
Novel Reversible Inhibitors of Xanthine Oxidase Targeting the Active Site of the Enzyme
 ,
,  , and
, and 
Abstract
:
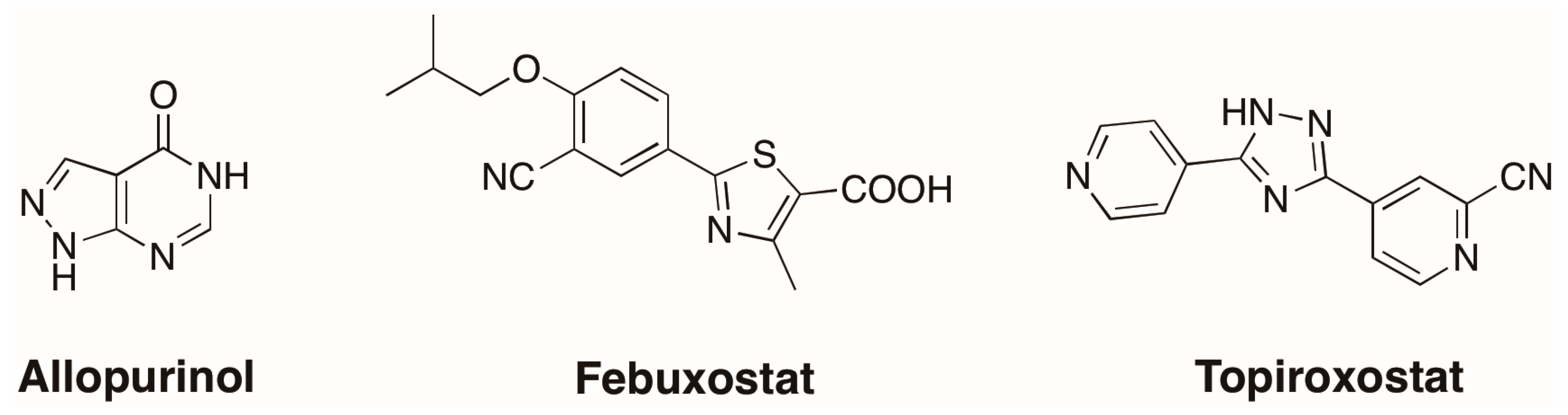
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Computational Methods
2.2.1. Protein and Ligand Preparation
2.2.2. Docking Simulations
2.3. Biochemical Methods
2.4. Statistical Analysis
3. Results and Discussion
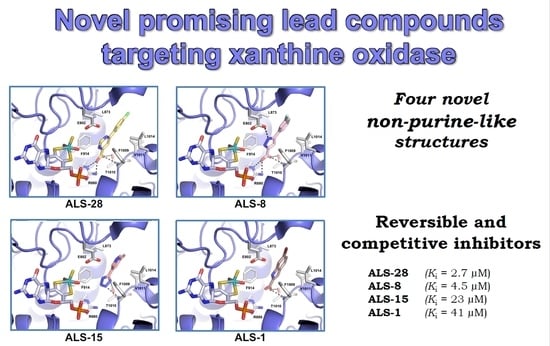
3.1. Novel Inhibitors of Xanthine Oxidase with Non-Purine-Like Structures
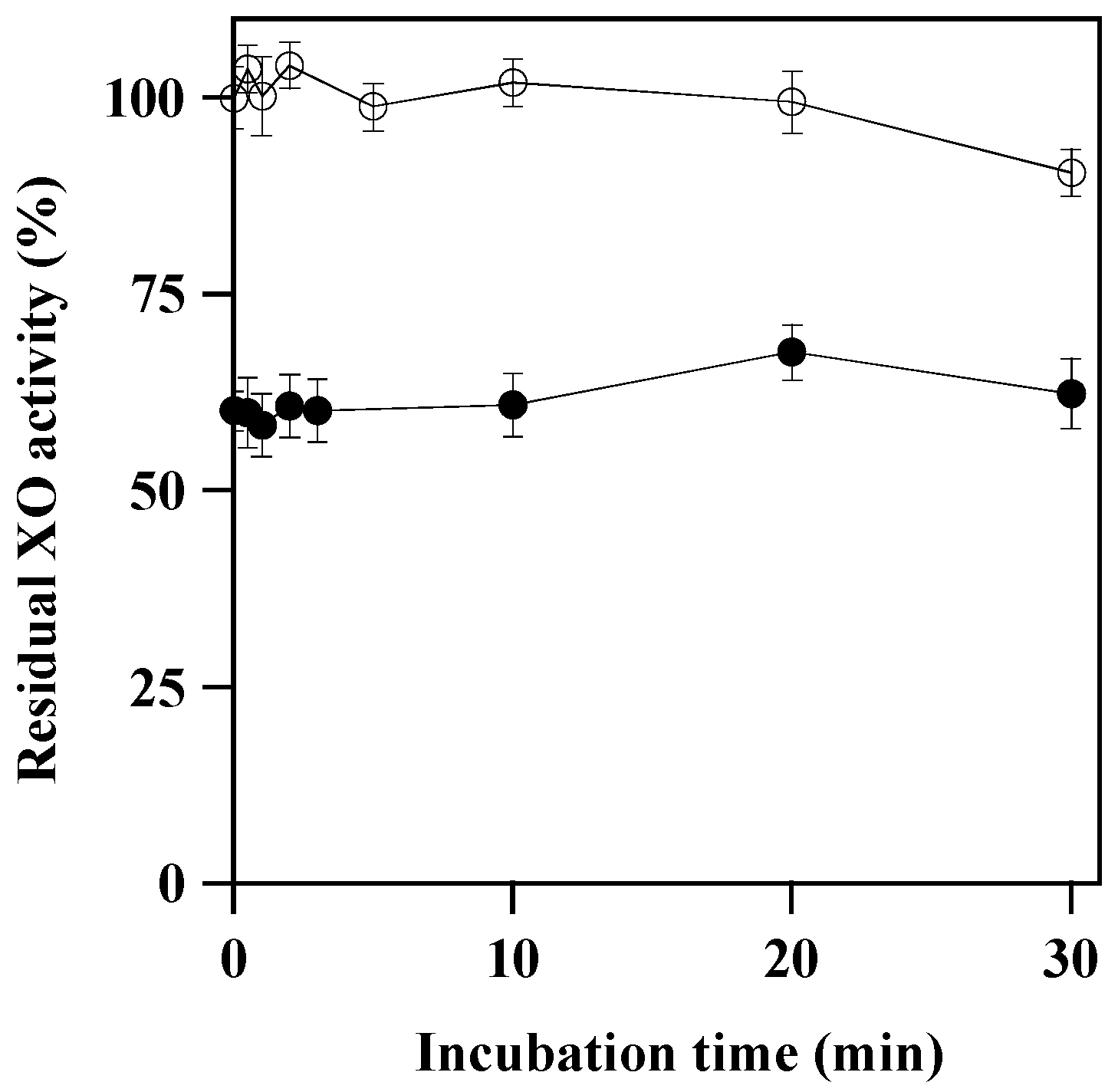
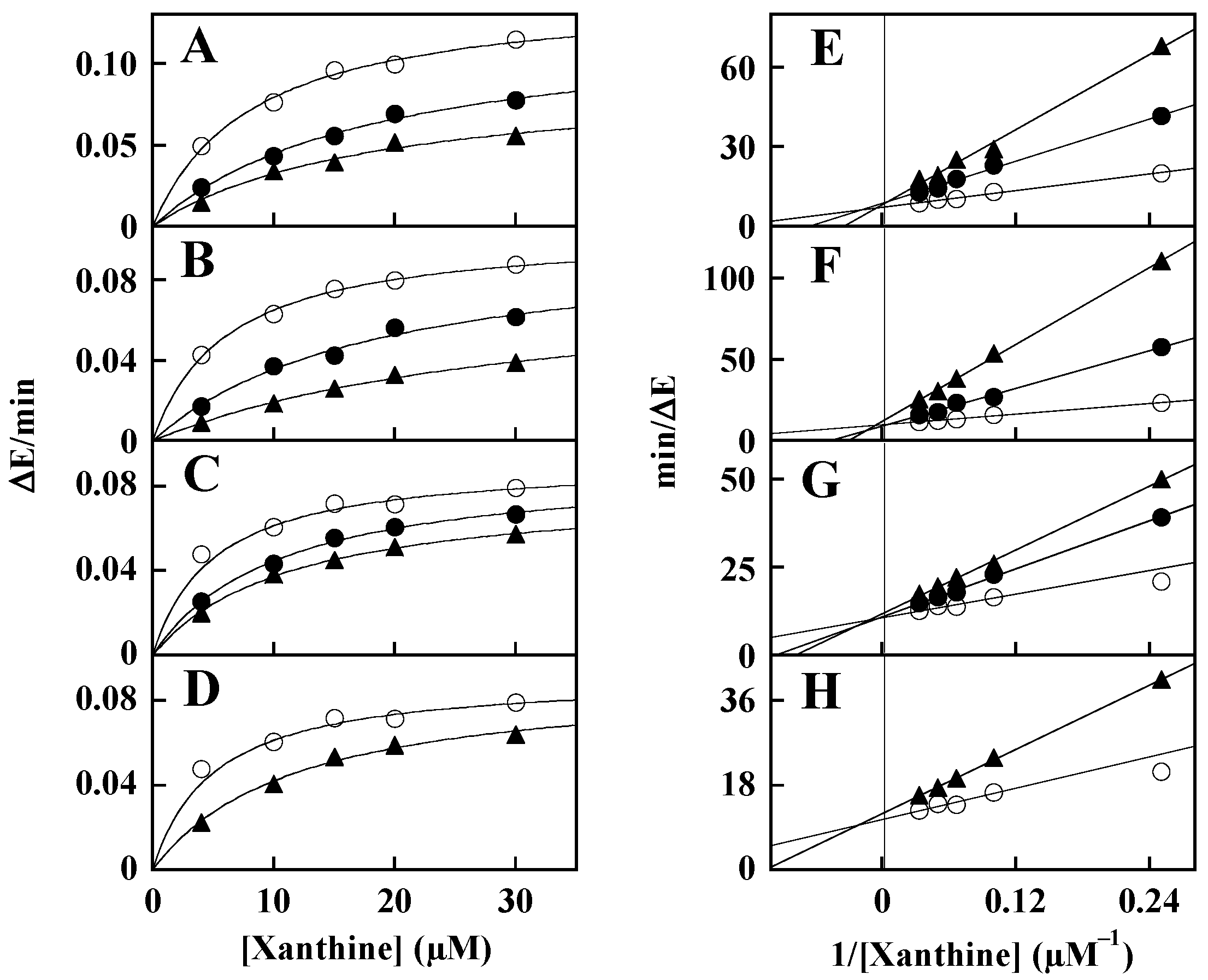
3.2. Mechanism of XO Inhibition
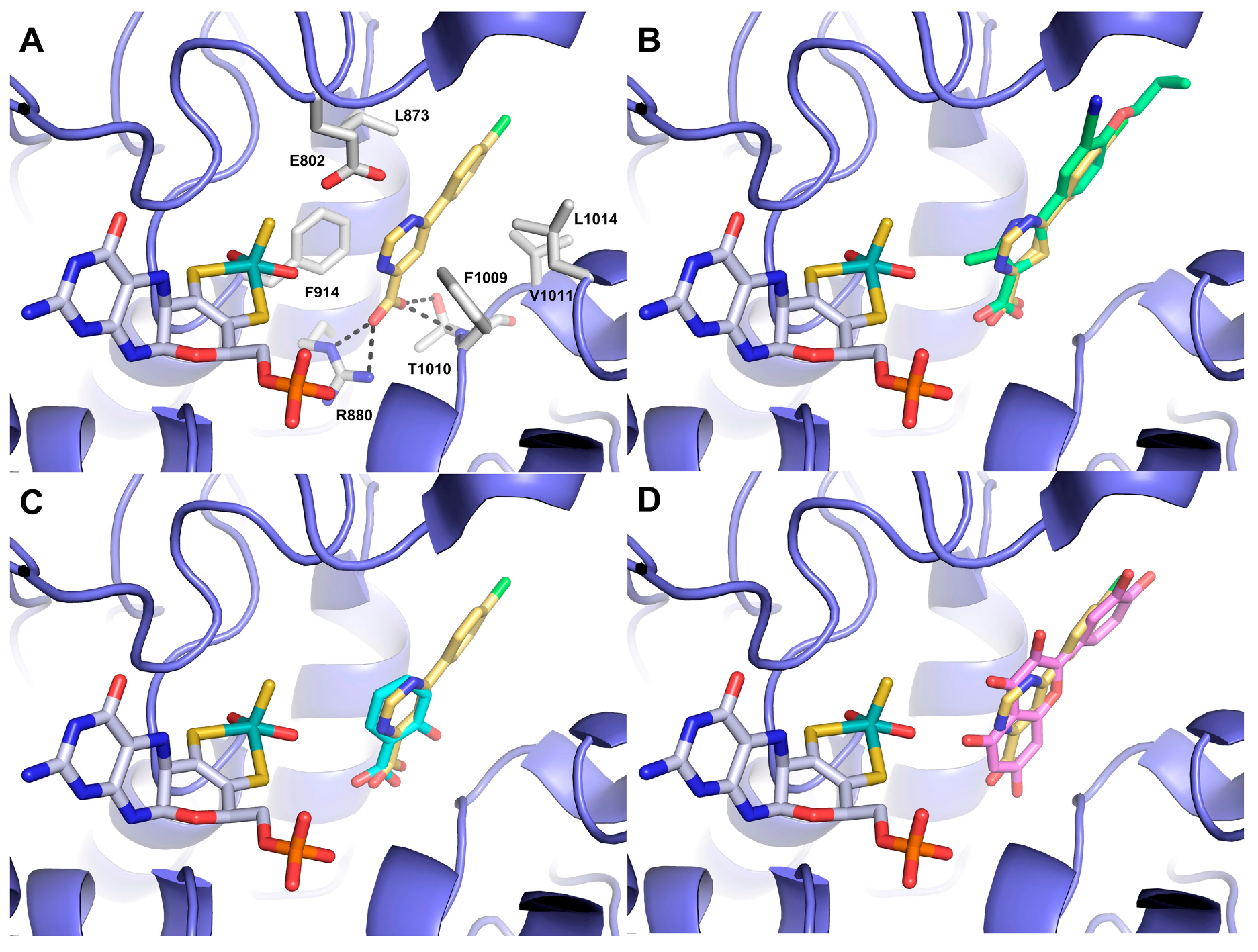
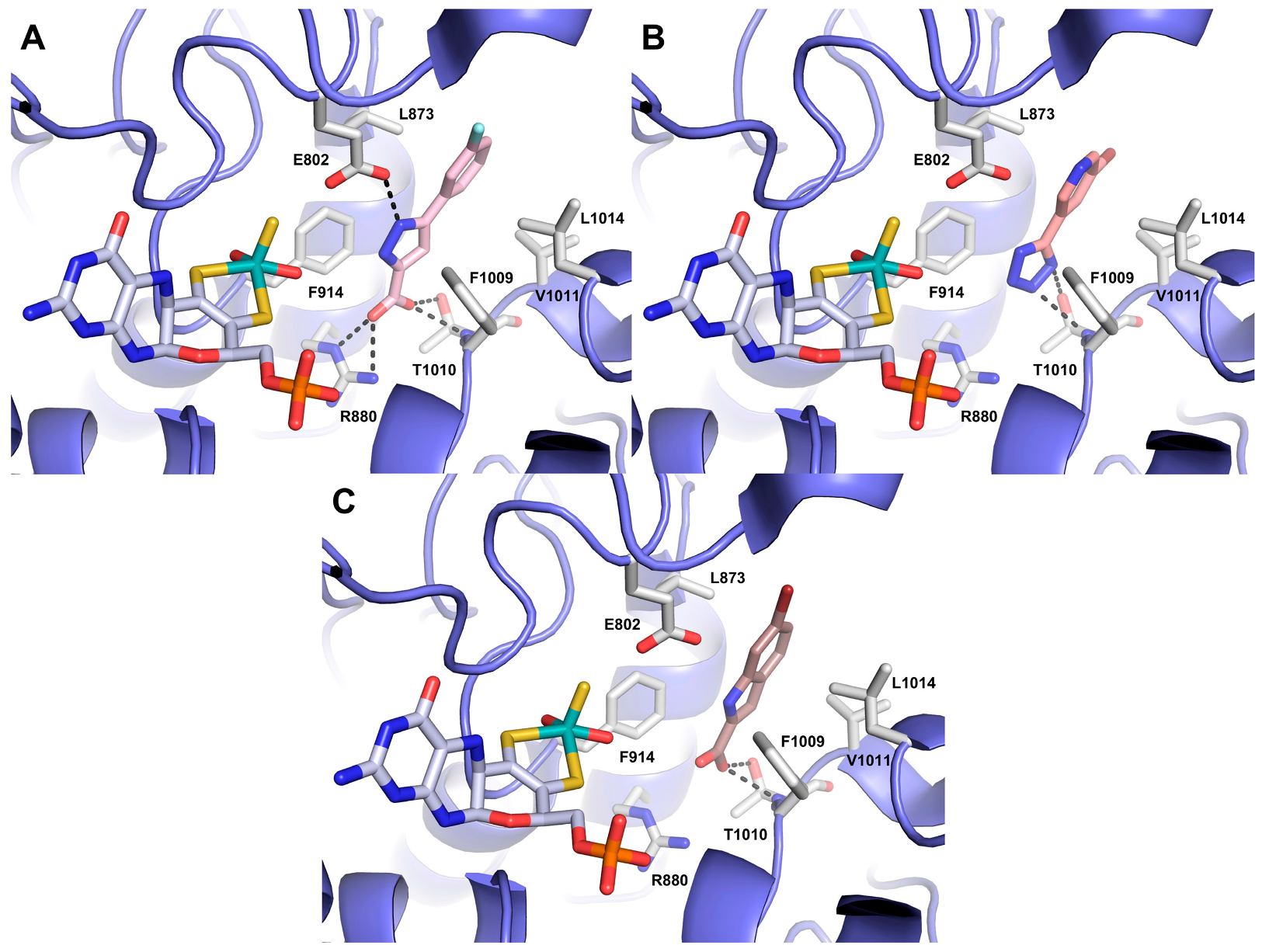
3.3. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parks, D.A.; Granger, D.N. Xanthine oxidase: Biochemistry, distribution and physiology. Acta Physiol. Scand. 1986, 126, 87–99. [Google Scholar]
- Krenitsky, T.A. Xanthine oxidase and aldehyde oxidase in purine and purine analogue metabolism. Adv. Exp. Med. Biol. 1973, 41, 57–64. [Google Scholar]
- Borges, F.; Fernandes, E.; Roleira, F. Progress Towards the Discovery of Xanthine Oxidase Inhibitors. Curr. Med. Chem. 2012, 9, 195–217. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Enroth, C.; Eger, B.T.; Okamoto, K.; Nishino, T.; Nishino, T.; Pai, E.F. Crystal structures of bovinemilk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc. Natl. Acad. Sci. USA 2000, 97, 10723–10728. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.M.; Massey, V. The oxidative half-reaction of xanthine dehydrogenase with NAD; reaction kinetics and steady-state mechanism. J. Biol. Chem. 1997, 272, 28335–28341. [Google Scholar] [CrossRef] [Green Version]
