
Antagonistic Interactions in Mitochondria ROS Signaling Responses to Manganese
 ,
,
Abstract
:
{kind=link}
{kind=link}
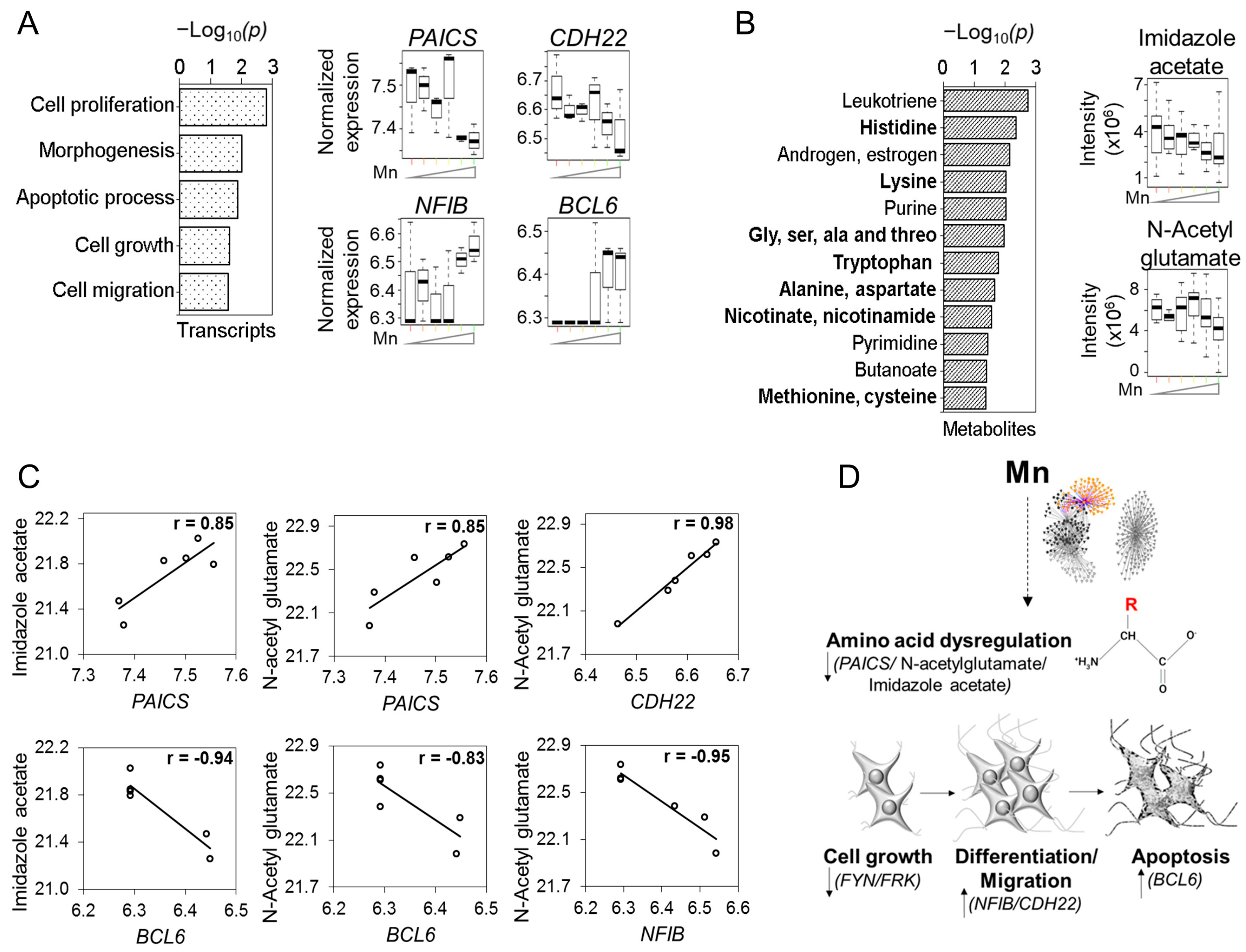{kind=link}
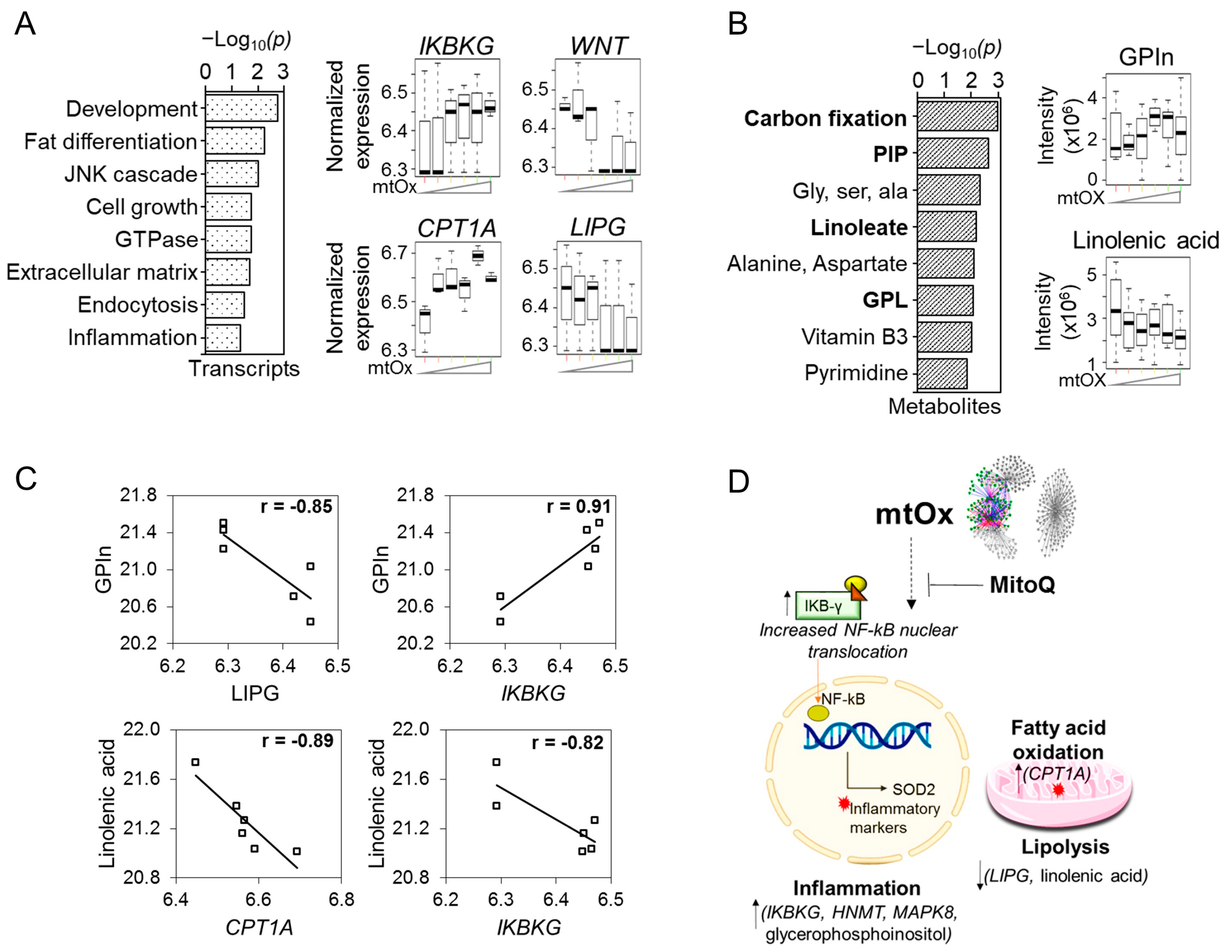{kind=link}
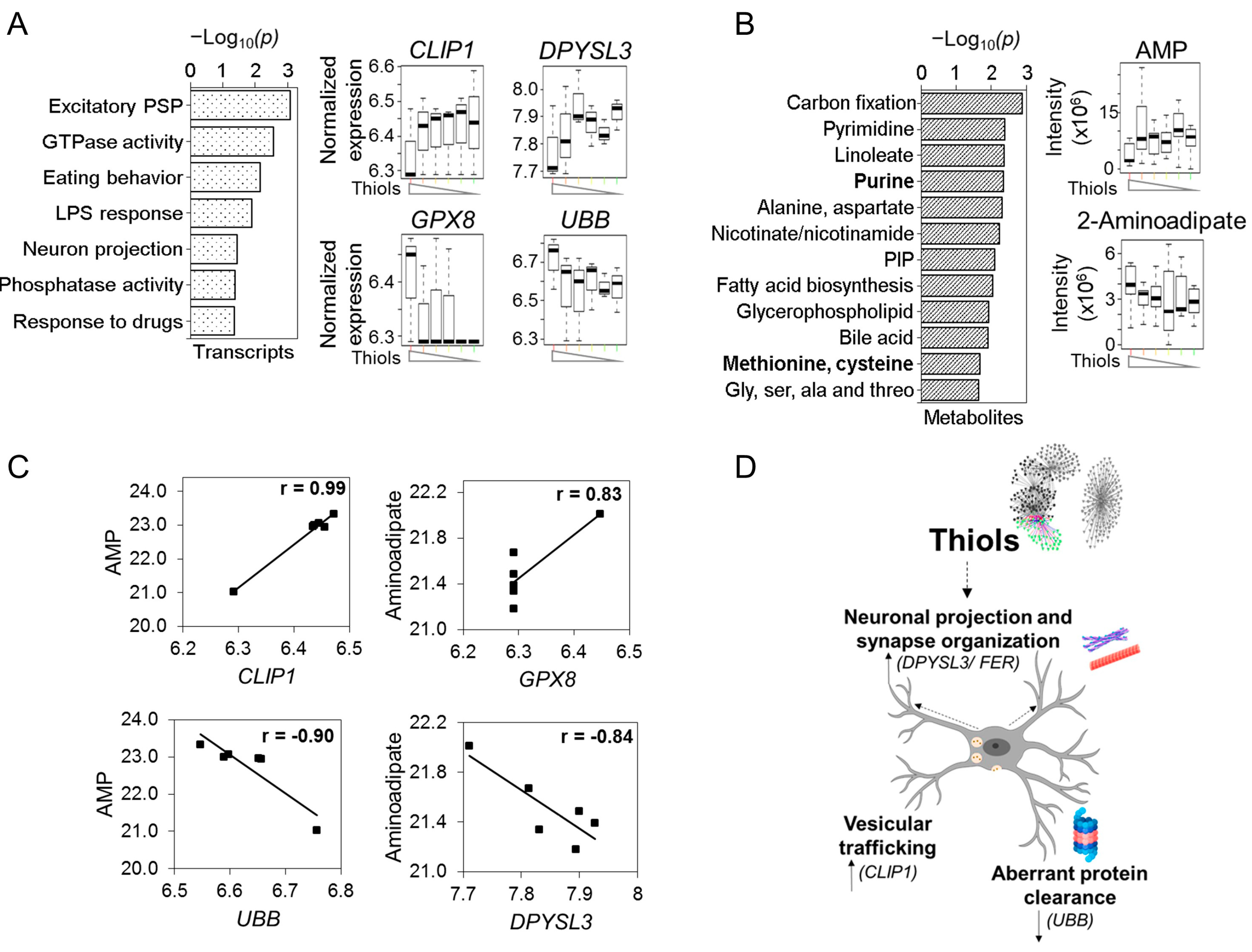{kind=link}
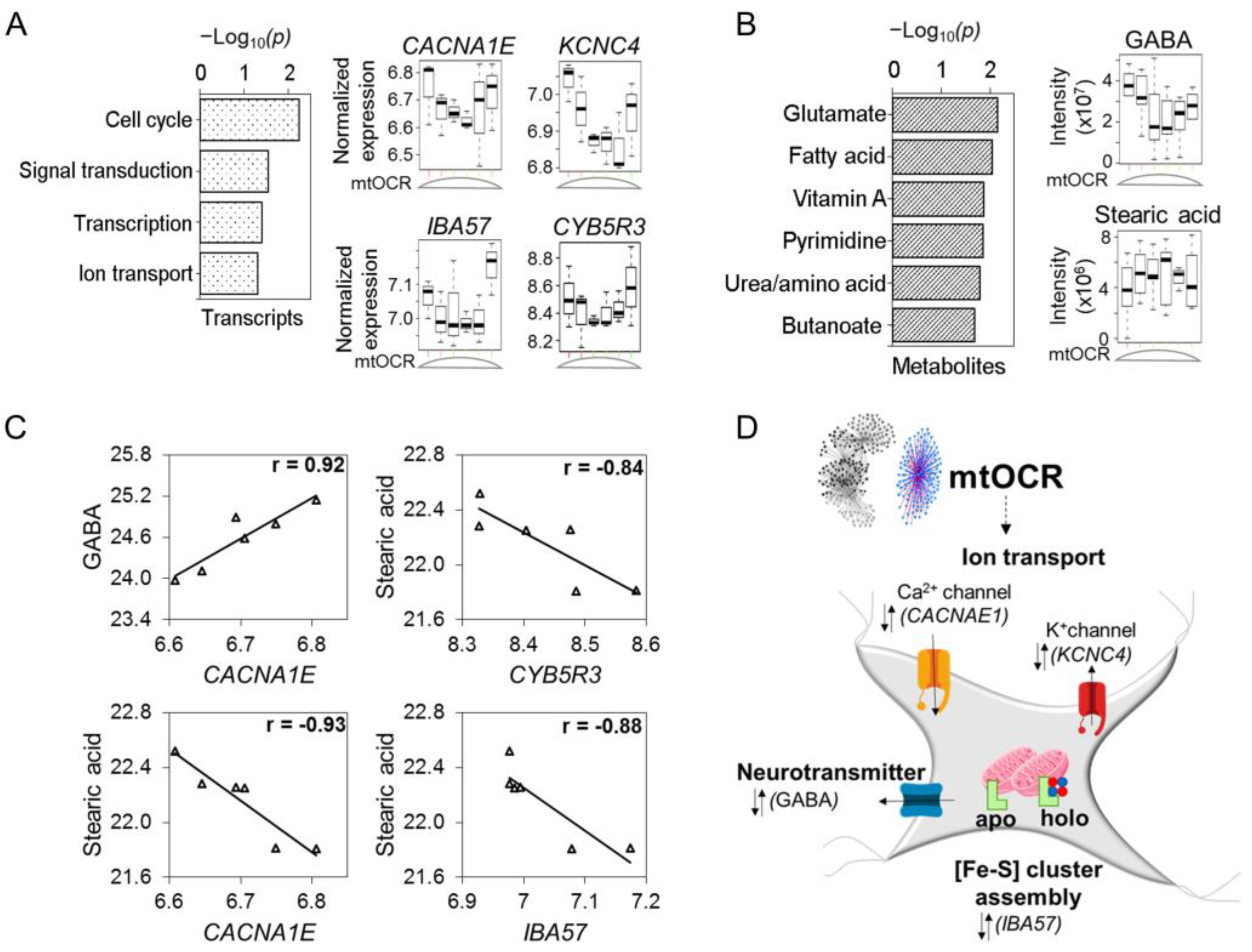{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Mn Dose Treatment, Big Data Acquisition
2.2. Integrated Multi-Omics Network Analysis
2.3. Pathway Analysis for Selected Metabolic Features and Genes
2.4. Metabolite Annotation and Identification
2.5. Subcellular Fractionation and Western Blotting
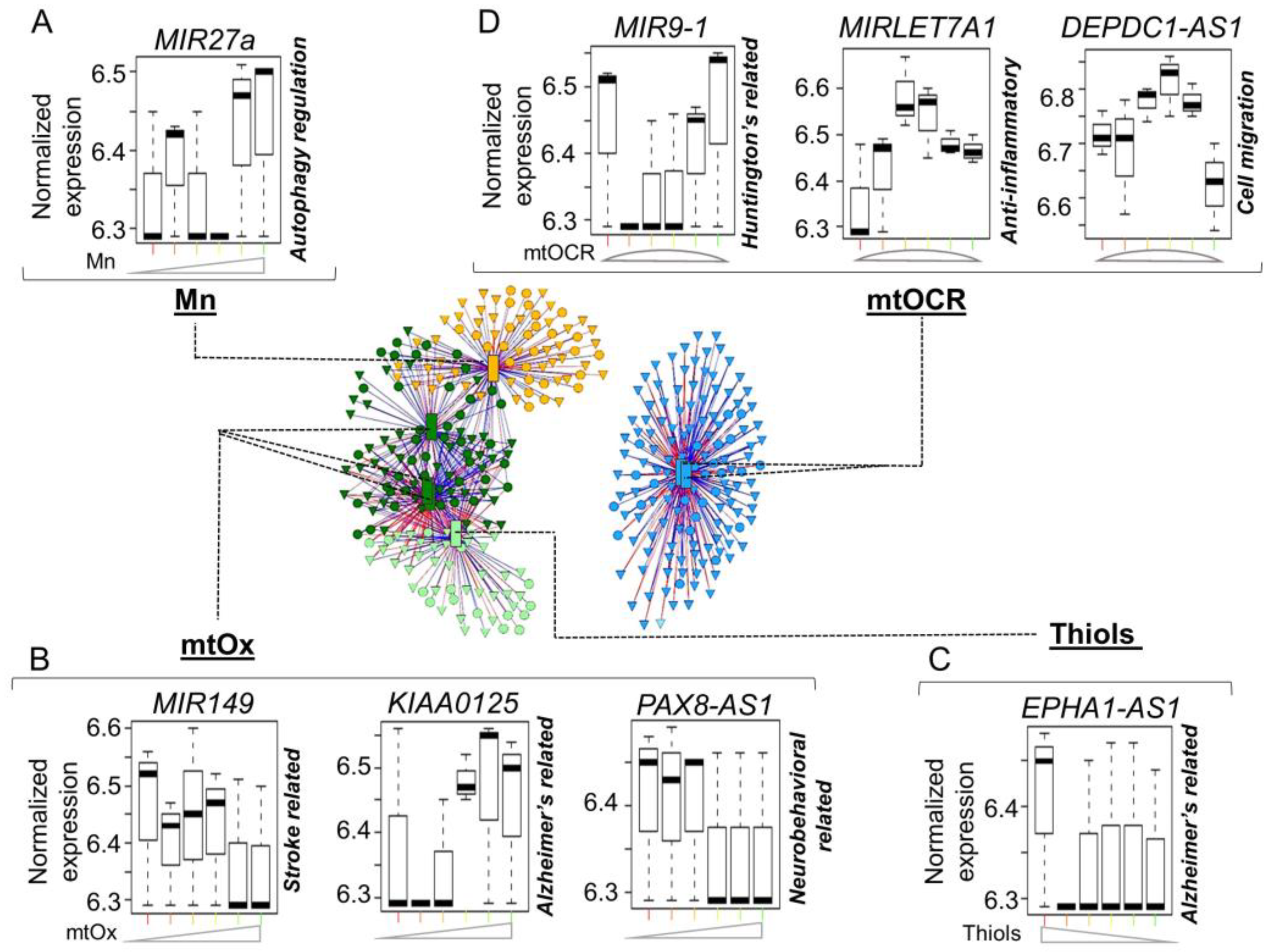
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef] [Green Version]
- Roede, J.R.; Uppal, K.; Park, Y.; Tran, V.; Jones, D.P. Transcriptome-metabolome wide association study (TMWAS) of maneb and paraquat neurotoxicity reveals network level interactions in toxicologic mechanism. Toxicol. Rep. 2014, 1, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sullivan, N.L.; Rouphael, N.; Yu, T.; Banton, S.; Maddur, M.S.; McCausland, M.; Chiu, C.; Canniff, J.; Dubey, S.; et al. Metabolic Phenotypes of Response to Vaccination in Humans. Cell 2017, 169, 862–877.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, J.D.; Hu, X.; Ko, E.J.; Park, S.; Lee, Y.T.; Orr, M.; Fernandes, J.; Uppal, K.; Kang, S.M.; Jones, D.P.; et al. Metabolic pathways of lung inflammation revealed by high-resolution metabolomics (HRM) of H1N1 influenza virus infection in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R906–R916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, O.B.; Aguirre-Gamboa, R.; Sanna, S.; Oosting, M.; Smeekens, S.P.; Jaeger, M.; Zorro, M.; Vosa, U.; Withoff, S.; Netea-Maier, R.T.; et al. Integration of multi-omics data and deep phenotyping enables prediction of cytokine responses. Nat. Immunol. 2018, 19, 776–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, Y.M.; Roede, J.R.; Orr, M.; Liang, Y.; Jones, D.P. Integrated redox proteomics and metabolomics of mitochondria to identify mechanisms of cd toxicity. Toxicol. Sci. 2014, 139, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.; Uppal, K.; Jones, D.P. Central Mitochondrial Signaling Mechanisms in Response to Environmental Agents. In Mitochondrial Dysfunction Caused by Drugs and Environmental Toxicants; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 639–654. [Google Scholar] [CrossRef]
