
How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
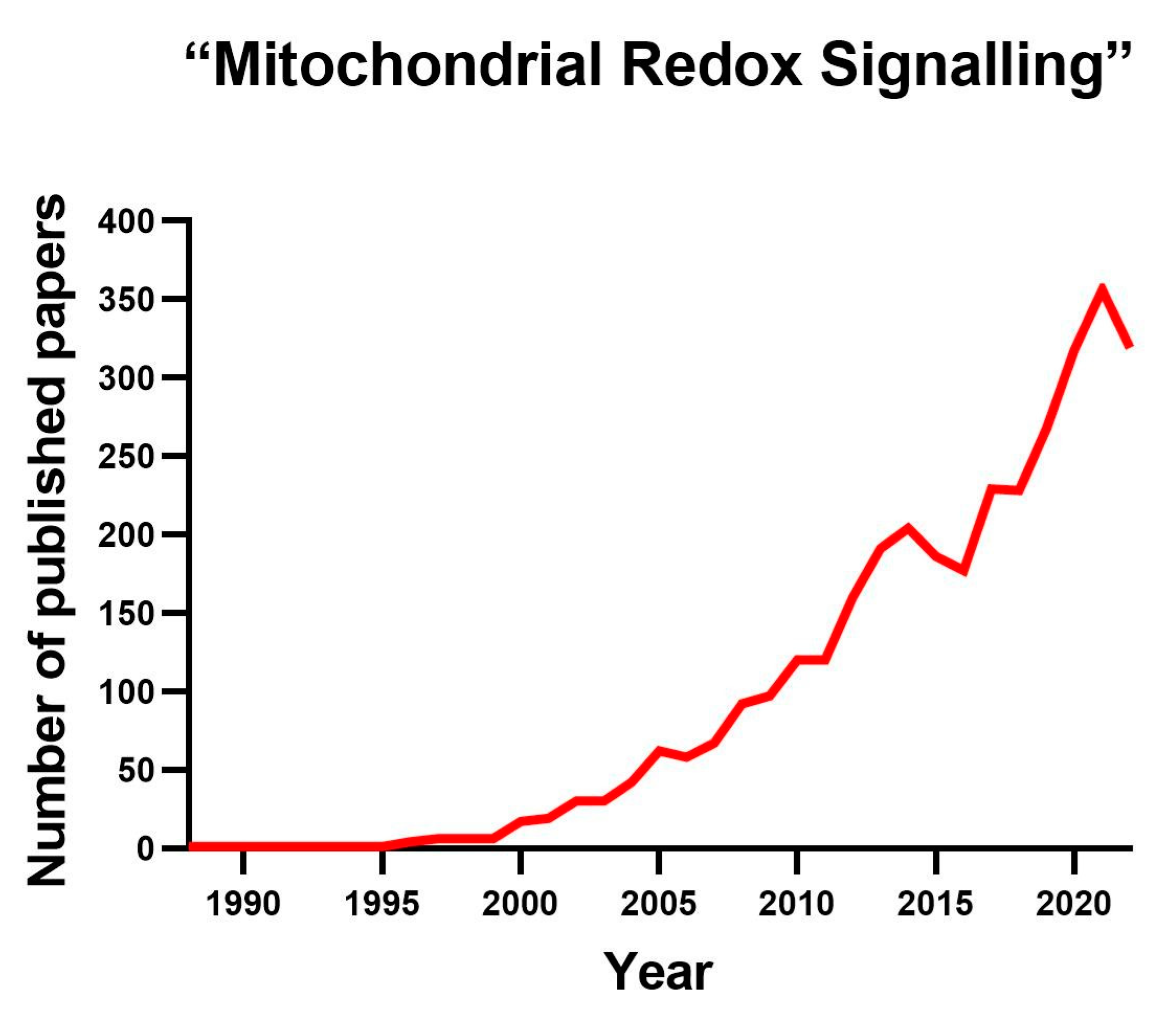
:1. Introduction
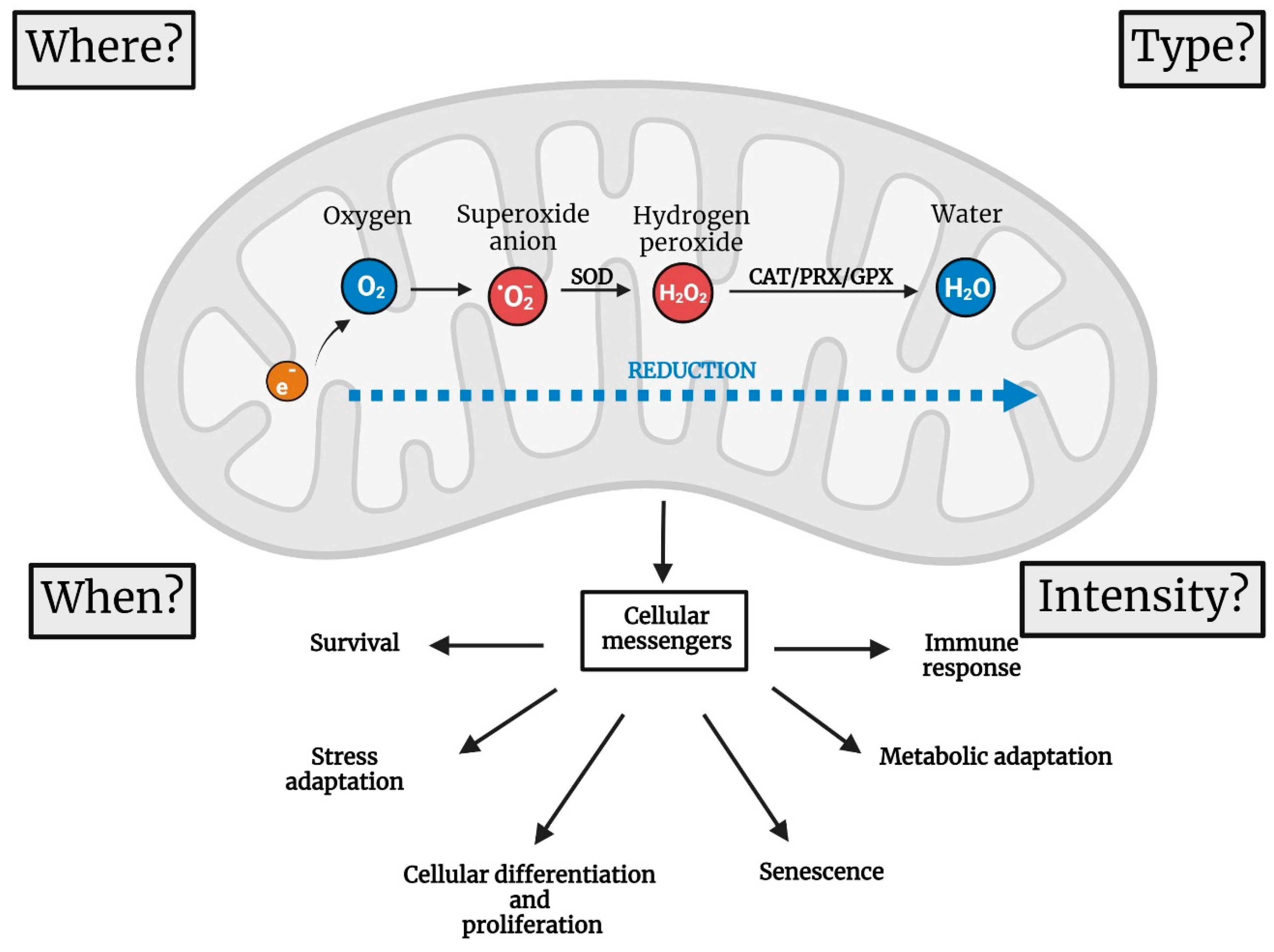
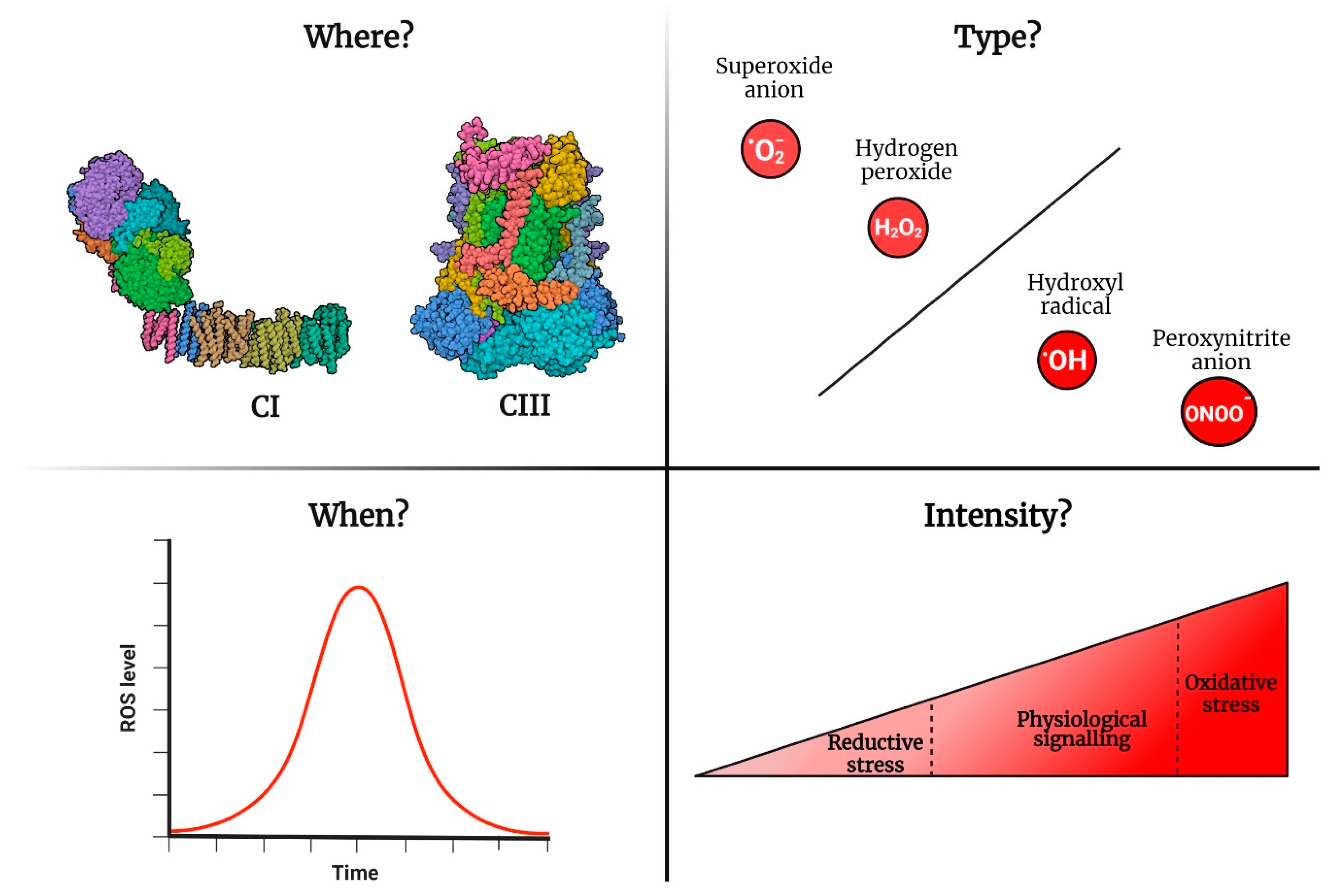
2. Four Parameters to Describe the Functions of mtROS
2.1. Where Are mtROS Produced?
2.2. When Are ROS Produced?
3. Pathological Role of mtROS
3.1. The Two Ways by Which ROS Cause Damage
3.2. The Role of mtROS in Ageing
3.3. Accumulation of Damaged Mitochondria during Ageing Causes an Interruption in Mitochondrial Redox Signalling
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AOX | Alternative oxidase |
| CoQ | Coenzyme Q |
| CI | Complex I |
| CII | Complex II |
| CIII | Complex III |
| CIV | Complex IV |
| CV | Complex V |
| ETC | Electron transport chain |
| HIF1a | Hypoxia-inducible factor 1-alpha |
| IF1 | ATPase inhibitory factor 1 |
| IL-6 | Interleukin 6 |
| MAPK | Mitogen-activated protein kinase |
| MFRTA | The mitochondrial free radical theory of ageing |
| mROS | Mitochondrial reactive oxygen species |
| NDI1 | NADH dehydrogenase internal 1 |
| NOTCH | Neurogenic locus notch homolog protein |
| NOX | NADPH oxidase |
| OXPHOS | Oxidative phosphorylation |
| PRDX2 | Peroxiredoxin-2 |
| pmf | Proton motive force |
| RET | Reverse electron transport |
| S1QELs | Suppressors of superoxide production in CI |
| S3QELs | Suppressors of superoxide production in CIII |
| SOD1 | Superoxide dismutase-1 |
| SOD2 | Superoxide dismutase-2 |
| TGF-beta | Transforming growth factor beta |
| TNF | Tumour necrosis factor |
References
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, Y.; Gerencser, A.A.; Brand, M.D. Effects of sugars, fatty acids and amino acids on cytosolic and mitochondrial hydrogen peroxide release from liver cells. Free Radic. Biol. Med. 2022, 188, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [Green Version]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Gorlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [Green Version]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Luzikov, V.N. Principles of control over formation of structures responsible for respiratory functions of mitochondria. Biochemistry 2009, 74, 1443–1456. [Google Scholar] [CrossRef]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19, 77–92. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flora, S.J. Structural, chemical and biological aspects of antioxidants for strategies against metal and metalloid exposure. Oxid. Med. Cell. Longev. 2009, 2, 191–206. [Google Scholar] [CrossRef] [Green Version]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.J.; Dong, W.; Stephanopoulos, G.N.; Sikes, H.D. Oxidative pentose phosphate pathway and glucose anaplerosis support maintenance of mitochondrial NADPH pool under mitochondrial oxidative stress. Bioeng. Transl. Med. 2020, 5, e10184. [Google Scholar] [CrossRef]