- Kelley, E.E.; Khoo, N.K.H.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Kusano, T.; Nishino, T. Chemical Nature and Reaction Mechanisms of the Molybdenum Cofactor of Xanthine Oxidoreductase. Curr. Pharm. Des. 2013, 19, 2606–2614. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Joshi, G.; Kler, H.; Kalra, S.; Kaur, M.; Arya, R. Toward an Understanding of Structural Insights of Xanthine and Aldehyde Oxidases: An Overview of their Inhibitors and Role in Various Diseases. Med. Res. Rev. 2018, 38, 1073–1125. [Google Scholar] [CrossRef]
- Della Corte, E.; Stirpe, F. The regulation of rat liver xanthine oxidase. Involvement of thiol groups in the conversion of the enzyme activity from dehydrogenase (type D) into oxidase (type O) and purification of the enzyme. Biochem. J. 1972, 126, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, T.; Nishino, T. The presence of desulfo xanthine dehydrogenase in purified and crude enzyme preparations from rat liver. Arch. Biochem. Biophys. 1986, 247, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Engerson, T.D.; McKelvey, G.; Rhyne, D.B.; Boggio, E.B.; Snyder, S.J.; Jones, H.P. Conversion of xanthine dehydrogenase to oxidase in ischemic rat tissues. J. Clin. Investig. 1987, 79, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nishino, T.; Tsushima, K. Interconversion between NAD-dependent and O2-dependent types of rat liver xanthine dehydrogenase and difference in kinetic and redox properties between them. Adv. Exp. Med. Biol. 1989, 253 B, 179–183. [Google Scholar]
- Amaya, Y.; Yamazaki, K.; Sato, M.; Noda, K.; Nishino, T.; Nishino, T. Proteolytic conversion of xanthine dehydrogenase from the NAD-dependent type to the O2-dependent type. Amino acid sequence of rat liver xanthine dehydrogenase and identification of the cleavage sites of the enzyme protein during irreversible conversion by. J. Biol. Chem. 1990, 265, 14170–14175. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef]
