
Activators of Nrf2 to Counteract Neurodegenerative Diseases



Abstract
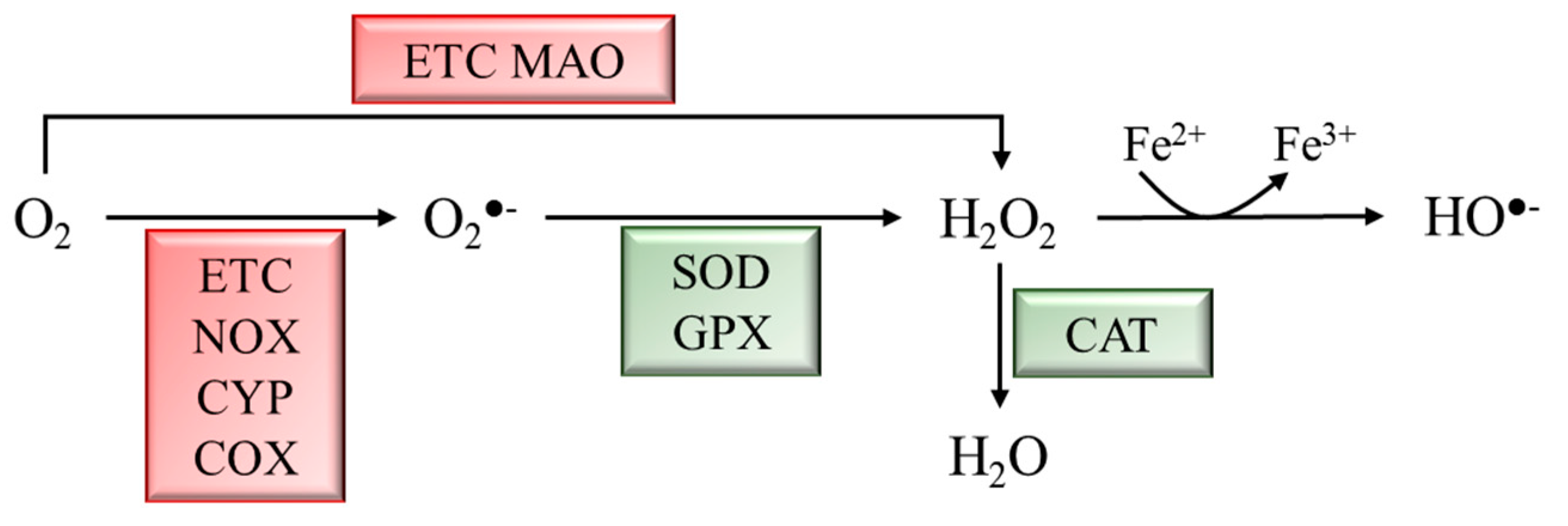
:1. Introduction
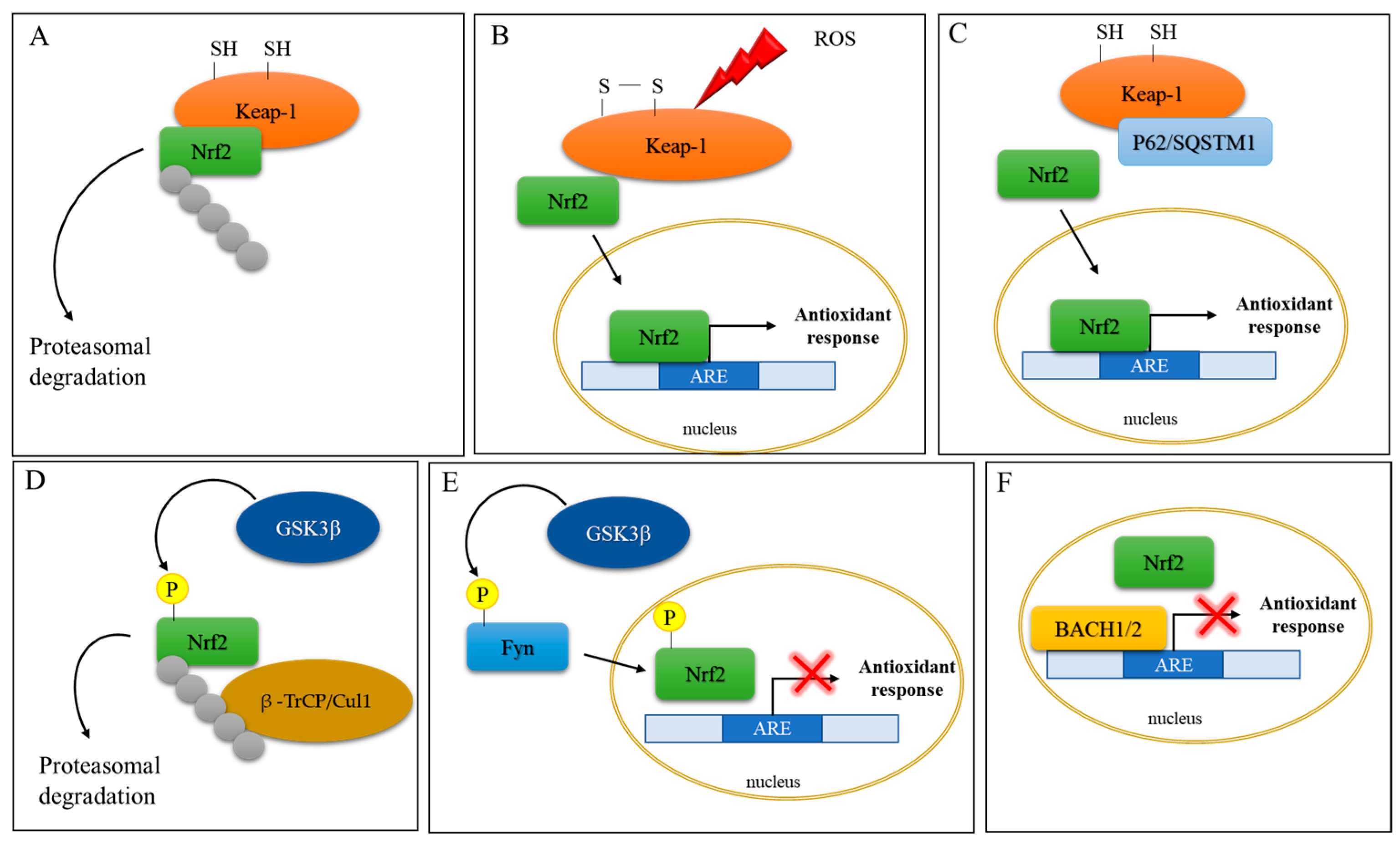
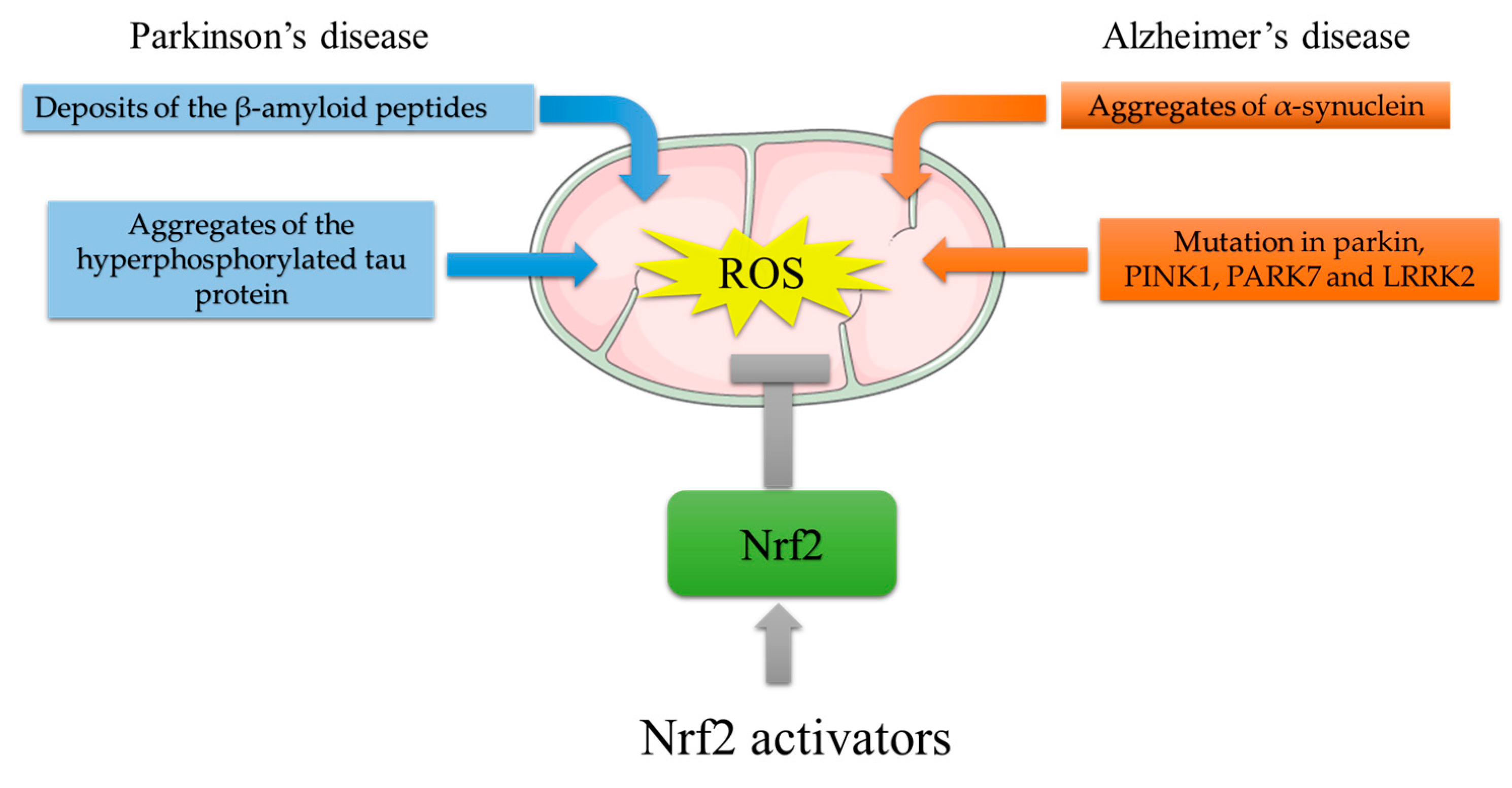
2. Implication of Oxidative Stress and Nrf2 Activation in Neurodegenerative Diseases
2.1. Nrf2 and Parkinson’s Disease
2.2. Nrf2 and Alzheimer’s Disease
3. Small Molecules Inducing Nrf2
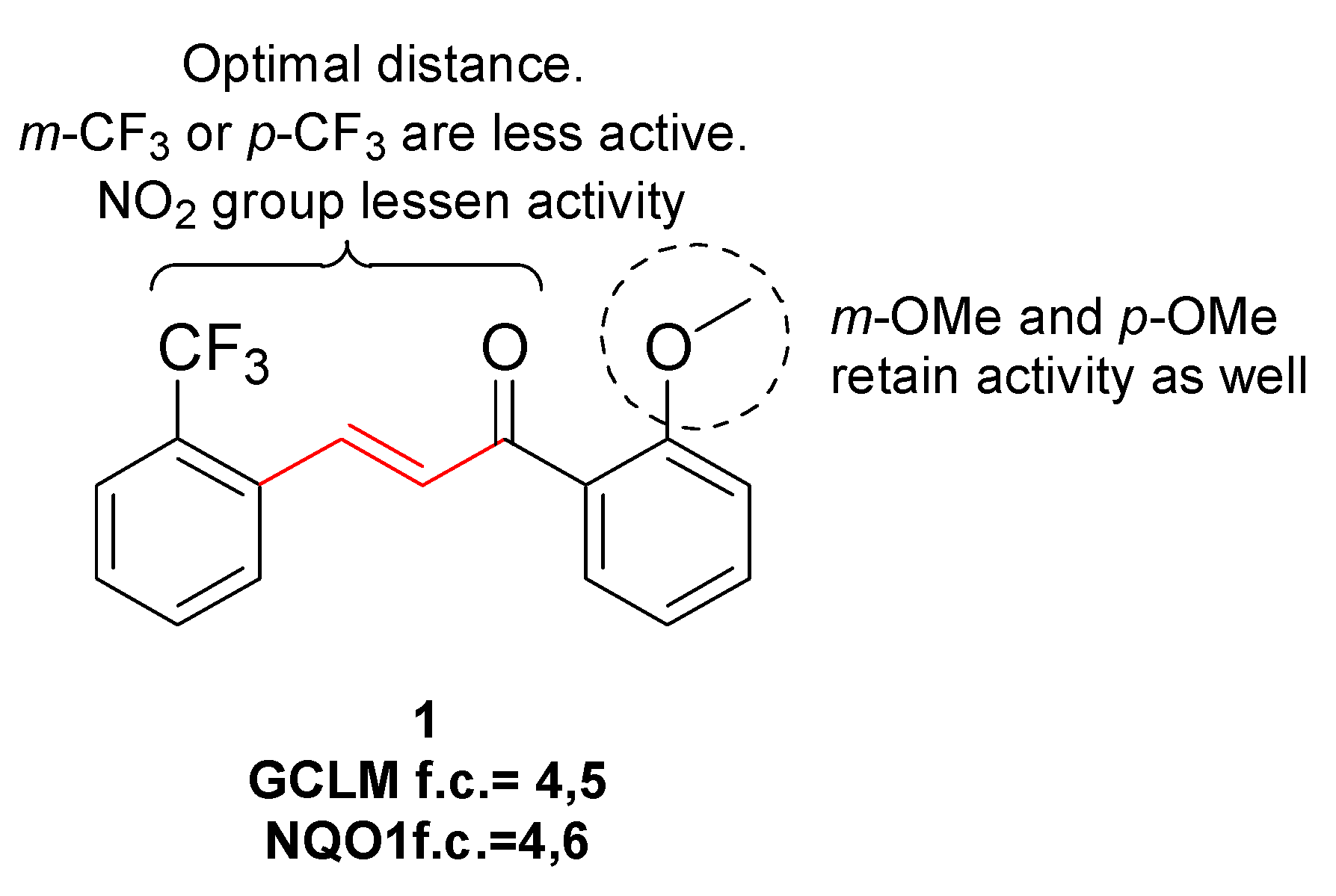
3.1. Electrophilic Activators
3.2. PPI Interfering Compounds: Azole-Based Compounds

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Activity | Cell Line | Ref. |
|---|---|---|---|
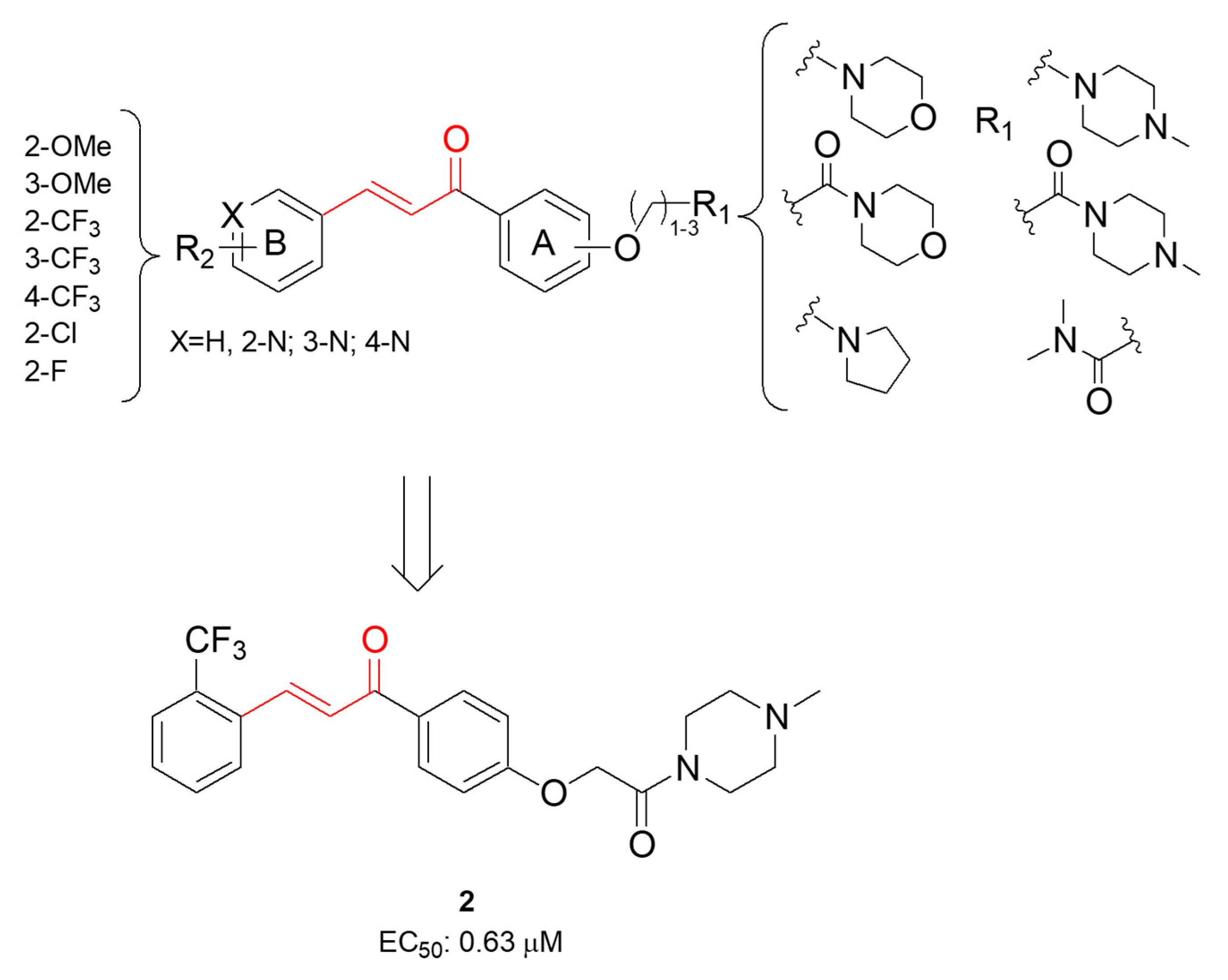
| 2 | Activation of Nrf2 nuclear translocation (EC50 = 0.63 μM) | BV-2 microglial cells | [68] |
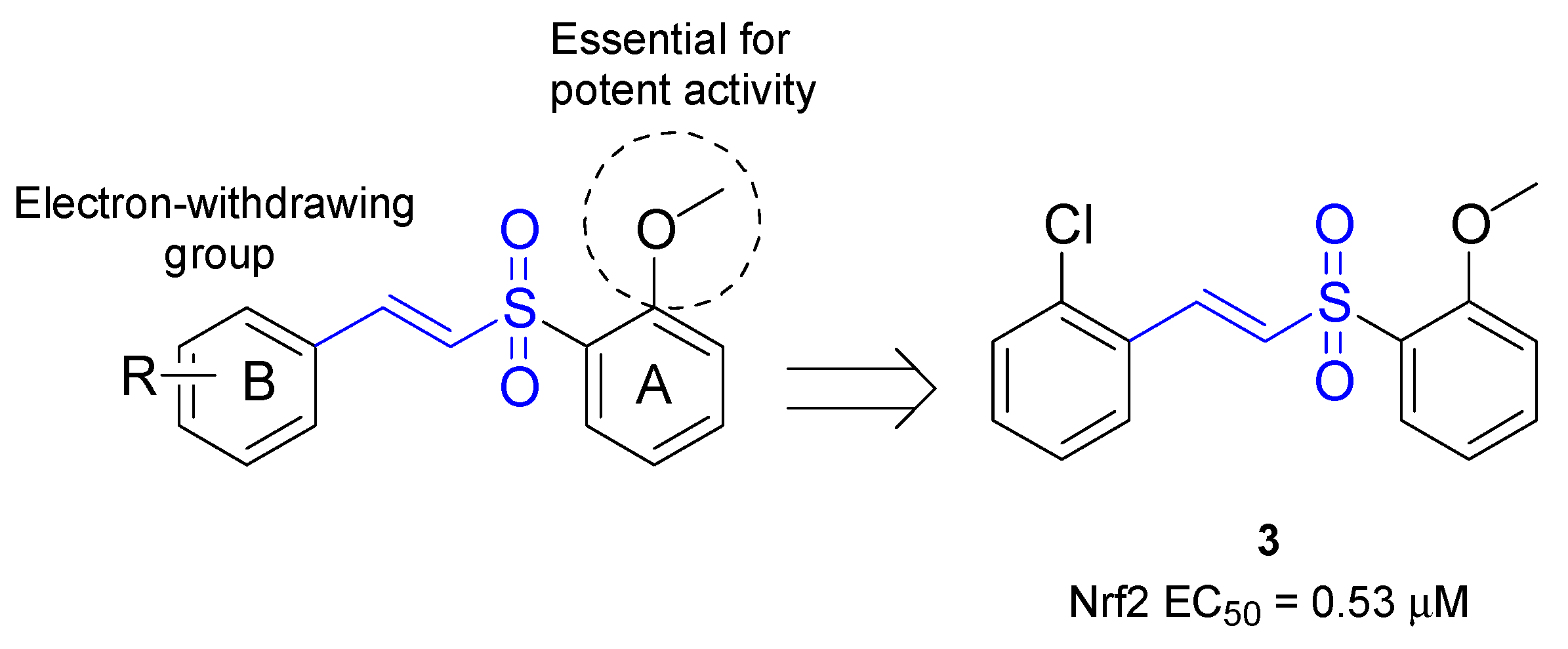
| 3 | Activation of Nrf2 nuclear translocation (EC50 = 0.53 μM) | CATH.a, mouse DAergic neuronal cell line | [70] |
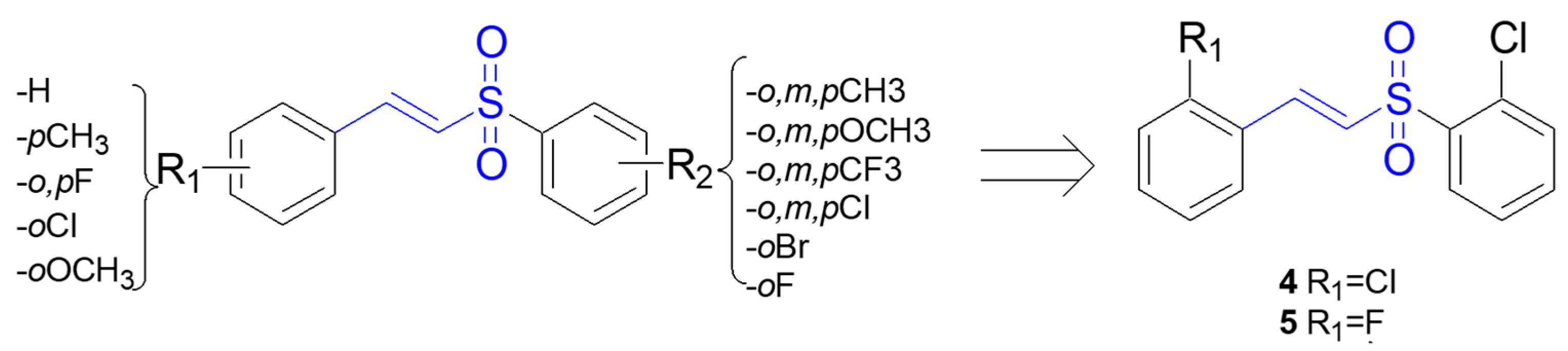
| 4, 5 | Activation of Nrf2 nuclear translocation; transcriptional activation of NRF2-responsive ARE genes | PC12 cell line | [71] |
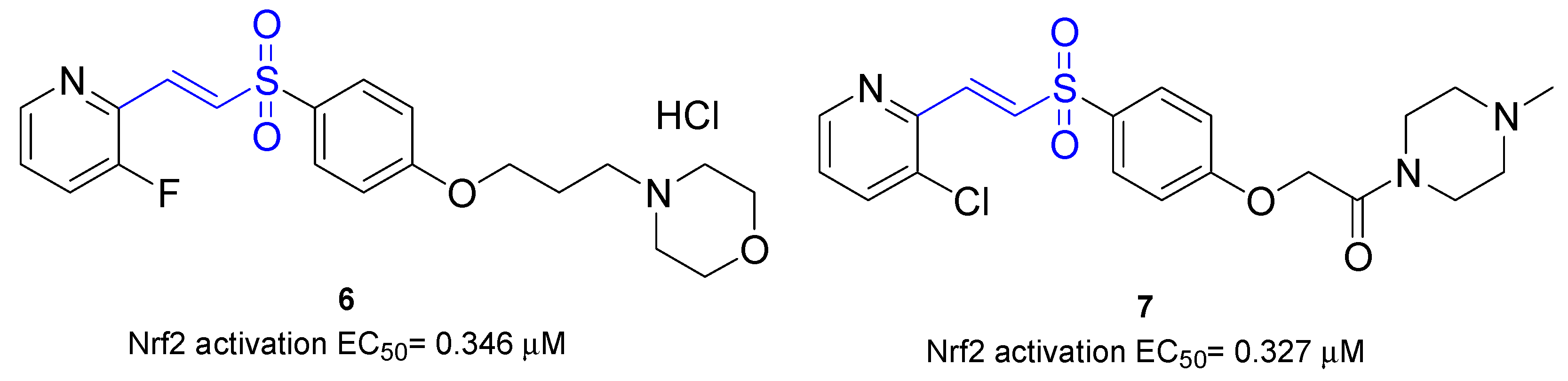
| 6 | Activation of Nrf2 nuclear translocation (EC50 = 0.346 μM) | BV-2 microglial cells | [72] |
| 7 | Activation of Nrf2 nuclear translocation (EC50 = 0.327 μM) | BV-2 microglial cells | [72] |
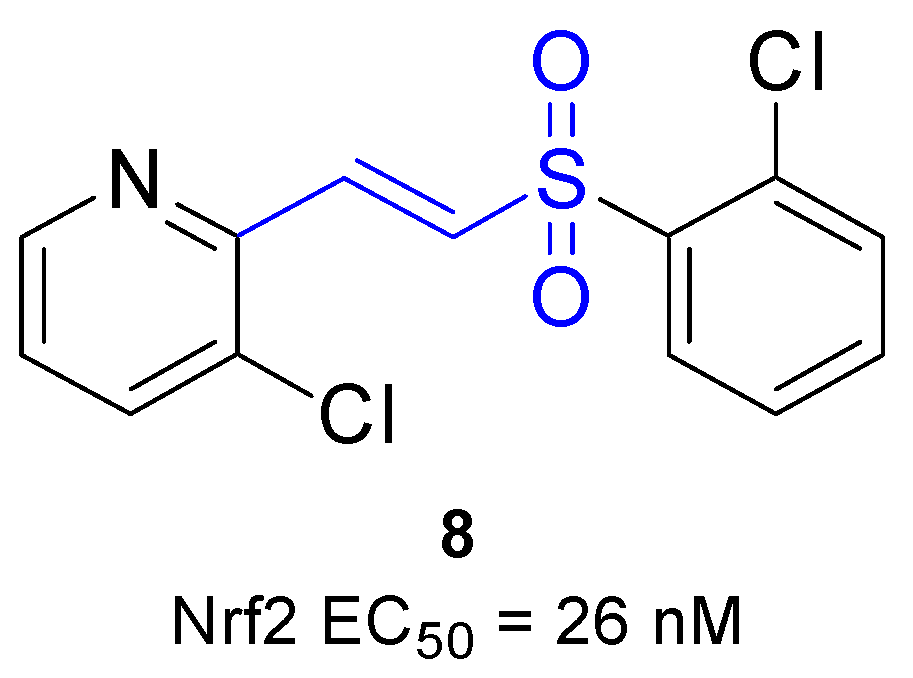
| 8 | Activation of Nrf2 nuclear translocation (EC50 = 0.327 μM) | U2OS cells and BV-2 microglial cells | [73] |
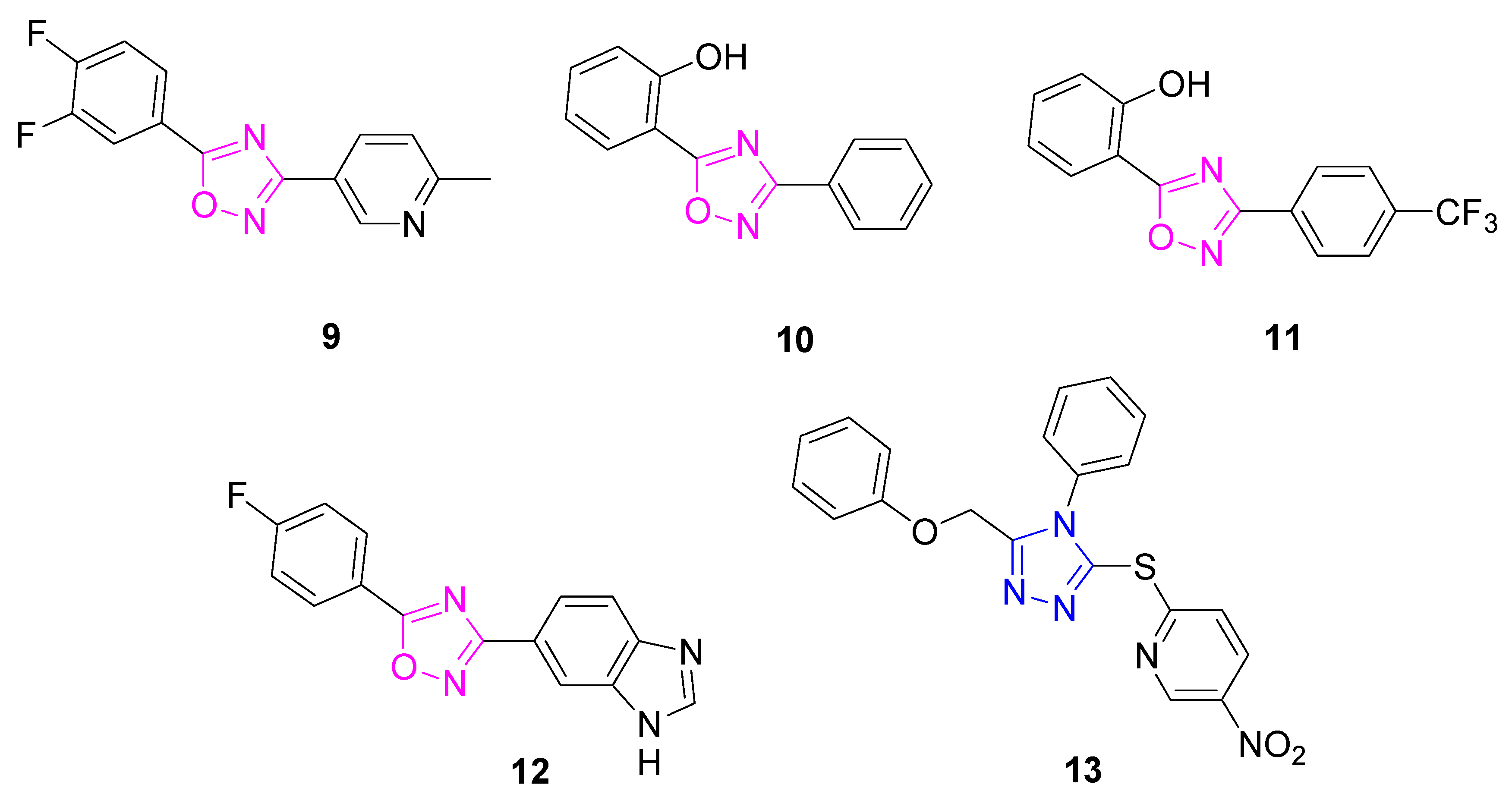
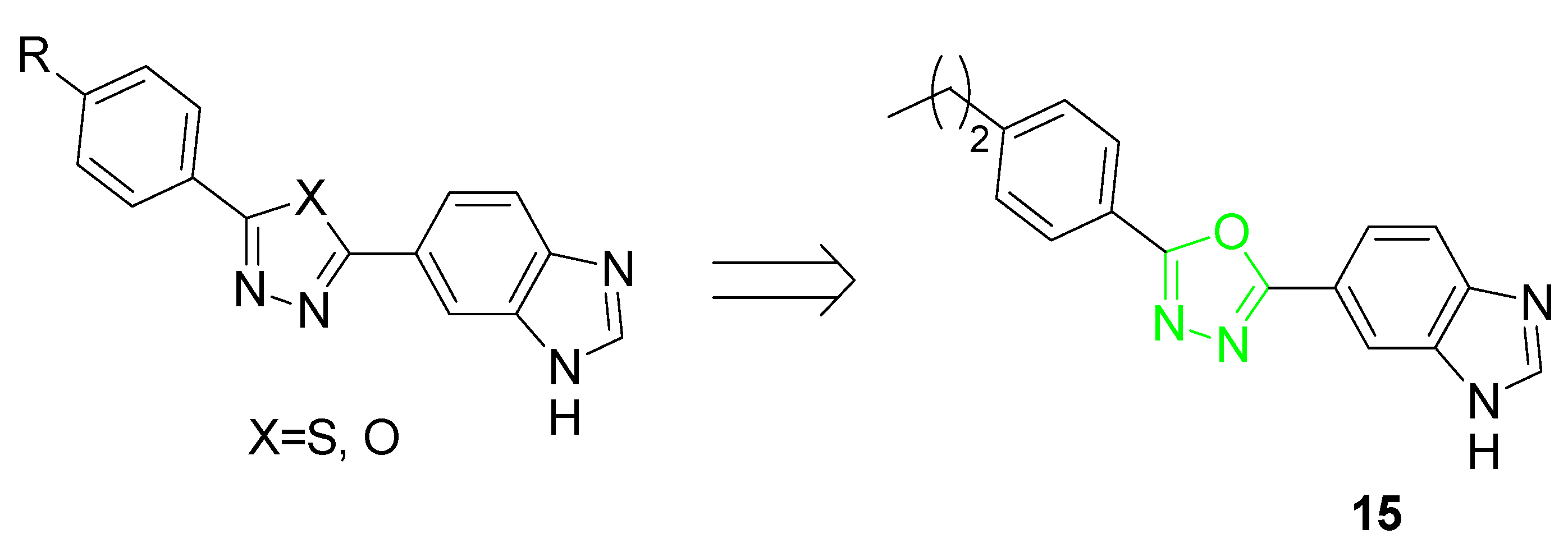
| 13 | Induction of NQO1 and GCLM proteins; transcriptional activation of NRF2-responsive ARE genes | Primary mouse neurons; primary corticostriatal neuronal cocultures | [77,78] |
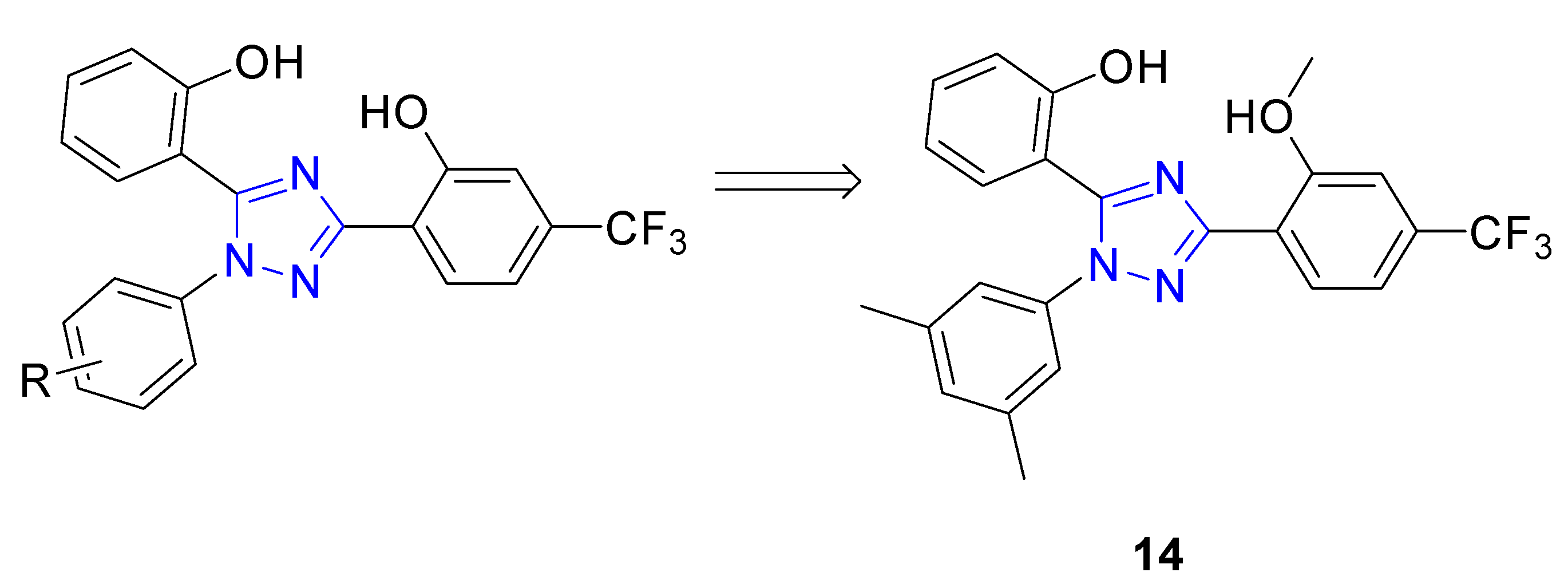
| 14 | Activation of Nrf2 nuclear translocation; induction of HO-1, NQO1, and GCLM proteins; Nrf2 displacement from the Keap1 Kealch domain (KD = 22.70 μM) | PC12 cell line | [79] |
| 15 | ARE inducing activity fold increase (f.i. = 1.8 @100 nM) | PC12 cell line | [80] |
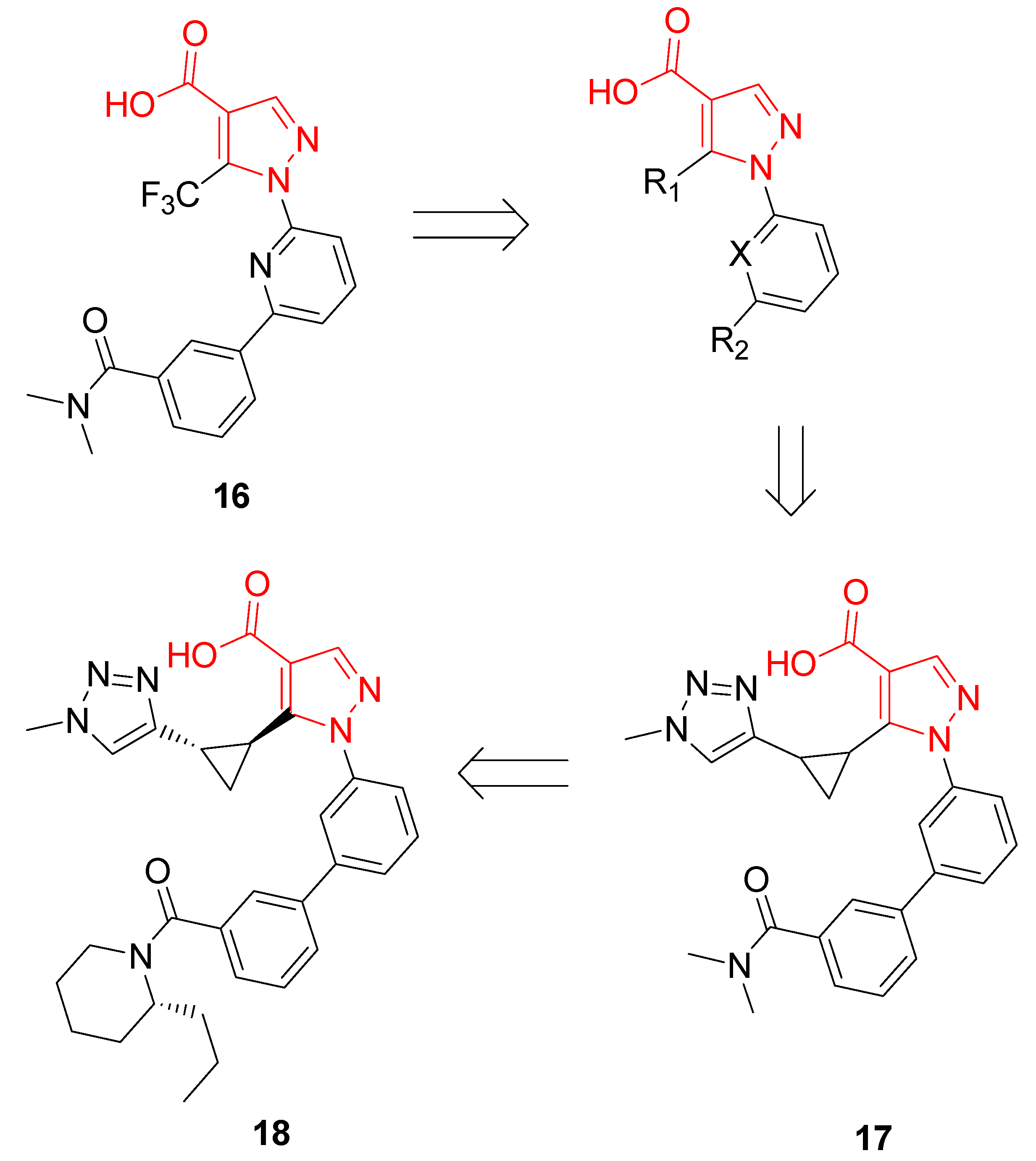
| 18 | Stimulation of NQO1 activity (EC50= 43 nM); Nrf2 displacement from the Keap1 Kealch domain (IC50 < 15 nM); transcriptional activation of NRF2-responsive ARE genes | BEAS-2B cells; normal human bronchial epithelial cells | [81] |
4. Natural Nrf2 Activators
5. Multitargeting Nrf2 Activators
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnham, K.; Masters, C.; Bush, A. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Maccallini, C.; Amoroso, R. Targeting neuronal nitric oxide synthase as a valuable strategy for the therapy of neurological disorders. Neural Regen. Res. 2016, 11, 1731–1734. [Google Scholar] [CrossRef]