- Dennis, K.K.; Jones, D.P. The Exposome: A New Frontier for Education. Am. Biol. Teach. 2016, 78, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Cortese-Krott, M.M.; Koning, A.; Kuhnle, G.G.C.; Nagy, P.; Bianco, C.L.; Pasch, A.; Wink, D.A.; Fukuto, J.M.; Jackson, A.A.; van Goor, H.; et al. The Reactive Species Interactome: Evolutionary Emergence, Biological Significance, and Opportunities for Redox Metabolomics and Personalized Medicine. Antioxid. Redox Signal. 2017, 27, 684–712. [Google Scholar] [CrossRef] [Green Version]
- Li-Pook-Than, J.; Snyder, M. iPOP goes the world: Integrated personalized Omics profiling and the road toward improved health care. Chem. Biol. 2013, 20, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Go, Y.M.; Fernandes, J.; Hu, X.; Uppal, K.; Jones, D.P. Mitochondrial network responses in oxidative physiology and disease. Free Radic Biol. Med. 2018, 116, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res. 2007, 35, 7475–7484. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.J.; Nguyen, A.Q. Antagonistic pleiotropy as a widespread mechanism for the maintenance of polymorphic disease alleles. BMC Med. Genet. 2011, 12, 160. [Google Scholar] [CrossRef] [Green Version]
- Austad, S.N.; Hoffman, J.M. Is antagonistic pleiotropy ubiquitous in aging biology? Evol. Med. Public Health 2018, 2018, 287–294. [Google Scholar] [CrossRef]
- Hashimoto, M.; Ho, G.; Takamatsu, Y.; Shimizu, Y.; Sugama, S.; Takenouchi, T.; Waragai, M.; Masliah, E. Evolvability and Neurodegenerative Disease: Antagonistic Pleiotropy Phenomena Derived from Amyloid Aggregates. J. Parkinsons Dis. 2018, 8, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Wang, M.C. Aging: Antagonistic Pleiotropy Supported by Gut Eating. Curr. Biol. 2018, 28, R890–R892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubev, A.; Hanson, A.D.; Gladyshev, V.N. A Tale of Two Concepts: Harmonizing the Free Radical and Antagonistic Pleiotropy Theories of Aging. Antioxid. Redox Signal. 2018, 29, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- ATSDR. Toxicological Profile for Manganese; United States Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2012. [Google Scholar]