- Fridovich, I. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J. Biol. Chem. 1970, 245, 4053–4057. [Google Scholar] [CrossRef]
- Choi, H.K.; Curhan, G. Gout: Epidemiology and lifestyle choices. Curr. Opin. Rheumatol. 2005, 17, 341–345. [Google Scholar]
- Lü, J.M.; Yao, Q.; Chen, C. 3,4-Dihydroxy-5-nitrobenzaldehyde (DHNB) is a potent inhibitor of xanthine oxidase: A potential therapeutic agent for treatment of hyperuricemia and gout. Biochem. Pharmacol. 2013, 86, 1328–1337. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.A.; Jolly, M. Hyperuricemia and Associated Diseases. Rheum. Dis. Clin. 2006, 32, 275–293. [Google Scholar] [CrossRef]
- Dehlin, M.; Jacobsson, L.; Roddy, E. Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 2020, 16, 380–390. [Google Scholar] [CrossRef]
- Rajendran, P.; Nandakumar, N.; Rengarajan, T.; Palaniswami, R.; Gnanadhas, E.N.; Lakshminarasaiah, U.; Gopas, J.; Nishigaki, I. Antioxidants and human diseases. Clin. Chim. Acta 2014, 436, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Kökoglu, E.; Belce, A.; Özyurt, E.; Tepeler, Z. Xanthine oxidase levels in human brain tumors. Cancer Lett. 1990, 50, 179–181. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Uric acid: A new look at an old risk marker for cardiovascular disease, metabolic syndrome, and type 2 diabetes mellitus: The urate redox shuttle. Nutr. Metab. 2004, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyva, F.; Anker, S.D.; Godsland, I.F.; Teixeira, M.; Hellewell, P.G.; Kox, W.J.; Poole-Wilson, P.A.; Coats, A.J.S. Uric acid in chronic heart failure: A marker of chronic inflammation. Eur. Heart J. 1998, 19, 1814–1822. [Google Scholar] [CrossRef]