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The Nrf2-Dependent Transcriptional Regulation of Antioxidant Defense Pathways: Relevance for Cell Type-Specific Vulnerability to Neurodegeneration and Therapeutic Intervention. Antioxidants 2021, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Heurtaux, T.; Bouvier, D.S.; Benani, A.; Helgueta Romero, S.; Frauenknecht, K.B.M.; Mittelbronn, M.; Sinkkonen, L. Normal and Pathological Nrf2 Signalling in the Central Nervous System. Antioxidants 2022, 11, 1426. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [Green Version]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, A9, 714370. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 250, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Ishii, T.; Wakabayashi, N.; Yamamoto, M. Regulatory mechanisms of cellular response to oxidative stress. Free Radic. Res. 1999, 31, 319–324. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 576. [Google Scholar]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Kanninen, K.; White, A.R.; Koistinaho, J.; Malm, T. Targeting Glycogen Synthase Kinase-3β for Therapeutic Benefit against Oxidative Stress in Alzheimer’s Disease: Involvement of the Nrf2-ARE Pathway. Int. J. Alzheimer’s Dis. 2011, 2011, 985085. [Google Scholar]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Matsumaru, D.; Motohashi, H. The KEAP1-Nrf2 System in Healthy Aging and Longevity. Antioxidants 2021, 10, 1929. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Acioglu, C.; Li, L.; Elkabes, S. Contribution of astrocytes to neuropathology of neurodegenerative diseases. Brain Res. 2021, 1758, 147291. [Google Scholar] [CrossRef]
- Lazdon, E.; Stolero, N.; Frenkel, D. Microglia and Parkinson’s disease: Footprints to pathology. J. Neural Transm. 2020, 127, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Todorovic, M.; Wood, S.A.; Mellick, G.D. Nrf2: A modulator of Parkinson’s disease? J. Neural Transm. 2016, 123, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Brown, D.R. α-Synuclein as a ferrireductase. Biochem. Soc. Trans. 2013, 41, 1513–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, J.R.; Caudle, W.M.; Guillot, T.S.; Watson, J.L.; Nakamaru-Ogiso, E.; Seo, B.B.; Sherer, T.B.; Greenamyre, J.T.; Yagi, T.; Matsuno-Yagi, A.; et al. Obligatory role for complex I inhibition in the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicol. Sci. 2007, 95, 196–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakkittukandiyil, A.; Sajini, D.V.; Karuppaiah, A.; Selvaraj, D. The principal molecular mechanisms behind the activation of Keap1/Nrf2/ARE pathway leading to neuroprotective action in Parkinson’s disease. Neurochem. Int. 2022, 156, 105325. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Sai, Y.; Zou, Z.; Peng, K.; Dong, Z. The Parkinson’s disease-related genes act in mitochondrial homeostasis. Neurosci. Biobehav. Rev. 2012, 36, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Kowalska, M.; Prendecki, M.; Piekut, T.; Kozłowska, J.; Kozubski, W. Oxidative stress factors in Parkinson’s disease. Neural Regen. Res. 2021, 16, 1383–1391. [Google Scholar] [CrossRef]
- Hermida-Ameijeiras, A.; Méndez-Alvarez, E.; Sánchez-Iglesias, S.; Sanmartín-Suárez, C.; Soto-Otero, R. Autoxidation and MAO-mediated metabolism of dopamine as a potential cause of oxidative stress: Role of ferrous and ferric ions. Neurochem. Int. 2004, 45, 103–116. [Google Scholar] [CrossRef]
- Innamorato, N.G.; Jazwa, A.; Rojo, A.I.; García, C.; Fernández-Ruiz, J.; Grochot-Przeczek, A.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Cuadrado, A. Different susceptibility to the Parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS ONE 2010, 5, e11838. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, J21, 3173–3192. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the Nrf2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal 2016, 25, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, M.; Ammal Kaidery, N.; Attucks, O.C.; McDade, E.; Hushpulian, D.M.; Gaisin, A.; Gaisina, I.; Ahn, Y.H.; Nikulin, S.; Poloznikov, A.; et al. Bach1 derepression is neuroprotective in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2111643118. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, F.; Imai, M.; Tamaki, S.; Ohta, E.; Kawashima, R.; Maekawa, T.; Kurosaki, Y.; Ohba, K.; Ichikawa, T. Nrf2 Expression Is Decreased in LRRK2 Transgenic Mouse Brain and LRRK2 Overexpressing SH-SY5Y Cells. Biol. Pharm. Bull. 2023, 46, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Gumeni, S.; Papanagnou, E.D.; Manola, M.S.; Trougakos, I.P. Nrf2 activation induces mitophagy and reverses Parkin/Pink1 knock down-mediated neuronal and muscle degeneration phenotypes. Cell Death Dis. 2021, 12, 671. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, K.; Cuvelier, E.; Destée, A.; Gressier, B.; Chartier-Harlin, M.C. NADPH oxidases in Parkinson’s disease: A systematic review. Mol. Neurodegener. 