- Lennicke, C.; Cocheme, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Kaya, A.; Lee, B.C.; Gladyshev, V.N. Regulation of protein function by reversible methionine oxidation and the role of selenoprotein MsrB1. Antioxid. Redox Signal. 2015, 23, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Nordzieke, D.E.; Medrano-Fernandez, I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants 2018, 7, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2017, 14, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Vivancos, A.P.; Castillo, E.A.; Biteau, B.; Nicot, C.; Ayte, J.; Toledano, M.B.; Hidalgo, E. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 8875–8880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Sanz, A.; Stefanatos, R.; McIlroy, G. Production of reactive oxygen species by the mitochondrial electron transport chain in Drosophila melanogaster. J. Bioenerg. Biomembr. 2010, 42, 135–142. [Google Scholar] [CrossRef]
- Keeley, T.P.; Mann, G.E. Defining Physiological Normoxia for Improved Translation of Cell Physiology to Animal Models and Humans. Physiol. Rev. 2019, 99, 161–234. [Google Scholar] [CrossRef] [Green Version]
- Maremonti, E.; Eide, D.M.; Rossbach, L.M.; Lind, O.C.; Salbu, B.; Brede, D.A. In vivo assessment of reactive oxygen species production and oxidative stress effects induced by chronic exposure to gamma radiation in Caenorhabditis elegans. Free. Radic. Biol. Med. 2020, 152, 583–596. [Google Scholar] [CrossRef]
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef]
- Scialo, F.; Sriram, A.; Stefanatos, R.; Spriggs, R.V.; Loh, S.H.Y.; Martins, L.M.; Sanz, A. Mitochondrial complex I derived ROS regulate stress adaptation in Drosophila melanogaster. Redox Biol. 2020, 32, 101450. [Google Scholar] [CrossRef]
- Scialo, F.; Sanz, A. Coenzyme Q redox signalling and longevity. Free Radic. Biol. Med. 2021, 164, 187–205. [Google Scholar] [CrossRef]
- Sanz, A.; Pamplona, R.; Barja, G. Is the mitochondrial free radical theory of aging intact? Antioxid. Redox Signal. 2006, 8, 582–599. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wong, H.S.; Brand, M.D. Production of superoxide and hydrogen peroxide in the mitochondrial matrix is dominated by site IQ of complex I in diverse cell lines. Redox Biol. 2020, 37, 101722. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.S.; Watson, M.A.; Wong, H.S.; Orr, A.L.; Brand, M.D. The use of site-specific suppressors to measure the relative contributions of different mitochondrial sites to skeletal muscle superoxide and hydrogen peroxide production. Redox Biol. 2020, 28, 101341. [Google Scholar] [CrossRef]
- de Cubas, L.; Pak, V.V.; Belousov, V.V.; Ayte, J.; Hidalgo, E. The Mitochondria-to-Cytosol H2O2 Gradient Is Caused by Peroxiredoxin-Dependent Cytosolic Scavenging. Antioxidants 2021, 10, 731. [Google Scholar] [CrossRef] [PubMed]