- Hare, J.M.; Johnson, R.J. Uric acid predicts clinical outcomes in heart failure: Insights regarding the role of xanthine oxidase and uric acid in disease pathophysiology. Circulation 2003, 107, 1951–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.J.; Kang, D.H.; Feig, D.; Kivlighn, S.; Kanellis, J.; Watanabe, S.; Tuttle, K.R.; Rodriguez-Iturbe, B.; Herrera-Acosta, J.; Mazzali, M. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 2003, 41, 1183–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.H.; Nakagawa, T.; Feng, L.; Watanabe, S.; Han, L.; Mazzali, M.; Truong, L.; Harris, R.; Johnson, R.J. A role for uric acid in the progression of renal disease. J. Am. Soc. Nephrol. 2002, 13, 2888–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7–11. [Google Scholar] [CrossRef]
- Kumar, R.; Darpan; Sharma, S.; Singh, R. Xanthine oxidase inhibitors: A patent survey. Expert Opin. Ther. Pat. 2011, 21, 1071–1108. [Google Scholar] [CrossRef]
- Chen, C.J.; Lü, J.M.; Yao, Q. Hyperuricemia-related diseases and xanthine oxidoreductase (XOR) inhibitors: An overview. Med. Sci. Monit. 2016, 22, 2501–2512. [Google Scholar] [CrossRef] [Green Version]
- Šmelcerović, A.; Tomović, K.; Šmelcerović, Ž.; Petronijević, Ž.; Kocić, G.; Tomašič, T.; Jakopin, Ž.; Anderluh, M. Xanthine oxidase inhibitors beyond allopurinol and febuxostat; an overview and selection of potential leads based on in silico calculated physico-chemical properties, predicted pharmacokinetics and toxicity. Eur. J. Med. Chem. 2017, 135, 491–516. [Google Scholar] [CrossRef]