2017, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.Y.; Ip, N.Y. The role of genetic risk factors of Alzheimer’s disease in synaptic dysfunction. Semin. Cell Dev. Biol. 2023, 139, 3–12. [Google Scholar] [CrossRef]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109 (Suppl. 1), 153–159. [Google Scholar] [CrossRef] [Green Version]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Hou, S.; Jiang, J.; Sekutowicz, M.; Kelly, J.; Bacskai, B.J. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 7904–7909. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Jara, C.; Aránguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol. 2018, 18, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, T.-C.; King, M.-E.; Kuret, J.; Berry, R.W.; Binder, L.I. Oxidative regulation of fatty acid-induced tau polymerization. Biochemistry 2000, 39, 14203–14210. [Google Scholar] [CrossRef]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Cente, M.; Filipcik, P.; Pevalova, M.; Novak, M. Expression of a truncated tau protein induces oxidative stress in a rodent model of tauopathy. Eur. J. Neurosci. 2006, 24, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.A.; Jové, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum. Mol. Genet. 2014, J23, 3716–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, A.I.; Pajares, M.; García-Yagüe, A.J.; Buendia, I.; Van Leuven, F.; Yamamoto, M.; López, M.G.; Cuadrado, A. Deficiency in the transcription factor Nrf2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. Nrf2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef]
- Ramesh Babu, J.; Lamar Seibenhener, M.; Peng, J.; Strom, A.-L.; Kemppainen, R.; Cox, N.; Zhu, H.; Wooten, M.-C.; Diaz-Meco, M.-T.; Moscat, J.; et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 2008, 106, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef]
- Uemura, K.; Kuzuya, A.; Shimozono, Y.; Aoyagi, N.; Ando, K.; Shimohama, S.; Kinoshita, A. GSK3beta activity modifies the localization and function of presenilin 1. J. Biol. Chem. 2007, 282, 15823–15832. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Kerns, J.K.; Callahan, J.F.; Moody, C.J. Keap calm, and carry on covalently. J. Med. Chem. 2013, 56, 7463–7476. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: Promises and perils of Nrf2. Pharmacol. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Hu, L. Nrf2 activation through the inhibition of Keap1–Nrf2 protein–protein interaction. Med. Chem. Res. 2020, 29, 846–867. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Hsieh, C.W.; Wu, C.C.; Wung, B.S. Chalcone inhibits the activation of NF-kappa B and STAT3 in endothelial cells via endogenous electrophile. Life Sci. 2007, 80, 1420–1430. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, S.; Hassan, M.; Wu, H.; Thimmulappa, R.K.; Kumar, A.; Sharma, S.K.; Parmar, V.S.; Biswal, S.; Malhotra, S.V. Novel chalcone derivatives as potent Nrf2 activators in mice and human lung epithelial cells. J. Med. Chem. 2011, 54, 4147–4159. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Jang, B.K.; Park, J.H.; Choi, J.W.; Park, S.J.; Byeon, S.R.; Pae, A.N.; Lee, Y.S.; Cheong, E.; Park, K.D. A novel chalcone derivative as Nrf2 activator attenuates learning and memory impairment in a scopolamine-induced mouse model. Eur. J. Med. Chem. 2022, 185, 111777. [Google Scholar] [CrossRef]
- Woo, S.Y.; Kim, J.H.; Moon, M.K.; Han, S.H.; Yeon, S.K.; Choi, J.W.; Jang, B.K.; Song, H.J.; Kang, Y.G.; Kim, J.W.; et al. Discovery of Vinyl Sulfones as a Novel Class of Neuroprotective Agents toward Parkinson’s Disease Therapy. J. Med. Chem. 2014, 57, 1473–1487. [Google Scholar] [CrossRef]
- Song, Z.L.; Hou, Y.; Bai, F.; Fang, J. Generation of potent Nrf2 activators via tuning the electrophilicity and steric hindrance of vinyl sulfones for neuroprotection. Bioorg. Chem. 2021, 107, 104520. [Google Scholar] [CrossRef]
- Choi, J.W.; Kim, S.; Park, J.H.; Kim, H.J.; Shin, S.J.; Kim, J.W.; Woo, S.Y.; Lee, C.; Han, S.M.; Lee, J.; et al. Optimization of Vinyl Sulfone Derivatives as Potent Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Activators for Parkinson’s Disease Therapy. J. Med. Chem. 2019, 62, 811–830. [Google Scholar] [CrossRef]
- Choi, J.W.; Kim, S.; Yoo, J.S.; Kim, H.J.; Kim, H.J.; Kim, B.E.; Lee, E.H.; Lee, Y.S.; Park, J.H.; Park, K.D. Development and optimization of halogenated vinyl sulfones as Nrf2 activators for the treatment of Parkinson’s disease. Eur. J. Med. Chem. 2021, 212, 113103. [Google Scholar] [CrossRef]
- Crisman, E.; Duarte, P.; Dauden, E.; Cuadrado, A.; Rodríguez-Franco, M.I.; López, M.G.; León, R. KEAP1-Nrf2 protein–protein interaction inhibitors: Design, pharmacological properties and therapeutic potential. Med. Res. Rev. 2023, 43, 237–287. [Google Scholar] [CrossRef]
- Xu, L.L.; Zhu, J.F.; Xu, X.L.; Zhu, J.; Li, L.; Xi, M.Y.; Jiang, Z.Y.; Zhang, M.Y.; Liu, F.; Lu, M.C.; et al. Discovery and modification of in vivo active Nrf2 activators with 1,2,4-oxadiazole core: Hits identification and structure-activity relationship study. J. Med. Chem. 2015, 58, 5419–5436. [Google Scholar] [CrossRef] [PubMed]