- Carmona, M.; de Cubas, L.; Bautista, E.; Moral-Blanch, M.; Medrano-Fernandez, I.; Sitia, R.; Boronat, S.; Ayte, J.; Hidalgo, E. Monitoring cytosolic H2O2 fluctuations arising from altered plasma membrane gradients or from mitochondrial activity. Nat. Commun. 2019, 10, 4526. [Google Scholar] [CrossRef] [Green Version]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2014, 33C, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. Commun. Free Radic. Res. 2020, 25, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Treberg, J.R.; Perevoshchikova, I.V.; Orr, A.L.; Brand, M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic. Biol. Med. 2012, 53, 1807–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Mezera, V.; Dighe, P.; Melov, S.; Gerencser, A.A.; Sweis, R.F.; Pliushchev, M.; Wang, Z.; Esbenshade, T.; McKibben, B.; et al. Superoxide produced by mitochondrial site IQ inactivates cardiac succinate dehydrogenase and induces hepatic steatosis in Sod2 knockout mice. Free Radic. Biol. Med. 2021, 164, 223–232. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolanos, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Judge, S.; Jang, Y.M.; Smith, A.; Selman, C.; Phillips, T.; Speakman, J.R.; Hagen, T.; Leeuwenburgh, C. Exercise by lifelong voluntary wheel running reduces subsarcolemmal and interfibrillar mitochondrial hydrogen peroxide production in the heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1564–R1572. [Google Scholar] [CrossRef] [Green Version]
- Owusu-Ansah, E.; Yavari, A.; Mandal, S.; Banerjee, U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 2008, 40, 356–361. [Google Scholar] [CrossRef]
- Lee, S.; Tak, E.; Lee, J.; Rashid, M.A.; Murphy, M.P.; Ha, J.; Kim, S.S. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 2011, 21, 817–834. [Google Scholar] [CrossRef] [Green Version]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernansanz-Agustin, P.; Ramos, E.; Navarro, E.; Parada, E.; Sanchez-Lopez, N.; Pelaez-Aguado, L.; Cabrera-Garcia, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Pina, T.; Moreno, L.; Izquierdo-Alvarez, A.; Cabrera-Garcia, J.D.; Cortes, A.; et al. Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291. [Google Scholar] [CrossRef]
- Fernandez-Aguera, M.C.; Gao, L.; Gonzalez-Rodriguez, P.; Pintado, C.O.; Arias-Mayenco, I.; Garcia-Flores, P.; Garcia-Perganeda, A.; Pascual, A.; Ortega-Saenz, P.; Lopez-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Arias-Mayenco, I.; Gonzalez-Rodriguez, P.; Torres-Torrelo, H.; Gao, L.; Fernandez-Aguera, M.C.; Bonilla-Henao, V.; Ortega-Saenz, P.; Lopez-Barneo, J. Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab. 2018, 28, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Graham, C.; Stefanatos, R.; Yek, A.E.H.; Spriggs, R.V.; Loh, S.H.Y.; Uribe, A.H.; Zhang, T.; Martins, L.M.; Maddocks, O.D.K.; Scialo, F.; et al. Mitochondrial ROS signalling requires uninterrupted electron flow and is lost during ageing in flies. Geroscience 2022, 44, 1961–1974. [Google Scholar] [CrossRef] [PubMed]