- Bredemeier, M.; Lopes, L.M.; Eisenreich, M.A.; Hickmann, S.; Bongiorno, G.K.; d’Avila, R.; Morsch, A.L.B.; da Silva Stein, F.; Campos, G.G.D. Xanthine oxidase inhibitors for prevention of cardiovascular events: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2018, 18, 24. [Google Scholar] [CrossRef]
- Ferguson, L.D.; Siebert, S.; McInnes, I.B.; Sattar, N. Cardiometabolic comorbidities in RA and PsA: Lessons learned and future directions. Nat. Rev. Rheumatol. 2019, 15, 461–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, R.J.; Ye, Y.Z.; Parks, D.A.; Beckman, J.S. Urate produced during hypoxia protects heart proteins from peroxynitrite-mediated protein nitration. Free Radic. Biol. Med. 2002, 33, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Spector, T.; Johns, D.G. Stoichiometric inhibition of reduced xanthine oxidase by hydroxypyrazolo [3,4-d]pyrimidines. J. Biol. Chem. 1970, 245, 5079–5085. [Google Scholar] [CrossRef] [PubMed]
- Elion, G. The purine path to chemotherapy. Science 1989, 244, 41–47. [Google Scholar] [CrossRef]
- Agarwal, V.; Hans, N.; Messerli, F.H. Effect of Allopurinol on Blood Pressure: A Systematic Review and Meta-Analysis. J. Clin. Hypertens. 2013, 15, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Garbe, E.; Suissa, S.; LeLorier, J. Exposure to allopurinol and the risk of cataract extraction in elderly patients. Arch. Ophthalmol. 1998, 116, 1652–1656. [Google Scholar] [CrossRef] [Green Version]
- Takano, Y.; Hase-Aoki, K.; Horiuchi, H.; Zhao, L.; Kasahara, Y.; Kondo, S.; Becker, M.A. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 2005, 76, 1835–1847. [Google Scholar] [CrossRef]