- Ayoup, M.S.; Abu-Serie, M.M.; Abdel-Hamid, H.; Teleb, M. Beyond direct Nrf2 activation; reinvestigating 1,2,4-oxadiazole scaffold as a master key unlocking the antioxidant cellular machinery for cancer therapy. Eur. J. Med. Chem. 2021, 220, 113475. [Google Scholar] [CrossRef] [PubMed]
- Quinti, L.; Casale, M.; Moniot, S.; Pais, T.F.; Van Kanegan, M.J.; Kaltenbach, L.S.; Pallos, J.; Lim, R.G.; Naidu, S.D.; Runne, H.; et al. SIRT2- and NRF2-Targeting Thiazole-Containing Compound with Therapeutic Activity in Huntington’s Disease Models. Cell Chem. Biol. 2016, 23, 849–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinti, L.; Dayalan, N.S.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted Nrf2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lao, Y.; Wang, Y.; Chen, J.; Huang, P.; Su, R.; Shi, J.; Jiang, C.; Zhang, J. Synthesis and biological evaluation of 1,2,4-triazole derivatives as potential Nrf2 activators for the treatment of cerebral ischemic injury. Eur. J. Med. Chem. 2022, 236, 114315. [Google Scholar] [CrossRef]
- Lin, H.; Qiao, Y.; Yang, H.; Li, Q.; Chen, Y.; Qu, W.; Liu, W.; Feng, F.; Sun, H. Design and evaluation of Nrf2 activators with 1,3,4-oxa/thiadiazole core as neuro-protective agents against oxidative stress in PC-12 cells. Bioorg. Med. Chem. Lett. 2020, 30, 126853. [Google Scholar] [CrossRef]
- Norton, D.; Bonnette, W.G.; Callahan, J.F.; Carr, M.G.; Griffiths-Jones, C.M.; Heightman, T.D.; Kerns, J.K.; Nie, H.; Rich, S.J.; Richardson, C.; et al. Fragment-Guided Discovery of Pyrazole Carboxylic Acid Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2 Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction. J. Med. Chem. 2021, 64, 15949–15972. [Google Scholar] [CrossRef]
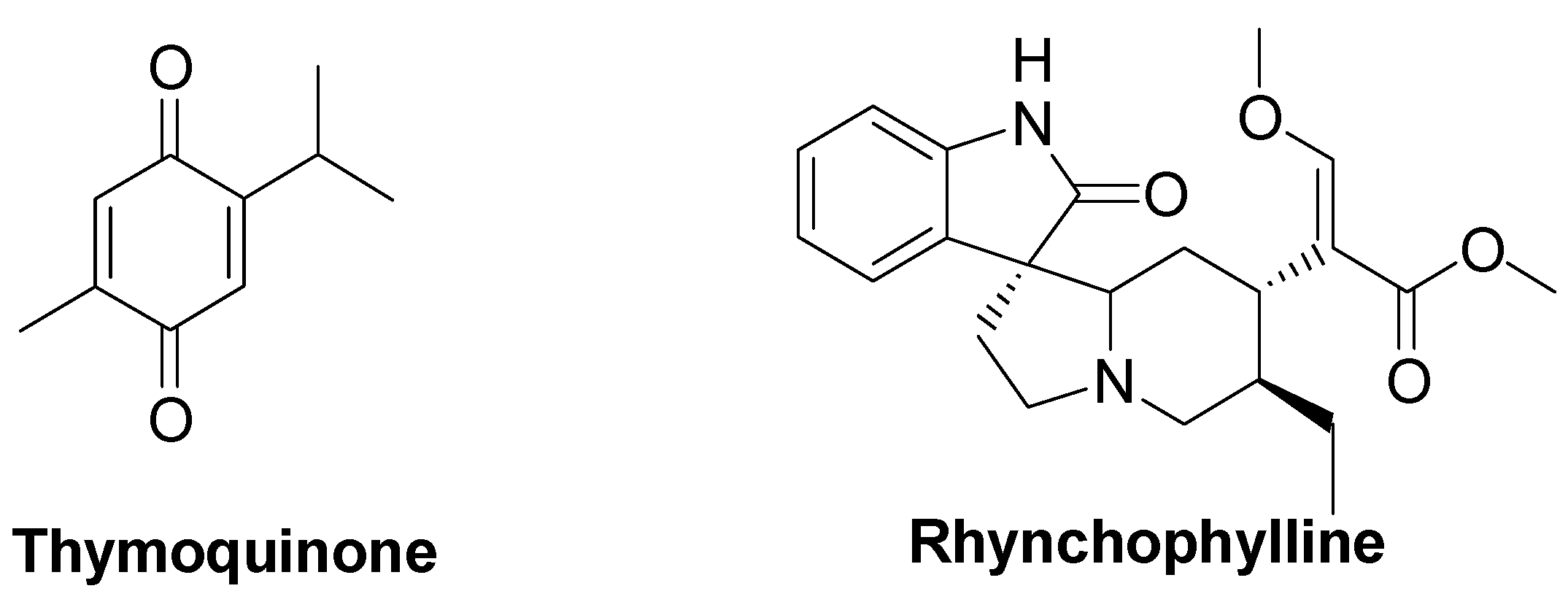
- Darakhshan, S.; Bidmeshki, P.A.; Hosseinzadeh, C.A.; Sisakhtnezhad, S. Thymoquinone and its therapeutic potentials. Pharmacol. Res. 2015, 95–96, 138–158. [Google Scholar] [CrossRef] [PubMed]
- Radad, K.; Moldzio, R.; Taha, M.; Rausch, W.D. Thymoquinone protects dopaminergic neurons against MPP+ and rotenone. Phytother. Res. 2009, 23, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Radad, K.S.; Al-Shraim, M.M.; Moustafa, M.F.; Rausch, W.D. Neuroprotective role of thymoquinone against 1-methyl-4-phenylpyridiniuminduced dopaminergic cell death in primary mesencephalic cell culture. Neurosciences 2015, 20, 10–16. [Google Scholar] [PubMed]
- Ardah, M.T.; Merghani, M.M.; Haque, M.E. Thymoquinone prevents neurodegeneration against MPTP in vivo and modulates α-synuclein aggregation in vitro. Neurochem. Int. 2019, 128, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Talebi, M.; Talebi, M.; Farkhondeh, T.; Samarghandian, S. Biological and therapeutic activities of thymoquinone: Focus on the Nrf2 signaling pathway. Phytother. Res. 2021, 35, 1739–1753. [Google Scholar] [CrossRef]