- Bouchard, M.F.; Sauve, S.; Barbeau, B.; Legrand, M.; Brodeur, M.E.; Bouffard, T.; Limoges, E.; Bellinger, D.C.; Mergler, D. Intellectual impairment in school-age children exposed to manganese from drinking water. Environ. Health Perspect. 2011, 119, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Bowler, R.M.; Mergler, D.; Sassine, M.P.; Larribe, F.; Hudnell, K. Neuropsychiatric effects of manganese on mood. Neurotoxicology 1999, 20, 367–378. [Google Scholar]
- Claus Henn, B.; Ettinger, A.S.; Schwartz, J.; Tellez-Rojo, M.M.; Lamadrid-Figueroa, H.; Hernandez-Avila, M.; Schnaas, L.; Amarasiriwardena, C.; Bellinger, D.C.; Hu, H.; et al. Early postnatal blood manganese levels and children’s neurodevelopment. Epidemiology 2010, 21, 433–439. [Google Scholar] [CrossRef]
- Kessissoglou, D.P. (Ed.) Manganese-Proteins and -Enzymes and Relevant Trinuclear Synthetic Complexes. In Bioinorganic Chemistry: An Inorganic Perspective of Life; Springer: Dordrecht, The Netherlands, 1995; pp. 299–320. [Google Scholar]
- Smith, M.R.; Fernandes, J.; Go, Y.M.; Jones, D.P. Redox dynamics of manganese as a mitochondrial life-death switch. Biochem. Biophys. Res. Commun. 2017, 482, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Fu, J.; Zhou, Z. In vitro effect of manganese chloride exposure on reactive oxygen species generation and respiratory chain complexes activities of mitochondria isolated from rat brain. Toxicol. In Vitro 2004, 18, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Finley, E.J.; Gavin, C.E.; Aschner, M.; Gunter, T.E. Manganese neurotoxicity and the role of reactive oxygen species. Free Radic Biol. Med. 2013, 62, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.; Hao, L.; Bijli, K.M.; Chandler, J.D.; Orr, M.; Hu, X.; Jones, D.P.; Go, Y.M. From the Cover: Manganese Stimulates Mitochondrial H2O2 Production in SH-SY5Y Human Neuroblastoma Cells over Physiologic as well as Toxicologic Range. Toxicol. Sci. 2017, 155, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Aschner, M.; Aschner, J.L. Manganese transport across the blood-brain barrier: Relationship to iron homeostasis. Brain Res. Bull. 1990, 24, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Gavin, C.E.; Gunter, K.K.; Gunter, T.E. Manganese and calcium transport in mitochondria: Implications for manganese toxicity. Neurotoxicology 1999, 20, 445–453. [Google Scholar]