- Bazopoulou, D.; Knoefler, D.; Zheng, Y.; Ulrich, K.; Oleson, B.J.; Xie, L.; Kim, M.; Kaufmann, A.; Lee, Y.T.; Dou, Y.; et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 2019, 576, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Obata, F.; Fons, C.O.; Gould, A.P. Early-life exposure to low-dose oxidants can increase longevity via microbiome remodelling in Drosophila. Nat. Commun. 2018, 9, 975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Marks, J.D.; Guzy, R.D.; Mungai, P.T.; Schriewer, J.M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 2013, 187, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Sommer, N.; Alebrahimdehkordi, N.; Pak, O.; Knoepp, F.; Strielkov, I.; Scheibe, S.; Dufour, E.; Andjelkovic, A.; Sydykov, A.; Saraji, A.; et al. Bypassing mitochondrial complex III using alternative oxidase inhibits acute pulmonary oxygen sensing. Sci. Adv. 2020, 6, eaba0694. [Google Scholar] [CrossRef] [Green Version]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Nuevo, A.; Torres-Sanchez, A.; Duran, J.M.; De Guirior, C.; Martinez-Zamora, M.A.; Boke, E. Oocytes maintain ROS-free mitochondrial metabolism by suppressing complex I. Nature 2022, 607, 756–761. [Google Scholar] [CrossRef]
- Han, Y.; Ishibashi, S.; Iglesias-Gonzalez, J.; Chen, Y.; Love, N.R.; Amaya, E. Ca2+-Induced Mitochondrial ROS Regulate the Early Embryonic Cell Cycle. Cell Rep. 2018, 22, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Hernansanz-Agustin, P.; Enriquez, J.A. Sodium in Mitochondrial Redox Signaling. Antioxid. Redox Signal. 2022, 37, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Soberanes, S.; Misharin, A.V.; Jairaman, A.; Morales-Nebreda, L.; McQuattie-Pimentel, A.C.; Cho, T.; Hamanaka, R.B.; Meliton, A.Y.; Reyfman, P.A.; Walter, J.M.; et al. Metformin Targets Mitochondrial Electron Transport to Reduce Air-Pollution-Induced Thrombosis. Cell Metab. 2019, 29, 335–347.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Esparza-Molto, P.B.; Romero-Carraminana, I.; Nunez de Arenas, C.; Pereira, M.P.; Blanco, N.; Pardo, B.; Bates, G.R.; Sanchez-Castillo, C.; Artuch, R.; Murphy, M.P.; et al. Generation of mitochondrial reactive oxygen species is controlled by ATPase inhibitory factor 1 and regulates cognition. PLoS Biol. 2021, 19, e3001252. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [Green Version]
- Menger, K.E.; James, A.M.; Cocheme, H.M.; Harbour, M.E.; Chouchani, E.T.; Ding, S.; Fearnley, I.M.; Partridge, L.; Murphy, M.P. Fasting, but Not Aging, Dramatically Alters the Redox Status of Cysteine Residues on Proteins in Drosophila melanogaster. Cell Rep. 2015, 11, 1856–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Oliver, C.N.; Starke-Reed, P.E.; Stadtman, E.R.; Liu, G.J.; Carney, J.M.; Floyd, R.A. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5144–5147. [Google Scholar] [CrossRef] [Green Version]
- Beach, T.E.; Prag, H.A.; Pala, L.; Logan, A.; Huang, M.M.; Gruszczyk, A.V.; Martin, J.L.; Mahbubani, K.; Hamed, M.O.; Hosgood, S.A.; et al. Targeting succinate dehydrogenase with malonate ester prodrugs decreases renal ischemia reperfusion injury. Redox Biol. 2020, 36, 101640. [Google Scholar] [CrossRef]