- Hosoya, T.; Ohno, I.; Nomura, S.; Hisatome, I.; Uchida, S.; Fujimori, S.; Yamamoto, T.; Hara, S. Effects of topiroxostat on the serum urate levels and urinary albumin excretion in hyperuricemic stage 3 chronic kidney disease patients with or without gout. Clin. Exp. Nephrol. 2014, 18, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Kondo, S.; Pai, E.F.; Nishino, T. An extremely potent inhibitor of xanthine oxidoreductase: Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J. Biol. Chem. 2003, 278, 1848–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Matsumoto, K.; Hille, R.; Eger, B.T.; Pai, E.F.; Nishino, T. The crystal structure of xanthine oxidoreductase during catalysis: Implications for reaction mechanism and enzyme inhibition. Proc. Natl. Acad. Sci. USA 2004, 101, 7931–7936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marino, S.; Festa, C.; Zollo, F.; Nini, A.; Antenucci, L.; Raimo, G.; Iorizzi, M. Antioxidant Activity and Chemical Components as Potential Anticancer Agents in the Olive Leaf (Olea europaea L. cv Leccino.) Decoction. Anticancer. Agents Med. Chem. 2014, 14, 1376–1385. [Google Scholar] [CrossRef]
- Vitale, R.M.; Antenucci, L.; Gavagnin, M.; Raimo, G.; Amodeo, P. Structure–activity relationships of fraxamoside as an unusual xanthine oxidase inhibitor. J. Enzyme Inhib. Med. Chem. 2017, 32, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavecchia, A.; Di Giovanni, C. Virtual Screening Strategies in Drug Discovery: A Critical Review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Cerchia, C.; Roscetto, E.; Nasso, R.; Catania, M.R.; De Vendittis, E.; Lavecchia, A.; Masullo, M.; Rullo, R. In Silico Identification of Novel Inhibitors Targeting the Homodimeric Interface of Superoxide Dismutase from the Dental Pathogen Streptococcus mutans. Antioxidants 2022, 11, 785. [Google Scholar] [CrossRef]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavecchia, A. Deep learning in drug discovery: Opportunities, challenges and future prospects. Drug Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef]
