James, A.M.; Cocheme, H.M.; Harbour, M.E.; Chouchani, E.T.; Ding, S.; Fearnley, I.M.; Partridge, L.; Murphy, M.P. Fasting, but Not Aging, Dramatically Alters the Redox Status of Cysteine Residues on Proteins in Drosophila melanogaster. Cell Rep. 2015, 11, 1856–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Oliver, C.N.; Starke-Reed, P.E.; Stadtman, E.R.; Liu, G.J.; Carney, J.M.; Floyd, R.A. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5144–5147. [Google Scholar] [CrossRef] [Green Version]
- Beach, T.E.; Prag, H.A.; Pala, L.; Logan, A.; Huang, M.M.; Gruszczyk, A.V.; Martin, J.L.; Mahbubani, K.; Hamed, M.O.; Hosgood, S.A.; et al. Targeting succinate dehydrogenase with malonate ester prodrugs decreases renal ischemia reperfusion injury. Redox Biol. 2020, 36, 101640. [Google Scholar] [CrossRef]
- Martin, J.L.; Costa, A.S.H.; Gruszczyk, A.V.; Beach, T.E.; Allen, F.M.; Prag, H.A.; Hinchy, E.C.; Mahbubani, K.; Hamed, M.; Tronci, L.; et al. Succinate accumulation drives ischaemia-reperfusion injury during organ transplantation. Nat. Metab. 2019, 1, 966–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Yasuda, K.; Akatsuka, A.; Hino, O.; Hartman, P.S.; Ishii, N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005, 65, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, N.; Long, X.; Zhao, J.; Yang, J.; Du, X.; Yang, T.; Yuan, P.; Huang, X.; Zhang, J.; et al. SDHC-related deficiency of SDH complex activity promotes growth and metastasis of hepatocellular carcinoma via ROS/NFkappaB signaling. Cancer Lett. 2019, 461, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Tantray, I.; Rimal, S.; Mitra, S.; Cheshier, S.; Lu, B. Regulation of reverse electron transfer at mitochondrial complex I by unconventional Notch action in cancer stem cells. Dev. Cell 2022, 57, 260–276.e9. [Google Scholar] [CrossRef]
- Chen, W.L.; Sheu, J.R.; Chen, R.J.; Hsiao, S.H.; Hsiao, C.J.; Chou, Y.C.; Chung, C.L.; Hsiao, G. Mycobacterium tuberculosis Upregulates TNF-alpha Expression via TLR2/ERK Signaling and Induces MMP-1 and MMP-9 Production in Human Pleural Mesothelial Cells. PLoS ONE 2015, 10, e0137979. [Google Scholar] [CrossRef] [Green Version]
- Roca, F.J.; Whitworth, L.J.; Prag, H.A.; Murphy, M.P.; Ramakrishnan, L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science 2022, 376, eabh2841. [Google Scholar] [CrossRef]
- Soberanes, S.; Urich, D.; Baker, C.M.; Burgess, Z.; Chiarella, S.E.; Bell, E.L.; Ghio, A.J.; De Vizcaya-Ruiz, A.; Liu, J.; Ridge, K.M.; et al. Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J. Biol. Chem. 2009, 284, 2176–2186. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Barja, G. Free radicals and aging. Trends Neurosci. 2004, 27, 595–600. [Google Scholar] [CrossRef]
- Hohn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; Konig, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Ku, H.H.; Agarwal, S.; Forster, M.J.; Lal, H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech. Ageing Dev. 1994, 74, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barja, G.; Herrero, A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Sanz, A.; Stefanatos, R.K. The mitochondrial free radical theory of aging: A critical view. Curr. Aging Sci. 2008, 1, 10–21. [Google Scholar] [CrossRef]
- Gems, D.; Doonan, R. Antioxidant defense and aging in C. elegans: Is the oxidative damage theory of aging wrong? Cell Cycle 2009, 8, 1681–1687. [Google Scholar] [CrossRef] [Green Version]
- Perez, V.I.; Van Remmen, H.; Bokov, A.; Epstein, C.J.; Vijg, J.; Richardson, A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 2009, 8, 73–75. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.C.; Perez, V.I.; Song, W.; Lustgarten, M.S.; Salmon, A.B.; Mele, J.; Qi, W.; Liu, Y.; Liang, H.; Chaudhuri, A.; et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [Green Version]