- Martin, J.L.; Costa, A.S.H.; Gruszczyk, A.V.; Beach, T.E.; Allen, F.M.; Prag, H.A.; Hinchy, E.C.; Mahbubani, K.; Hamed, M.; Tronci, L.; et al. Succinate accumulation drives ischaemia-reperfusion injury during organ transplantation. Nat. Metab. 2019, 1, 966–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Yasuda, K.; Akatsuka, A.; Hino, O.; Hartman, P.S.; Ishii, N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005, 65, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, N.; Long, X.; Zhao, J.; Yang, J.; Du, X.; Yang, T.; Yuan, P.; Huang, X.; Zhang, J.; et al. SDHC-related deficiency of SDH complex activity promotes growth and metastasis of hepatocellular carcinoma via ROS/NFkappaB signaling. Cancer Lett. 2019, 461, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Tantray, I.; Rimal, S.; Mitra, S.; Cheshier, S.; Lu, B. Regulation of reverse electron transfer at mitochondrial complex I by unconventional Notch action in cancer stem cells. Dev. Cell 2022, 57, 260–276.e9. [Google Scholar] [CrossRef]
- Chen, W.L.; Sheu, J.R.; Chen, R.J.; Hsiao, S.H.; Hsiao, C.J.; Chou, Y.C.; Chung, C.L.; Hsiao, G. Mycobacterium tuberculosis Upregulates TNF-alpha Expression via TLR2/ERK Signaling and Induces MMP-1 and MMP-9 Production in Human Pleural Mesothelial Cells. PLoS ONE 2015, 10, e0137979. [Google Scholar] [CrossRef] [Green Version]
- Roca, F.J.; Whitworth, L.J.; Prag, H.A.; Murphy, M.P.; Ramakrishnan, L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science 2022, 376, eabh2841. [Google Scholar] [CrossRef]
- Soberanes, S.; Urich, D.; Baker, C.M.; Burgess, Z.; Chiarella, S.E.; Bell, E.L.; Ghio, A.J.; De Vizcaya-Ruiz, A.; Liu, J.; Ridge, K.M.; et al. Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J. Biol. Chem. 2009, 284, 2176–2186. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Barja, G. Free radicals and aging. Trends Neurosci. 2004, 27, 595–600. [Google Scholar] [CrossRef]
- Hohn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; Konig, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Ku, H.H.; Agarwal, S.; Forster, M.J.; Lal, H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech. Ageing Dev. 1994, 74, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barja, G.; Herrero, A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Sanz, A.; Stefanatos, R.K. The mitochondrial free radical theory of aging: A critical view. Curr. Aging Sci. 2008, 1, 10–21. [Google Scholar] [CrossRef]
- Gems, D.; Doonan, R. Antioxidant defense and aging in C. elegans: Is the oxidative damage theory of aging wrong? Cell Cycle 2009, 8, 1681–1687. [Google Scholar] [CrossRef] [Green Version]
- Perez, V.I.; Van Remmen, H.; Bokov, A.; Epstein, C.J.; Vijg, J.; Richardson, A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 2009, 8, 73–75. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.C.; Perez, V.I.; Song, W.; Lustgarten, M.S.; Salmon, A.B.; Mele, J.; Qi, W.; Liu, Y.; Liang, H.; Chaudhuri, A.; et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [Green Version]
