
Nutriepigenomics in Environmental-Associated Oxidative Stress
 ,
,  , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
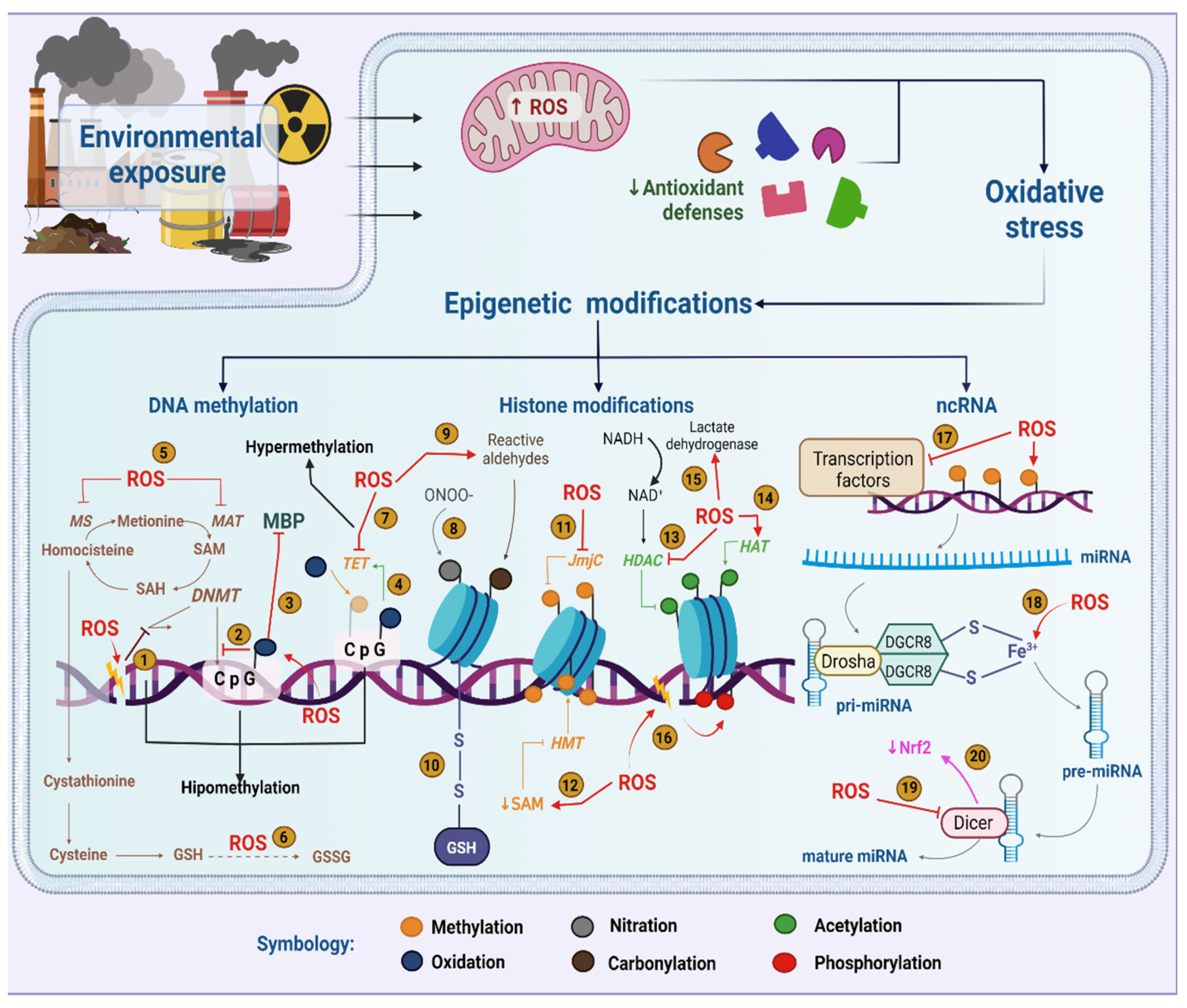
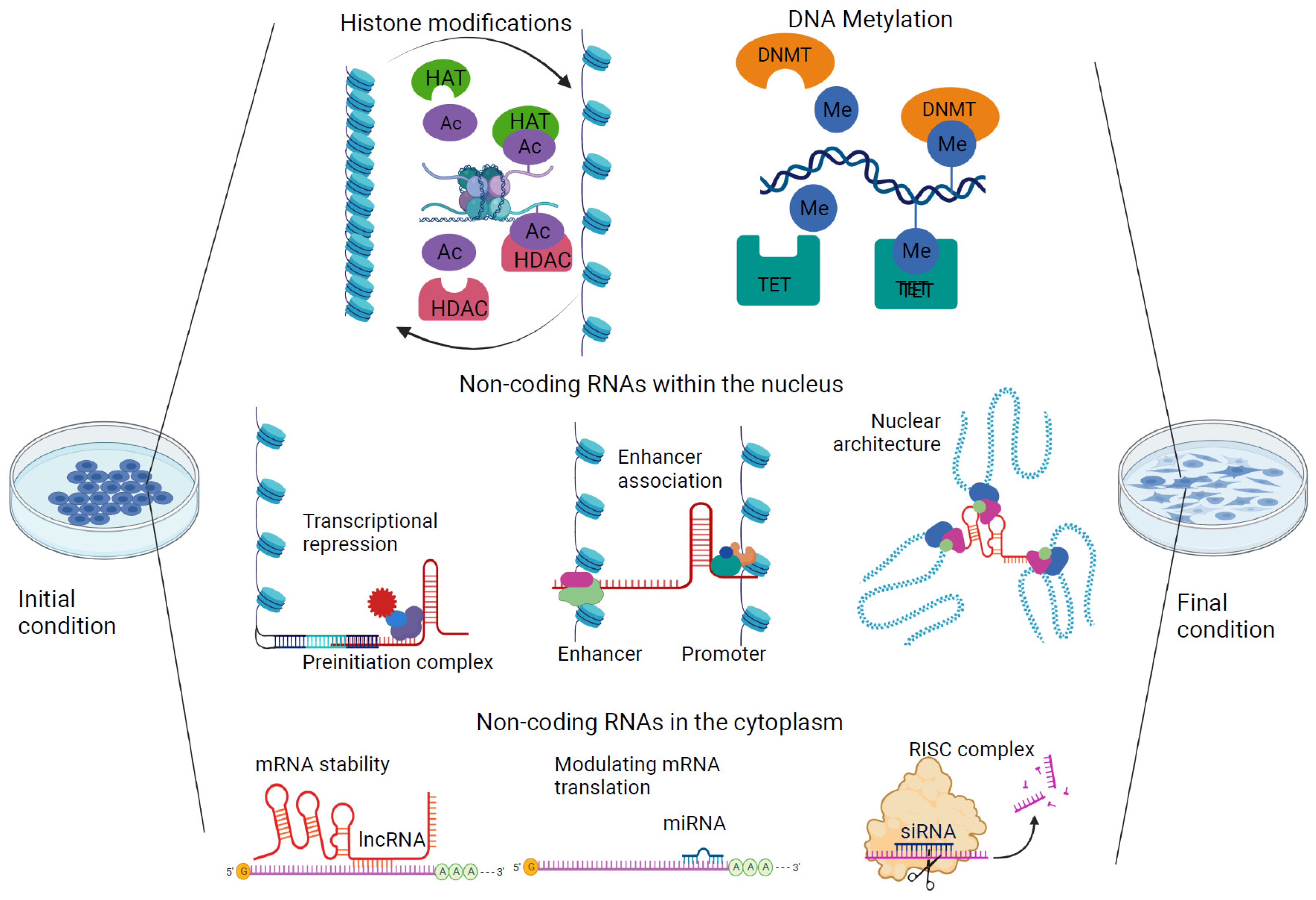
2. Epigenetics in the Context of Environmental Exposure
3. Relationship between Epigenetics and Oxidative Stress in the Context of Environmental Exposure
3.1. DNA Methylation and Oxidative Stress
3.2. Histone Modifications and Oxidative Stress
3.3. Noncoding RNAs and Oxidative Stress
4. Microenvironment and Nutritional Influence on the Preservation of Epigenetic Marks
4.1. Epigenetic Response to Culture Conditions
4.2. The Role of Extracellular Vesicles and Additional Cofactors
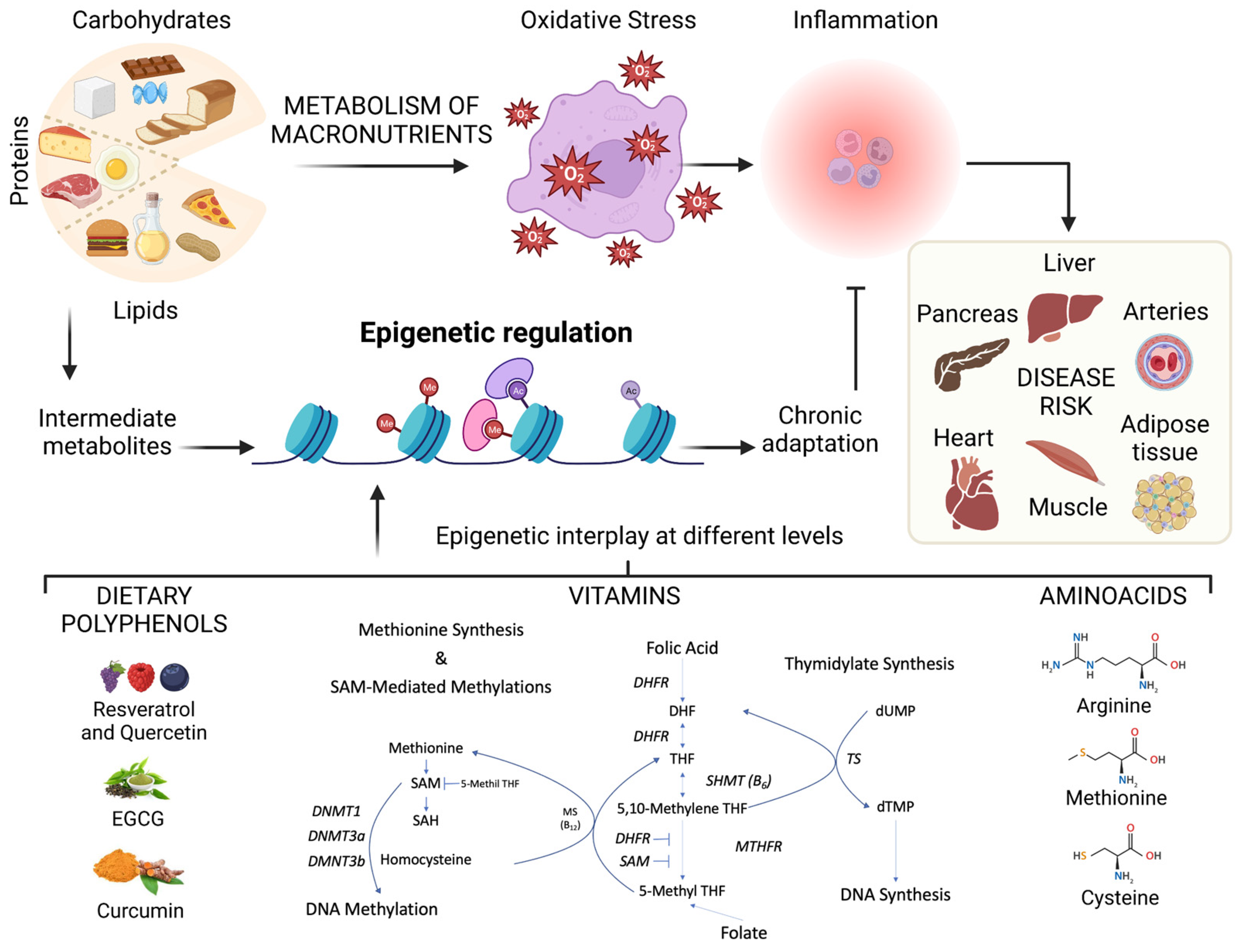
4.3. Nutrigenomics Impacts Epigenomics
5. Effects of Dietary Nutrients on the Epigenome