- Pinsino, A.; Roccheri, M.C.; Costa, C.; Matranga, V. Manganese interferes with calcium, perturbs ERK signaling, and produces embryos with no skeleton. Toxicol. Sci. 2011, 123, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Gavin, C.E.; Gunter, K.K.; Gunter, T.E. Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol. Appl. Pharmacol. 1992, 115, 1–5. [Google Scholar] [CrossRef]
- Galvani, P.; Fumagalli, P.; Santagostino, A. Vulnerability of mitochondrial complex I in PC12 cells exposed to manganese. Eur. J. Pharmacol. 1995, 293, 377–383. [Google Scholar] [CrossRef]
- Fernandes, J.; Chandler, J.D.; Lili, L.N.; Uppal, K.; Hu, X.; Hao, L.; Go, Y.M.; Jones, D.P. Transcriptome Analysis Reveals Distinct Responses to Physiologic versus Toxic Manganese Exposure in Human Neuroblastoma Cells. Front. Genet. 2019, 10, 676. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.; Chandler, J.D.; Liu, K.H.; Uppal, K.; Hao, L.; Hu, X.; Go, Y.M.; Jones, D.P. Metabolomic Responses to Manganese Dose in SH-SY5Y Human Neuroblastoma Cells. Toxicol. Sci. 2019, 169, 89–94. [Google Scholar] [CrossRef]
- Uppal, K.; Ma, C.; Go, Y.M.; Jones, D.P.; Wren, J. xMWAS: A data-driven integration and differential network analysis tool. Bioinformatics 2018, 34, 701–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Park, Y.; Johnson, J.M.; Jones, D.P. apLCMS—adaptive processing of high-resolution LC/MS data. Bioinformatics 2009, 25, 1930–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppal, K.; Soltow, Q.A.; Strobel, F.H.; Pittard, W.S.; Gernert, K.M.; Yu, T.; Jones, D.P. xMSanalyzer: Automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data. BMC Bioinform. 2013, 14, 15. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef] [Green Version]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [Green Version]
- Uppal, K.; Walker, D.I.; Jones, D.P. xMSannotator: An R Package for Network-Based Annotation of High-Resolution Metabolomics Data. Anal. Chem. 2017, 89, 1063–1067. [Google Scholar] [CrossRef] [Green Version]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saarimaki-Vire, J.; Alitalo, A.; Partanen, J. Analysis of Cdh22 expression and function in the developing mouse brain. Dev. Dyn. 2011, 240, 1989–2001. [Google Scholar] [CrossRef]