- Mockett, R.J.; Bayne, A.C.; Kwong, L.K.; Orr, W.C.; Sohal, R.S. Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radic. Biol. Med. 2003, 34, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genom. 2003, 16, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ikeno, Y.; Qi, W.; Chaudhuri, A.; Li, Y.; Bokov, A.; Thorpe, S.R.; Baynes, J.W.; Epstein, C.; Richardson, A.; et al. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1212–1220. [Google Scholar] [CrossRef]
- Jang, Y.C.; Remmen, V.H. The mitochondrial theory of aging: Insight from transgenic and knockout mouse models. Exp. Gerontol. 2009, 44, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Radyuk, S.N.; Rebrin, I.; Klichko, V.I.; Sohal, B.H.; Michalak, K.; Benes, J.; Sohal, R.S.; Orr, W.C. Mitochondrial peroxiredoxins are critical for the maintenance of redox state and the survival of adult Drosophila. Free Radic. Biol. Med. 2010, 49, 1892–1902. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Ayala, D.J.; Sanz, A.; Vartiainen, S.; Kemppainen, K.K.; Babusiak, M.; Mustalahti, E.; Costa, R.; Tuomela, T.; Zeviani, M.; Chung, J.; et al. Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab. 2009, 9, 449–460. [Google Scholar] [CrossRef]
- Sanz, A.; Fernandez-Ayala, D.J.; Stefanatos, R.K.; Jacobs, H.T. Mitochondrial ROS production correlates with, but does not directly regulate lifespan in Drosophila. Aging 2010, 2, 200–223. [Google Scholar] [CrossRef]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, M.; Priebe, S.; Groth, M.; Hartmann, N.; Menzel, U.; Pandolfini, L.; Koch, P.; Felder, M.; Ristow, M.; Englert, C.; et al. Longitudinal RNA-Seq Analysis of Vertebrate Aging Identifies Mitochondrial Complex I as a Small-Molecule-Sensitive Modifier of Lifespan. Cell Syst. 2016, 2, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Copeland, J.M.; Cho, J.; Lo, T., Jr.; Hur, J.H.; Bahadorani, S.; Arabyan, T.; Rabie, J.; Soh, J.; Walker, D.W. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 2009, 19, 1591–1598. [Google Scholar] [CrossRef] [Green Version]
- Owusu-Ansah, E.; Song, W.; Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 2013, 155, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimal, S.; Tantray, I.; Li, Y.; Pal Khaket, T.; Li, Y.; Bhurtel, S.; Li, W.; Zeng, C.; Lu, B. Reverse electron transfer is activated during aging and contributes to aging and age-related disease. EMBO Rep. 2023, e55548. [Google Scholar] [CrossRef]
- Sanz, A.; Soikkeli, M.; Portero-Otin, M.; Wilson, A.; Kemppainen, E.; McIlroy, G.; Ellila, S.; Kemppainen, K.K.; Tuomela, T.; Lakanmaa, M.; et al. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc. Natl. Acad. Sci. USA 2010, 107, 9105–9110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahadorani, S.; Cho, J.; Lo, T.; Contreras, H.; Lawal, H.O.; Krantz, D.E.; Bradley, T.J.; Walker, D.W. Neuronal expression of a single-subunit yeast NADH-ubiquinone oxidoreductase (Ndi1) extends Drosophila lifespan. Aging Cell 2010, 9, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahadorani, S.; Hur, J.H.; Lo, T., Jr.; Vu, K.; Walker, D.W. Perturbation of mitochondrial complex V alters the response to dietary restriction in Drosophila. Aging Cell 2010, 9, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wheeler, C.T.; Yolitz, J.; Laslo, M.; Alberico, T.; Sun, Y.; Song, Q.; Zou, S. A mitochondrial ATP synthase subunit interacts with TOR signaling to modulate protein homeostasis and lifespan in Drosophila. Cell Rep. 2014, 8, 1781–1792. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, J.; Currais, A.; Prior, M.; Fischer, W.; Chiruta, C.; Ratliff, E.; Daugherty, D.; Dargusch, R.; Finley, K.; Esparza-Molto, P.B.; et al. The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell 2018, 17, e12715. [Google Scholar] [CrossRef]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef] [Green Version]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.A.; Cerutti, R.; Beninca, C.; Brea-Calvo, G.; Jacobs, H.T.; Zeviani, M.; Szibor, M.; Viscomi, C. Perturbed Redox Signaling Exacerbates a Mitochondrial Myopathy. Cell Metab. 2018, 28, 764–775.e765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Cocheme, H.M.; Quin, C.; McQuaker, S.J.; Cabreiro, F.; Logan, A.; Prime, T.A.; Abakumova, I.; Patel, J.V.; Fearnley, I.M.; James, A.M.; et al. Measurement of H2O2 within living Drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metab. 2011, 13, 340–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Drew, B.; Phaneuf, S.; Dirks, A.; Selman, C.; Gredilla, R.; Lezza, A.; Barja, G.; Leeuwenburgh, C. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R474–R480. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, M.; Mockett, R.J.; Shen, Y.; Orr, W.C.; Sohal, R.S. Age-associated decline in mitochondrial respiration and electron transport in Drosophila melanogaster. Biochem. J. 2005, 390, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Miwa, S.; Jow, H.; Baty, K.; Johnson, A.; Czapiewski, R.; Saretzki, G.; Treumann, A.; von Zglinicki, T. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat. Commun. 2014, 5, 3837. [Google Scholar] [CrossRef] [Green Version]
- Soro-Arnaiz, I.; Li, Q.O.Y.; Torres-Capelli, M.; Melendez-Rodriguez, F.; Veiga, S.; Veys, K.; Sebastian, D.; Elorza, A.; Tello, D.; Hernansanz-Agustin, P.; et al. Role of Mitochondrial Complex IV in Age-Dependent Obesity. Cell Rep. 2016, 16, 2991–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, A.; Boveris, A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1244–R1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabre, R.; Naudi, A.; Dominguez-Gonzalez, M.; Ayala, V.; Jove, M.; Mota-Martorell, N.; Pinol-Ripoll, G.; Gil-Villar, M.P.; Rue, M.; Portero-Otin, M.; et al. Sixty years old is the breakpoint of human frontal cortex aging. Free Radic. Biol. Med. 2017, 103, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoll, E.A.; Karapavlovic, N.; Rosa, H.; Woodmass, M.; Rygiel, K.; White, K.; Turnbull, D.M.; Faulkes, C.G. Naked mole-rats maintain healthy skeletal muscle and Complex IV mitochondrial enzyme function into old age. Aging 2016, 8, 3468–3485. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Yang, J.; Law, A.D.; Hendrix, D.A.; Kretzschmar, D.; Robinson, M.; Giebultowicz, J.M. Age-dependent effects of blue light exposure on lifespan, neurodegeneration, and mitochondria physiology in Drosophila melanogaster. NPJ Aging 2022, 8, 11. [Google Scholar] [CrossRef]