- Mockett, R.J.; Bayne, A.C.; Kwong, L.K.; Orr, W.C.; Sohal, R.S. Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radic. Biol. Med. 2003, 34, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genom. 2003, 16, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ikeno, Y.; Qi, W.; Chaudhuri, A.; Li, Y.; Bokov, A.; Thorpe, S.R.; Baynes, J.W.; Epstein, C.; Richardson, A.; et al. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1212–1220. [Google Scholar] [CrossRef]
- Jang, Y.C.; Remmen, V.H. The mitochondrial theory of aging: Insight from transgenic and knockout mouse models. Exp. Gerontol. 2009, 44, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Radyuk, S.N.; Rebrin, I.; Klichko, V.I.; Sohal, B.H.; Michalak, K.; Benes, J.; Sohal, R.S.; Orr, W.C. Mitochondrial peroxiredoxins are critical for the maintenance of redox state and the survival of adult Drosophila. Free Radic. Biol. Med. 2010, 49, 1892–1902. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Ayala, D.J.; Sanz, A.; Vartiainen, S.; Kemppainen, K.K.; Babusiak, M.; Mustalahti, E.; Costa, R.; Tuomela, T.; Zeviani, M.; Chung, J.; et al. Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab. 2009, 9, 449–460. [Google Scholar] [CrossRef]
- Sanz, A.; Fernandez-Ayala, D.J.; Stefanatos, R.K.; Jacobs, H.T. Mitochondrial ROS production correlates with, but does not directly regulate lifespan in Drosophila. Aging 2010, 2, 200–223. [Google Scholar] [CrossRef]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, M.; Priebe, S.; Groth, M.; Hartmann, N.; Menzel, U.; Pandolfini, L.; Koch, P.; Felder, M.; Ristow, M.; Englert, C.; et al. Longitudinal RNA-Seq Analysis of Vertebrate Aging Identifies Mitochondrial Complex I as a Small-Molecule-Sensitive Modifier of Lifespan. Cell Syst. 2016, 2, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Copeland, J.M.; Cho, J.; Lo, T., Jr.; Hur, J.H.; Bahadorani, S.; Arabyan, T.; Rabie, J.; Soh, J.; Walker, D.W. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 2009, 19, 1591–1598. [Google Scholar] [CrossRef] [Green Version]
- Owusu-Ansah, E.; Song, W.; Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 2013, 155, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimal, S.; Tantray, I.; Li, Y.; Pal Khaket, T.; Li, Y.; Bhurtel, S.; Li, W.; Zeng, C.; Lu, B. Reverse electron transfer is activated during aging and contributes to aging and age-related disease. EMBO Rep. 2023, e55548. [Google Scholar] [CrossRef]
- Sanz, A.; Soikkeli, M.; Portero-Otin, M.; Wilson, A.; Kemppainen, E.; McIlroy, G.; Ellila, S.; Kemppainen, K.K.; Tuomela, T.; Lakanmaa, M.; et al. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc. Natl. Acad. Sci. USA 2010, 107, 9105–9110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahadorani, S.; Cho, J.; Lo, T.; Contreras, H.; Lawal, H.O.; Krantz, D.E.; Bradley, T.J.; Walker, D.W. Neuronal expression of a single-subunit yeast NADH-ubiquinone oxidoreductase (Ndi1) extends Drosophila lifespan. Aging Cell 2010, 9, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahadorani, S.; Hur, J.H.; Lo, T., Jr.; Vu, K.; Walker, D.W. Perturbation of mitochondrial complex V alters the response to dietary restriction in Drosophila. Aging Cell 2010, 9, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wheeler, C.T.; Yolitz, J.; Laslo, M.; Alberico, T.; Sun, Y.; Song, Q.; Zou, S. A mitochondrial ATP synthase subunit interacts with TOR signaling to modulate protein homeostasis and lifespan in Drosophila. Cell Rep. 2014, 8, 1781–1792. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, J.; Currais, A.; Prior, M.; Fischer, W.; Chiruta, C.; Ratliff, E.; Daugherty, D.; Dargusch, R.; Finley, K.; Esparza-Molto, P.B.; et al. The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell 2018, 17, e12715. [Google Scholar] [CrossRef]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.A.; Cerutti, R.; Beninca, C.; Brea-Calvo, G.; Jacobs, H.T.; Zeviani, M.; Szibor, M.; Viscomi, C. Perturbed Redox Signaling Exacerbates a Mitochondrial Myopathy. Cell Metab. 2018, 28, 764–775.e765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Cocheme, H.M.; Quin, C.; McQuaker, S.J.; Cabreiro, F.; Logan, A.; Prime, T.A.; Abakumova, I.; Patel, J.V.; Fearnley, I.M.; James, A.M.; et al. Measurement of H2O2 within living Drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metab. 2011, 13, 340–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Drew, B.; Phaneuf, S.; Dirks, A.; Selman, C.; Gredilla, R.; Lezza, A.; Barja, G.; Leeuwenburgh, C. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R474–R480. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, M.; Mockett, R.J.; Shen, Y.; Orr, W.C.; Sohal, R.S. Age-associated decline in mitochondrial respiration and electron transport in Drosophila melanogaster. Biochem. J. 2005, 390, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Miwa, S.; Jow, H.; Baty, K.; Johnson, A.; Czapiewski, R.; Saretzki, G.; Treumann, A.; von Zglinicki, T. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat. Commun. 2014, 5, 3837. [Google Scholar] [CrossRef] [Green Version]
- Soro-Arnaiz, I.; Li, Q.O.Y.; Torres-Capelli, M.; Melendez-Rodriguez, F.; Veiga, S.; Veys, K.; Sebastian, D.; Elorza, A.; Tello, D.; Hernansanz-Agustin, P.; et al. Role of Mitochondrial Complex IV in Age-Dependent Obesity. Cell Rep. 2016, 16, 2991–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, A.; Boveris, A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1244–R1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabre, R.; Naudi, A.; Dominguez-Gonzalez, M.; Ayala, V.; Jove, M.; Mota-Martorell, N.; Pinol-Ripoll, G.; Gil-Villar, M.P.; Rue, M.; Portero-Otin, M.; et al. Sixty years old is the breakpoint of human frontal cortex aging. Free Radic. Biol. Med. 2017, 103, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoll, E.A.; Karapavlovic, N.; Rosa, H.; Woodmass, M.; Rygiel, K.; White, K.; Turnbull, D.M.; Faulkes, C.G. Naked mole-rats maintain healthy skeletal muscle and Complex IV mitochondrial enzyme function into old age. Aging 2016, 8, 3468–3485. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Yang, J.; Law, A.D.; Hendrix, D.A.; Kretzschmar, D.; Robinson, M.; Giebultowicz, J.M. Age-dependent effects of blue light exposure on lifespan, neurodegeneration, and mitochondria physiology in Drosophila melanogaster. NPJ Aging 2022, 8, 11. [Google Scholar] [CrossRef]
- Weinrich, T.W.; Coyne, A.; Salt, T.E.; Hogg, C.; Jeffery, G. Improving mitochondrial function significantly reduces metabolic, visual, motor and cognitive decline in aged Drosophila melanogaster. Neurobiol. Aging 2017, 60, 34–43. [Google Scholar] [CrossRef]
- Martini, H.; Passos, J.F. Cellular senescence: All roads lead to mitochondria. FEBS J. 2022, 290, 1186–1202. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Sun, J.; Folk, D.; Bradley, T.J.; Tower, J. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics 2002, 161, 661–672. [Google Scholar]
- Esposito, G.; Vos, M.; Vilain, S.; Swerts, J.; De Sousa Valadas, J.; Van Meensel, S.; Schaap, O.; Verstreken, P. Aconitase causes iron toxicity in Drosophila pink1 mutants. PLoS Genet. 2013, 9, e1003478. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, P.; Mooradian, A.D. Demystifying Oxidative Stress. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland. [CrossRef]