- Harris, L.; Genovesi, L.A.; Gronostajski, R.M.; Wainwright, B.J.; Piper, M. Nuclear factor one transcription factors: Divergent functions in developmental versus adult stem cell populations. Dev. Dyn. 2015, 244, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Bai, M.; Agnantis, N.J.; Skyrlas, A.; Tsanou, E.; Kamina, S.; Galani, V.; Kanavaros, P. Increased expression of the bcl6 and CD10 proteins is associated with increased apoptosis and proliferation in diffuse large B-cell lymphomas. Mod. Pathol. 2003, 16, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Anneren, C.; Lindholm, C.K.; Kriz, V.; Welsh, M. The FRK/RAK-SHB signaling cascade: A versatile signal-transduction pathway that regulates cell survival, differentiation and proliferation. Curr. Mol. Med. 2003, 3, 313–324. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [Green Version]
- Imamura, I.; Watanabe, T.; Hase, Y.; Sakamoto, Y.; Fukuda, Y.; Yamamoto, H.; Tsuruhara, T.; Wada, H. Histamine metabolism in patients with histidinemia: Determination of urinary levels of histamine, N tau-methylhistamine, imidazole acetic acid, and its conjugate(s). J. Biochem. 1984, 96, 1925–1929. [Google Scholar] [CrossRef]
- Shi, D.; Caldovic, L.; Tuchman, M. Sources and Fates of Carbamyl Phosphate: A Labile Energy-Rich Molecule with Multiple Facets. Biology 2018, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Maubach, G.; Schmadicke, A.C.; Naumann, M. NEMO Links Nuclear Factor-kappaB to Human Diseases. Trends Mol. Med. 2017, 23, 1138–1155. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.Z.; Shang, Y.C.; Hou, J.; Maiese, K. Wnt1 neuroprotection translates into improved neurological function during oxidant stress and cerebral ischemia through AKT1 and mitochondrial apoptotic pathways. Oxidative Med. Cell. Longev. 2010, 3, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.E.; Marubio, L.M.; Deal, C.R.; Hans, M.; Brust, P.F.; Philipson, L.H.; Miller, R.J.; Johnson, E.C.; Harpold, M.M.; Ellis, S.B. Structure and functional characterization of neuronal alpha 1E calcium channel subtypes. J. Biol. Chem. 1994, 269, 22347–22357. [Google Scholar] [CrossRef] [PubMed]