- Weinrich, T.W.; Coyne, A.; Salt, T.E.; Hogg, C.; Jeffery, G. Improving mitochondrial function significantly reduces metabolic, visual, motor and cognitive decline in aged Drosophila melanogaster. Neurobiol. Aging 2017, 60, 34–43. [Google Scholar] [CrossRef]
- Martini, H.; Passos, J.F. Cellular senescence: All roads lead to mitochondria. FEBS J. 2022, 290, 1186–1202. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Sun, J.; Folk, D.; Bradley, T.J.; Tower, J. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics 2002, 161, 661–672. [Google Scholar]
- Esposito, G.; Vos, M.; Vilain, S.; Swerts, J.; De Sousa Valadas, J.; Van Meensel, S.; Schaap, O.; Verstreken, P. Aconitase causes iron toxicity in Drosophila pink1 mutants. PLoS Genet. 2013, 9, e1003478. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, P.; Mooradian, A.D. Demystifying Oxidative Stress. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland. [CrossRef]
- Landis, G.; Shen, J.; Tower, J. Gene expression changes in response to aging compared to heat stress, oxidative stress and ionizing radiation in Drosophila melanogaster. Aging 2012, 4, 768–789. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983.e24. [Google Scholar] [CrossRef]
- Sohal, R.S.; Arnold, L.; Orr, W.C. Effect of age on superoxide dismutase, catalase, glutathione reductase, inorganic peroxides, TBA-reactive material, GSH/GSSG, NADPH/NADP+ and NADH/NAD+ in Drosophila melanogaster. Mech. Ageing Dev. 1990, 56, 223–235. [Google Scholar] [CrossRef] [PubMed]
- de la Asuncion, J.G.; Millan, A.; Pla, R.; Bruseghini, L.; Esteras, A.; Pallardo, F.V.; Sastre, J.; Vina, J. Mitochondrial glutathione oxidation correlates with age-associated oxidative damage to mitochondrial DNA. FASEB J. 1996, 10, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Kempf, A.; Song, S.M.; Talbot, C.B.; Miesenbock, G. A potassium channel beta-subunit couples mitochondrial electron transport to sleep. Nature 2019, 568, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.; Keene, A.C. Molecular mechanisms of age-related sleep loss in the fruit fly—A mini-review. Gerontology 2013, 59, 334–339. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castejon-Vega, B.; Cordero, M.D.; Sanz, A. How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing. Antioxidants 2023, 12, 831. https://doi.org/10.3390/antiox12040831
Castejon-Vega B, Cordero MD, Sanz A. How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing. Antioxidants. 2023; 12(4):831. https://doi.org/10.3390/antiox12040831
Chicago/Turabian StyleCastejon-Vega, Beatriz, Mario D. Cordero, and Alberto Sanz. 2023. "How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing" Antioxidants 12, no. 4: 831. https://doi.org/10.3390/antiox12040831