- Fujiwara, H.; Iwasaki, K.; Furukawa, K.; Seki, T.; He, M.; Maruyama, M.; Tomita, N.; Kudo, Y.; Higuchi, M.; Saido, T.C.; et al. Uncaria rhynchophylla, a Chinese medicinal herb, has potent antiaggregation effects on Alzheimer’s β-amyloid proteins. J. Neurosci. Res. 2006, 84, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, J.; Xu, J.; Song, X.; Huang, H.; Feng, Y.; Fu, C. Rhynchophylline loaded-mPEG-PLGA nanoparticles coated with Tween-80 for preliminary study in Alzheimer’s disease. Int. J. Nanomed. 2020, 15, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.J.; Cui, L.Q.; Li, P.; Wang, Y.B.; Zhang, X.Z.; Guo, M.L. Rhynchophylline ameliorates myocardial ischemia/reperfusion injury through the modulation of mitochondrial mechanisms to mediate myocardial apoptosis. Mol. Med. Rep. 2019, 19, 2581–2590. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Chen, L.; Xu, J.; Liu, W.; Feng, F.; Qu, W. Neuroprotective Effects of Rhynchophylline Against Aβ1–42-Induced Oxidative Stress, Neurodegeneration, and Memory Impairment Via Nrf2–ARE Activation. Neurochem. Res. 2021, 46, 2439–2450. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Carlo Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, J.; Wei, X.; Niu, C.; Jia, M.; Qinhan Li, Q.; Meng, D. Bach1: Function, regulation, and involvement in disease. Oxid. Med. Cell. Longev. 2018, 2018, 1347969. [Google Scholar] [CrossRef]
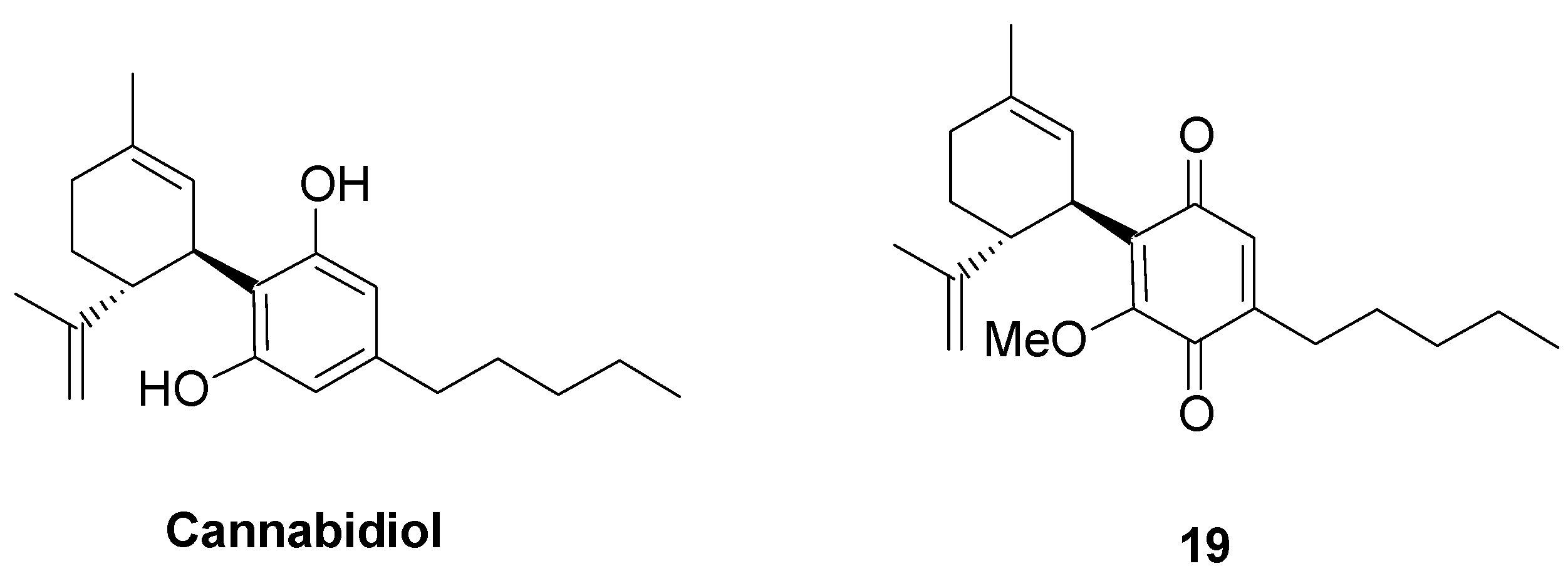
- Casares, L.; Unciti-Broceta, J.D.; Prados, M.E.; Caprioglio, D.; Mattoteia, D.; Higgins, M.; Apendino, G.; Dinkova-Kostova, A.T.; Munoz, E.; de la Vega, L. Isomeric O-methyl cannabidiolquinones with dual BACH1/Nrf2 activity. Redox Biol. 2020, 37, 101689. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef] [Green Version]
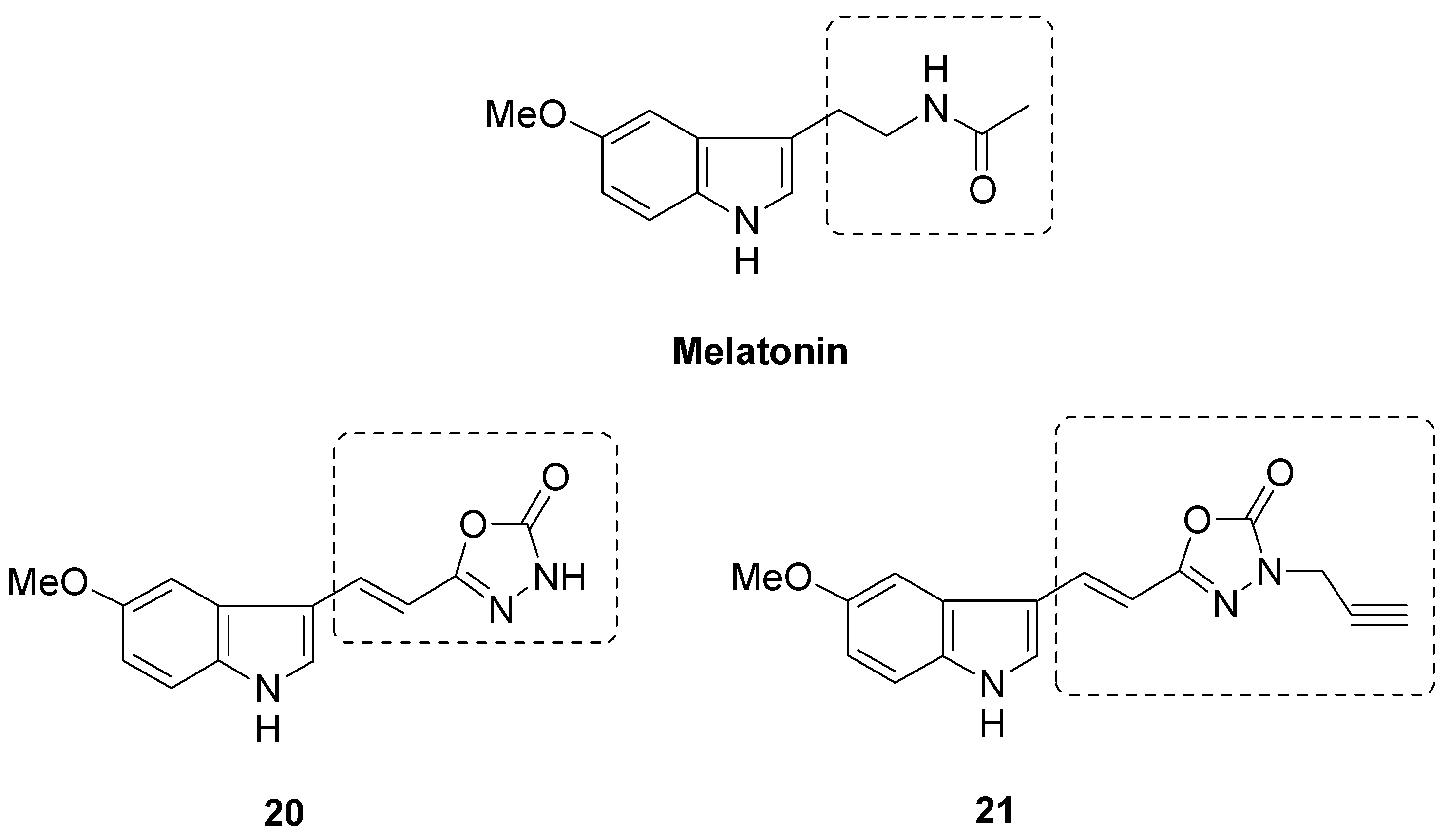
- Nosjean, O.; Ferro, M.; Cogé, F.; Beauverger, P.; Henlin, J.-M.; Lefoulon, F.; Fauchère, J.-L.; Delagrange, P.; Canet, E.; Boutin, J.A. Identification of the melatonin-binding site MT 3 as the quinone reductase 2. J. Biol. Chem. 2000, 275, 31311–31317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Arozamena, C.; Estrada-Valencia, M.; Pérez, C.; Lagartera, L.; Morales-García, J.A.; Pérez-Castillo, A.; Franco-Gonzalez, J.F.; Michalska, P.; Duarte, P.; Leòn, R.; et al. Tuning melatonin receptor subtype selectivity in oxadiazolone-based analogues: Discovery of QR2 ligands and Nrf2 activators with neurogenic properties. Eur. J. Med. Chem. 2020, 190, 112090. [Google Scholar] [CrossRef] [PubMed]