- Cerchia, C.; Lavecchia, A. New avenues in artificial-intelligence-assisted drug discovery. Drug Discov. Today 2023, 28, 103516. [Google Scholar] [CrossRef]
- Luna, G.; Dolzhenko, A.V.; Mancera, R.L. Inhibitors of Xanthine Oxidase: Scaffold Diversity and Structure-Based Drug Design. ChemMedChem 2019, 14, 714–743. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Bergmeyer, H.U.; Gawehn, K.; Grassl, M. Enzymes as Biochemical Reagents; Bergmeyer, H.U., Ed.; Academic Press: New York, NY, USA, 1974; Volume 1. [Google Scholar]
- Sohn, J.; Kiburz, B.; Li, Z.; Deng, L.; Safi, A.; Pirrung, M.C.; Rudolph, J. Inhibition of Cdc25 phosphatases by indolyldihydroxyquinones. J. Med. Chem. 2003, 46, 2580–2588. [Google Scholar] [CrossRef]
- Lavecchia, A.; Di Giovanni, C.; Pesapane, A.; Montuori, N.; Ragno, P.; Martucci, N.M.; Masullo, M.; De Vendittis, E.; Novellino, E. Discovery of new inhibitors of Cdc25B dual specificity phosphatases by structure-based virtual screening. J. Med. Chem. 2012, 55, 4142–4158. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, P.; Cariati, L.; Desiderio, D.; Sgammato, R.; Lamberti, A.; Arcone, R.; Salerno, R.; Nardi, M.; Masullo, M.; Oliverio, M. Design, Synthesis, and Evaluation of Donepezil-Like Compounds as AChE and BACE-1 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 470–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Dello Russo, A.; Rullo, R.; Nitti, G.; Masullo, M.; Bocchini, V. Iron superoxide dismutase from the archaeon Sulfolobus solfataricus: Average hydrophobicity and amino acid weight are involved in the adaptation of proteins to extreme environments. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1997, 1343, 23–30. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Structure | Steady-State Determination (a) | |