- Nyholt, D.R.; LaForge, K.S.; Kallela, M.; Alakurtti, K.; Anttila, V.; Farkkila, M.; Hamalainen, E.; Kaprio, J.; Kaunisto, M.A.; Heath, A.C.; et al. A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum. Mol. Genet. 2008, 17, 3318–3331. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, A.; D’Onofrio, M.; Buzzi, M.G.; Arisi, I.; Grieco, G.S.; Pierelli, F.; Santorelli, F.M.; Schoenen, J. Possible Involvement of the CACNA1E Gene in Migraine: A Search for Single Nucleotide Polymorphism in Different Clinical Phenotypes. Headache 2017, 57, 1136–1144. [Google Scholar] [CrossRef]
- Pascual, J.M.; Carceller, F.; Roda, J.M.; Cerdan, S. Glutamate, glutamine, and GABA as substrates for the neuronal and glial compartments after focal cerebral ischemia in rats. Stroke 1998, 29, 1048–1056; discussion 1056–1047. [Google Scholar] [CrossRef] [Green Version]
- Kamal, M.A.; Mushtaq, G.; Greig, N.H. Current Update on Synopsis of miRNA Dysregulation in Neurological Disorders. CNS Neurol. Disord. Drug Targets 2015, 14, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhuang, Y.; Zhao, X.; Li, X. Long Non-coding RNA in Neuronal Development and Neurological Disorders. Front. Genet. 2018, 9, 744. [Google Scholar] [CrossRef] [Green Version]
- Stern, M. Evidence that a mitochondrial death spiral underlies antagonistic pleiotropy. Aging Cell 2017, 16, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Mauck, K.E.; Chesnais, Q.; Shapiro, L.R. Evolutionary Determinants of Host and Vector Manipulation by Plant Viruses. Adv. Virus Res. 2018, 101, 189–250. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chiao, Y.A.; Martin, G.M.; Marcinek, D.J.; Basisty, N.; Quarles, E.K.; Rabinovitch, P.S. Mitochondrial-Targeted Catalase: Extended Longevity and the Roles in Various Disease Models. Prog. Mol. Biol. Transl. 2017, 146, 203–241. [Google Scholar] [CrossRef]
- Plachez, C.; Lindwall, C.; Sunn, N.; Piper, M.; Moldrich, R.X.; Campbell, C.E.; Osinski, J.M.; Gronostajski, R.M.; Richards, L.J. Nuclear factor I gene expression in the developing forebrain. J. Comp. Neurol. 2008, 508, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.D.; Nukala, V.N.; Sullivan, P.G. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenergetics 2013, 5, 10. [Google Scholar] [CrossRef]