- Landis, G.; Shen, J.; Tower, J. Gene expression changes in response to aging compared to heat stress, oxidative stress and ionizing radiation in Drosophila melanogaster. Aging 2012, 4, 768–789. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983.e24. [Google Scholar] [CrossRef]
- Sohal, R.S.; Arnold, L.; Orr, W.C. Effect of age on superoxide dismutase, catalase, glutathione reductase, inorganic peroxides, TBA-reactive material, GSH/GSSG, NADPH/NADP+ and NADH/NAD+ in Drosophila melanogaster. Mech. Ageing Dev. 1990, 56, 223–235. [Google Scholar] [CrossRef] [PubMed]
- de la Asuncion, J.G.; Millan, A.; Pla, R.; Bruseghini, L.; Esteras, A.; Pallardo, F.V.; Sastre, J.; Vina, J. Mitochondrial glutathione oxidation correlates with age-associated oxidative damage to mitochondrial DNA. FASEB J. 1996, 10, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Kempf, A.; Song, S.M.; Talbot, C.B.; Miesenbock, G. A potassium channel beta-subunit couples mitochondrial electron transport to sleep. Nature 2019, 568, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.; Keene, A.C. Molecular mechanisms of age-related sleep loss in the fruit fly—A mini-review. Gerontology 2013, 59, 334–339. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Number of published papers containing the expression “Mitochondrial Redox Signalling” in their titles or abstracts, as extracted from the Scopus database. The y-axis indicates the number of publications per year, and the x-axis shows the years included in the search. The data provide insights into the growing popularity of redox biology within the scientific community.
Figure 1.
Number of published papers containing the expression “Mitochondrial Redox Signalling” in their titles or abstracts, as extracted from the Scopus database. The y-axis indicates the number of publications per year, and the x-axis shows the years included in the search. The data provide insights into the growing popularity of redox biology within the scientific community.

Figure 2.
Mitochondrial reactive oxygen species (ROS) are important cellular messengers, but understanding their role in cellular physiology requires consideration of four questions: (1) Where is the signal produced?; (2) When is the signal produced?; (3) What type of signal is produced?; and (4) What is the intensity of the signal? These four characteristics of an mtROS signal determine the downstream effects it can induce. Antioxidants act as essential regulators of ROS signalling by either transforming one signal into another, regulating the intensity of the signal or turning off the signal. The figure depicts some examples of the processes controlled by mtROS signalling. For more information, please refer to the main text.
Figure 2.
Mitochondrial reactive oxygen species (ROS) are important cellular messengers, but understanding their role in cellular physiology requires consideration of four questions: (1) Where is the signal produced?; (2) When is the signal produced?; (3) What type of signal is produced?; and (4) What is the intensity of the signal? These four characteristics of an mtROS signal determine the downstream effects it can induce. Antioxidants act as essential regulators of ROS signalling by either transforming one signal into another, regulating the intensity of the signal or turning off the signal. The figure depicts some examples of the processes controlled by mtROS signalling. For more information, please refer to the main text.

Figure 3.