- Harald Hampel, H.; Robert Vassar, R.; Bart De Strooper, B.; John Hardy, J.; Michael Willem, M.; Neeraj Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
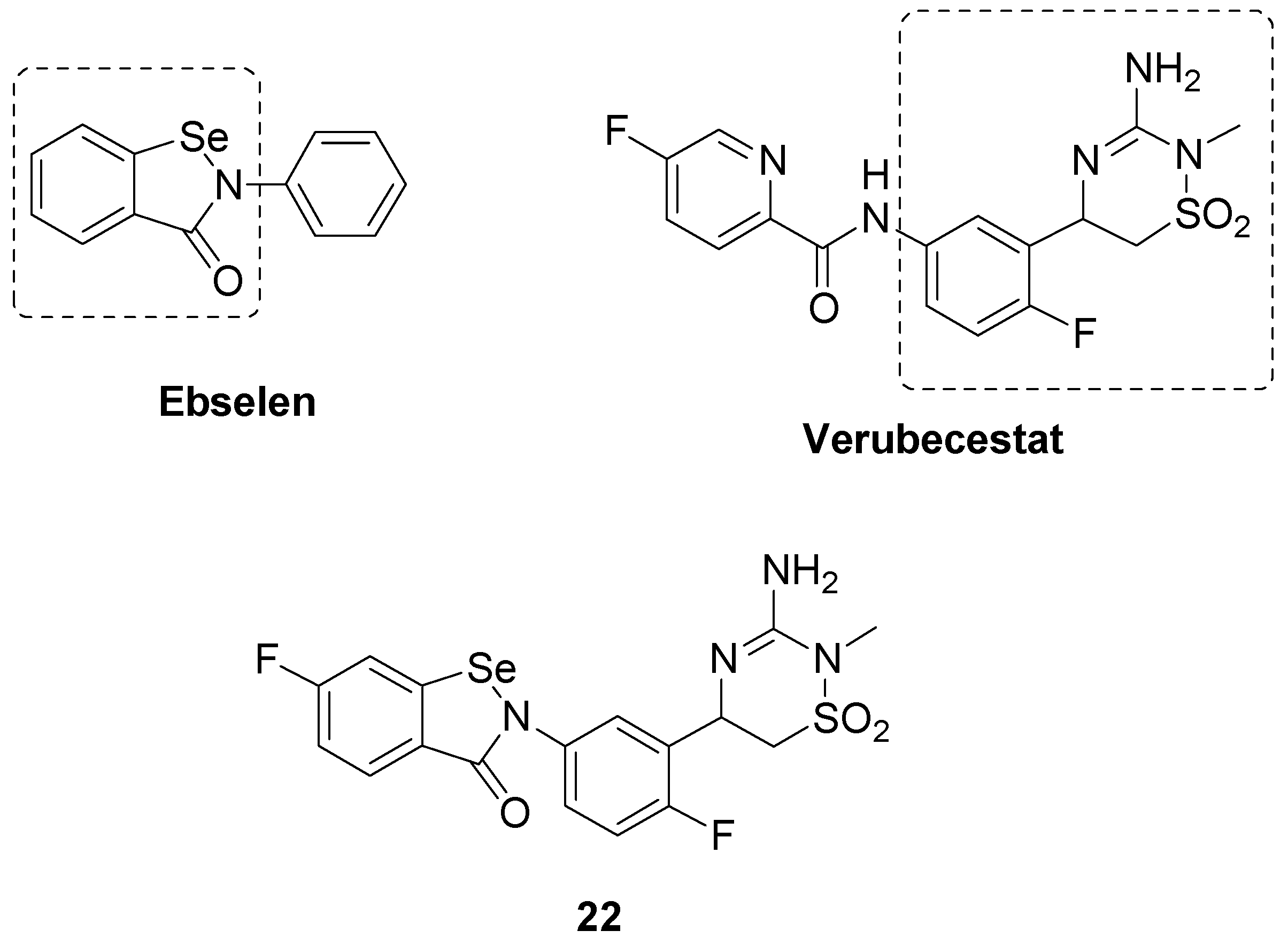
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8, 363ra150. [Google Scholar] [CrossRef]
- Qu, L.; Ji, L.; Wang, C.; Luo, H.; Li, S.; Peng, W.; Yin, F.; Lu, D.; Liu, X.; Kong, L.; et al. Synthesis and evaluation of multi-target-directed ligands with BACE-1 inhibitory and Nrf2 agonist activities as potential agents against Alzheimer’s disease. Eur. J. Med. Chem. 2021, 219, 113441. [Google Scholar] [CrossRef]
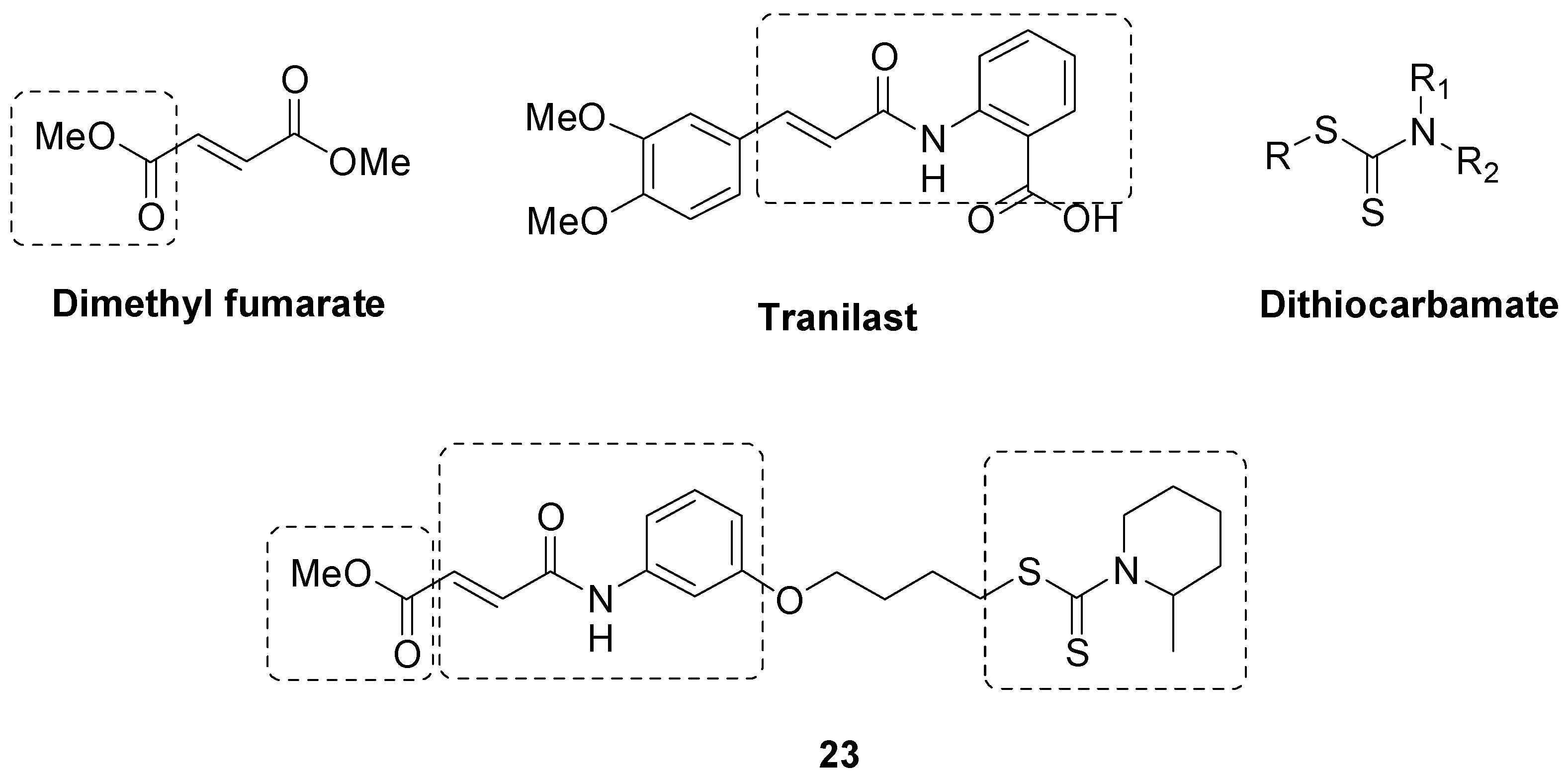
- Guo, J.; Cheng, M.; Liu, P.; Cao, D.; Luo, J.; Wan, Y.; Fang, Y.; Jin, Y.; Xie, S.-S.; Liu, J. A multi-target directed ligands strategy for the treatment of Alzheimer’s disease: Dimethyl fumarate plus Tranilast modified Dithiocarbate as AchE inhibitor and Nrf2 activator. Eur. J. Med. Chem. 2022, 242, 114630. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Beschea Chiriac, S.I.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s Disease Pharmacotherapy in relation to cholinergic system involvement. Biomolecules 2020, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapak, P.; Bishnoi, M.; Sharma, S.S. Tranilast, a Transient Receptor Potential Vanilloid 2 Channel (TRPV2) inhibitor attenuates amyloid β-induced cognitive impairment: Possible mechanisms. Neuromol. Med. 2022, 24, 183–194. [Google Scholar] [CrossRef] [PubMed]
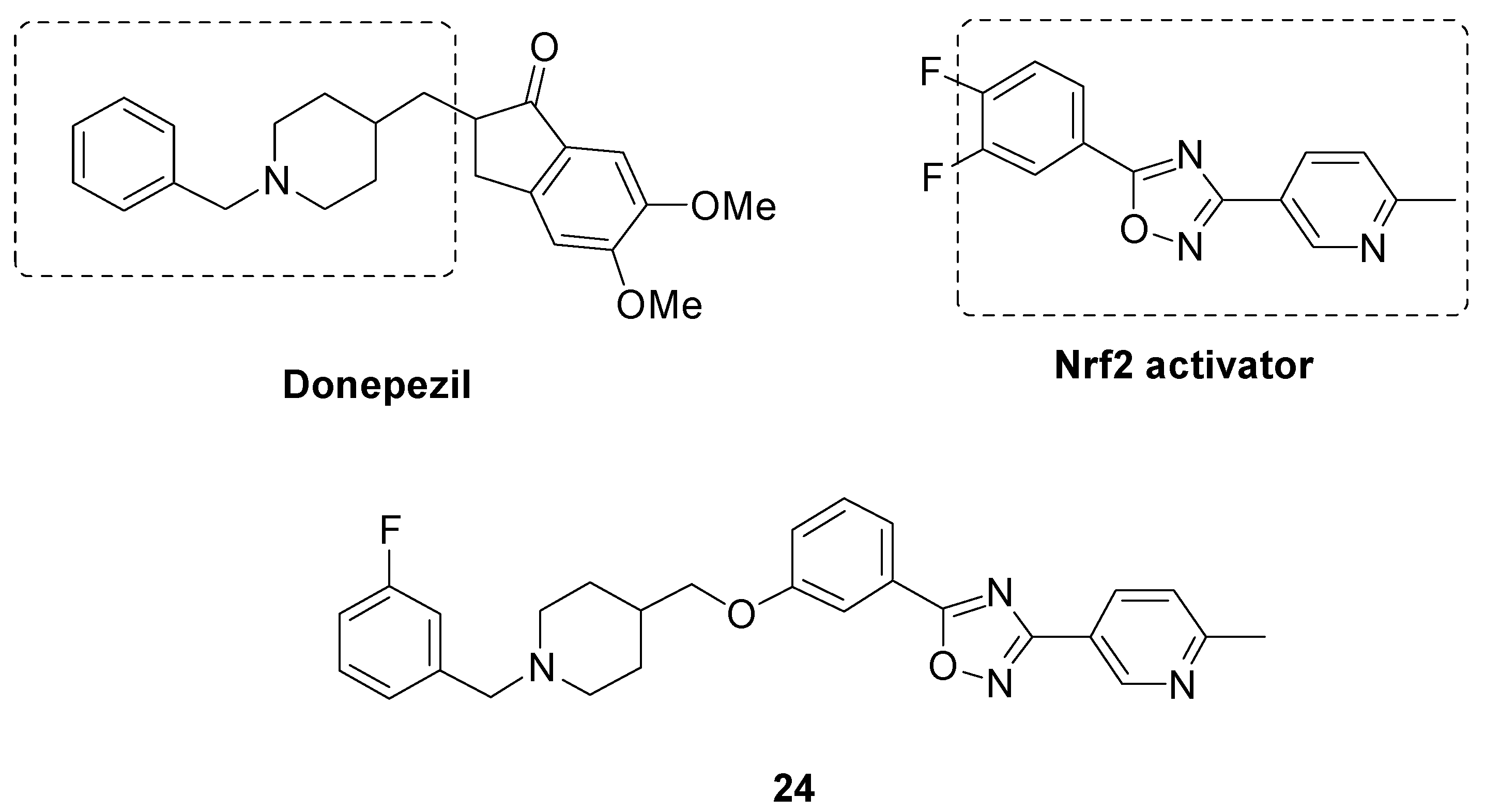
- Wang, Y.; Xiong, B.; Lin, H.; Li, Q.; Yang, H.; Qiao, Y.; Li, Q.; Xu, Z.; Lyu, W.; Qu, W.; et al. Design, synthesis and evaluation of fused hybrids with acetylcholinesterase inhibiting and Nrf2 activating functions for Alzheimer’s disease. Eur. J. Med. Chem. 2022, 244, 114806. [Google Scholar] [CrossRef] [PubMed]



















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amoroso, R.; Maccallini, C.; Bellezza, I. Activators of Nrf2 to Counteract Neurodegenerative Diseases. Antioxidants 2023, 12, 778. https://doi.org/10.3390/antiox12030778
Amoroso R, Maccallini C, Bellezza I. Activators of Nrf2 to Counteract Neurodegenerative Diseases. Antioxidants. 2023; 12(3):778. https://doi.org/10.3390/antiox12030778
Chicago/Turabian StyleAmoroso, Rosa, Cristina Maccallini, and Ilaria Bellezza. 2023. "Activators of Nrf2 to Counteract Neurodegenerative Diseases" Antioxidants 12, no. 3: 778. https://doi.org/10.3390/antiox12030778