- Debray, F.G.; Stumpfig, C.; Vanlander, A.V.; Dideberg, V.; Josse, C.; Caberg, J.H.; Boemer, F.; Bours, V.; Stevens, R.; Seneca, S.; et al. Mutation of the iron-sulfur cluster assembly gene IBA57 causes fatal infantile leukodystrophy. J. Inherit. Metab. Dis. 2015, 38, 1147–1153. [Google Scholar] [CrossRef]
- Chugunova, A.; Loseva, E.; Mazin, P.; Mitina, A.; Navalayeu, T.; Bilan, D.; Vishnyakova, P.; Marey, M.; Golovina, A.; Serebryakova, M.; et al. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 4940–4945. [Google Scholar] [CrossRef] [Green Version]
- Strittmatter, P.; Spatz, L.; Corcoran, D.; Rogers, M.J.; Setlow, B.; Redline, R. Purification and properties of rat liver microsomal stearyl coenzyme A desaturase. Proc. Natl. Acad. Sci. USA 1974, 71, 4565–4569. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, H.M.; Swerdlow, R.H. Relationships Between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Curr. Top. Med. Chem. 2016, 16, 849–857. [Google Scholar] [CrossRef]
- Verri, M.; Pastoris, O.; Dossena, M.; Aquilani, R.; Guerriero, F.; Cuzzoni, G.; Venturini, L.; Ricevuti, G.; Bongiorno, A.I. Mitochondrial alterations, oxidative stress and neuroinflammation in Alzheimer’s disease. Int. J. Immunopathol. Pharmacol. 2012, 25, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Peruzzotti-Jametti, L.; Pluchino, S. Targeting Mitochondrial Metabolism in Neuroinflammation: Towards a Therapy for Progressive Multiple Sclerosis. Trends Mol. Med. 2018, 24, 838–855. [Google Scholar] [CrossRef] [Green Version]
- Bavner, A.; Shafaati, M.; Hansson, M.; Olin, M.; Shpitzen, S.; Meiner, V.; Leitersdorf, E.; Bjorkhem, I. On the mechanism of accumulation of cholestanol in the brain of mice with a disruption of sterol 27-hydroxylase. J. Lipid Res. 2010, 51, 2722–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, S.; Chen, G.; Cao, X.; Zhang, Y. Cerebrotendinous xanthomatosis: A comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet. J. Rare Dis. 2014, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Lund, E.G.; Horton, J.D.; Leitersdorf, E.; Russell, D.W.; Dietschy, J.M.; Turley, S.D. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. J. Biol. Chem. 2000, 275, 39685–39692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balcerczyk, A.; Bartosz, G. Thiols are main determinants of total antioxidant capacity of cellular homogenates. Free Radic Res. 2003, 37, 537–541. [Google Scholar] [CrossRef]