|---|---|---|---|
| Concentration Interval of the Inhibitor | IC50 µM | ||
| ALS-28 |  | 2–40 µM | 18 |
| ALS-8 |  | 3–40 µM | 30 |
| ALS-15 |  | 5–60 µM | 64 |
| ALS-1 |  | 10–100 µM | 82 |
| Inhibitor | Kinetic Determinationa | |||
|---|---|---|---|---|
| Concentration of the Inhibitor (µM) | KM (µM) | Vmax (ΔE/min) | Ki (µM) | |
| None | 5.9 ± 1.2 (n = 8) | 0.107 ± 0.021 (n = 8) | ||
| ALS-28 | 3 | 16.8 ± 1.5 (n = 2) | 0.113 ± 0.007 (n = 4) | 2.7 ± 1.5 (n = 4) |
| 10 | 23.2 ± 5.1 (n = 2) | |||
| ALS-8 | 7.5 | 19.8 ± 1.4 (n = 2) | 0.093 ± 0.007 (n = 4) | 4.5 ± 1.5 (n = 4) |
| 25 | 31.8 ± 2.0 (n = 2) | |||
| ALS-15 | 12 | 10.3 ± 2.0 (n = 2) | 0.086 ± 0.003 (n = 4) | 23 ± 9 (n = 4) |
| 30 | 11.9 ± 0.8 (n = 2) | |||
| ALS-1 | 30 | 10.5 ± 1.1 (n = 2) | 0.087 ± 0.004 (n = 2) | 41 ± 14 (n = 2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rullo, R.; Cerchia, C.; Nasso, R.; Romanelli, V.; Vendittis, E.D.; Masullo, M.; Lavecchia, A. Novel Reversible Inhibitors of Xanthine Oxidase Targeting the Active Site of the Enzyme. Antioxidants 2023, 12, 825. https://doi.org/10.3390/antiox12040825
Rullo R, Cerchia C, Nasso R, Romanelli V, Vendittis ED, Masullo M, Lavecchia A. Novel Reversible Inhibitors of Xanthine Oxidase Targeting the Active Site of the Enzyme. Antioxidants. 2023; 12(4):825. https://doi.org/10.3390/antiox12040825
Chicago/Turabian StyleRullo, Rosario, Carmen Cerchia, Rosarita Nasso, Virgilio Romanelli, Emmanuele De Vendittis, Mariorosario Masullo, and Antonio Lavecchia. 2023. "Novel Reversible Inhibitors of Xanthine Oxidase Targeting the Active Site of the Enzyme" Antioxidants 12, no. 4: 825. https://doi.org/10.3390/antiox12040825