The four aspects of an mtROS signal that impact its downstream effects. “Where” indicates that ROS produced by complexes I and III participate in various physiological and pathological processes. “When” refers to the temporal dynamics of mtROS signalling and highlights the importance of understanding how the signal is initiated and terminated using kinetic measurements rather than a single-point measurement. “Type” explains that mtROS signalling is mainly mediated by low reactive species, such as superoxide and hydrogen peroxide, while highly reactive species, such as hydroxyl radical, are responsible for oxidative damage. Finally, “Intensity” emphasizes that the intensity of mtROS signals needs to be within an appropriate physiological range to avoid disrupting cellular homeostasis by triggering oxidative or reductive stress. The figure illustrates the interplay between these four aspects of mtROS signalling and their downstream effects.
Figure 3.
The four aspects of an mtROS signal that impact its downstream effects. “Where” indicates that ROS produced by complexes I and III participate in various physiological and pathological processes. “When” refers to the temporal dynamics of mtROS signalling and highlights the importance of understanding how the signal is initiated and terminated using kinetic measurements rather than a single-point measurement. “Type” explains that mtROS signalling is mainly mediated by low reactive species, such as superoxide and hydrogen peroxide, while highly reactive species, such as hydroxyl radical, are responsible for oxidative damage. Finally, “Intensity” emphasizes that the intensity of mtROS signals needs to be within an appropriate physiological range to avoid disrupting cellular homeostasis by triggering oxidative or reductive stress. The figure illustrates the interplay between these four aspects of mtROS signalling and their downstream effects.

Figure 4.
Mitochondrial redox signalling is altered during ageing, with young mitochondria producing ROS only in response to specific stimuli, thereby maintaining low total ROS levels. Conversely, ageing mitochondria continually produce high levels of ROS and are unresponsive to external stimuli, resulting in oxidative stress and damage. Antioxidants alone are not effective in increasing lifespan, as, while they can reduce oxidative damage, they cannot restore redox signalling. Effective antiageing therapies require not only a reduction in oxidative damage but also restoration of mitochondrial ROS (mtROS) signalling. The figure illustrates the contrasting differences between young and ageing mitochondria, highlighting the importance of restoring mtROS signalling for healthy ageing. For the sake of clarity, we only depicted CI-generated ROS in young mitochondria. Nevertheless, it is evident that CIII also plays a significant role in generating ROS in young and healthy individuals. In aged mitochondria, both CI and CIII are displayed to indicate the likelihood that multiple ROS generators (both specific and non-specific) are concurrently active.
Figure 4.
Mitochondrial redox signalling is altered during ageing, with young mitochondria producing ROS only in response to specific stimuli, thereby maintaining low total ROS levels. Conversely, ageing mitochondria continually produce high levels of ROS and are unresponsive to external stimuli, resulting in oxidative stress and damage. Antioxidants alone are not effective in increasing lifespan, as, while they can reduce oxidative damage, they cannot restore redox signalling. Effective antiageing therapies require not only a reduction in oxidative damage but also restoration of mitochondrial ROS (mtROS) signalling. The figure illustrates the contrasting differences between young and ageing mitochondria, highlighting the importance of restoring mtROS signalling for healthy ageing. For the sake of clarity, we only depicted CI-generated ROS in young mitochondria. Nevertheless, it is evident that CIII also plays a significant role in generating ROS in young and healthy individuals. In aged mitochondria, both CI and CIII are displayed to indicate the likelihood that multiple ROS generators (both specific and non-specific) are concurrently active.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Castejon-Vega, B.; Cordero, M.D.; Sanz, A. How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing. Antioxidants 2023, 12, 831. https://doi.org/10.3390/antiox12040831
AMA Style
Castejon-Vega B, Cordero MD, Sanz A. How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing. Antioxidants. 2023; 12(4):831. https://doi.org/10.3390/antiox12040831
Chicago/Turabian StyleCastejon-Vega, Beatriz, Mario D. Cordero, and Alberto Sanz. 2023. "How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing" Antioxidants 12, no. 4: 831. https://doi.org/10.3390/antiox12040831
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.