- Go, Y.M.; Orr, M.; Jones, D.P. Actin cytoskeleton redox proteome oxidation by cadmium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L831–L843. [Google Scholar] [CrossRef] [Green Version]
- Rosslenbroich, V.; Dai, L.; Baader, S.L.; Noegel, A.A.; Gieselmann, V.; Kappler, J. Collapsin response mediator protein-4 regulates F-actin bundling. Exp. Cell Res. 2005, 310, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Shapovalova, Z.; Tabunshchyk, K.; Greer, P.A. The Fer tyrosine kinase regulates an axon retraction response to Semaphorin 3A in dorsal root ganglion neurons. BMC Dev. Biol. 2007, 7, 133. [Google Scholar] [CrossRef] [Green Version]
- Larti, F.; Kahrizi, K.; Musante, L.; Hu, H.; Papari, E.; Fattahi, Z.; Bazazzadegan, N.; Liu, Z.; Banan, M.; Garshasbi, M.; et al. A defect in the CLIP1 gene (CLIP-170) can cause autosomal recessive intellectual disability. Eur. J. Hum. Genet. 2015, 23, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, J.A.; Sunderram, J. Oxygen-sensing neurons in the central nervous system. J. Appl. Physiol. 2004, 96, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppal, K.; Walker, D.I.; Liu, K.; Li, S.; Go, Y.M.; Jones, D.P. Computational Metabolomics: A Framework for the Million Metabolome. Chem. Res. Toxicol. 2016, 29, 1956–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Go, Y.M.; Jones, D.P. Omics Integration for Mitochondria Systems Biology. Antioxid. Redox Signal. 2020, 32, 853–872. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, J.; Uppal, K.; Liu, K.H.; Hu, X.; Orr, M.; Tran, V.; Go, Y.-M.; Jones, D.P. Antagonistic Interactions in Mitochondria ROS Signaling Responses to Manganese. Antioxidants 2023, 12, 804. https://doi.org/10.3390/antiox12040804
Fernandes J, Uppal K, Liu KH, Hu X, Orr M, Tran V, Go Y-M, Jones DP. Antagonistic Interactions in Mitochondria ROS Signaling Responses to Manganese. Antioxidants. 2023; 12(4):804. https://doi.org/10.3390/antiox12040804
Chicago/Turabian StyleFernandes, Jolyn, Karan Uppal, Ken H. Liu, Xin Hu, Michael Orr, ViLinh Tran, Young-Mi Go, and Dean P. Jones. 2023. "Antagonistic Interactions in Mitochondria ROS Signaling Responses to Manganese" Antioxidants 12, no. 4: 804. https://doi.org/10.3390/antiox12040804