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, M.K. Environmental Epigenetics and a Unified Theory of the Molecular Aspects of Evolution: A Neo-Lamarckian Concept that Facilitates Neo-Darwinian Evolution. Genome Biol. Evol. 2015, 7, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Doonan, S.R.; Ordog, T.; Bailey, R.C. Translational Opportunities for Microfluidic Technologies to Enable Precision Epigenomics. Anal. Chem. 2020, 92, 7989–7997. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Rubio, K.; Dobersch, S.; Barreto, G. Functional interactions between scaffold proteins, noncoding RNAs, and genome loci induce liquid-liquid phase separation as organizing principle for 3-dimensional nuclear architecture: Implications in cancer. FASEB J. 2019, 33, 5814–5822. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Contreras, A.; Cordero, J.; Rubio, K.; Dobersch, S.; Günther, S.; Jeratsch, S.; Mehta, A.; Krüger, M.; Graumann, J.; et al. MiCEE is a ncRNA-protein complex that mediates epigenetic silencing and nucleolar organization. Nat. Genet. 2018, 50, 990–1001. [Google Scholar] [CrossRef]
- Dobersch, S.; Rubio, K.; Singh, I.; Günther, S.; Graumann, J.; Cordero, J.; Castillo-Negrete, R.; Huynh, M.B.; Mehta, A.; Braubach, P.; et al. Positioning of nucleosomes containing gamma-H2AX precedes active DNA demethylation and transcription initiation. Nat. Commun. 2021, 12, 1072. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [Green Version]
- Gardiner-Garden, M.; Frommer, M. CpG Islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Barreto, G.; Schäfer, A.; Marhold, J.; Stach, D.; Swaminathan, S.K.; Handa, V.; Döderlein, G.; Maltry, N.; Wu, W.; Lyko, F.; et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 2007, 445, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meehan, R.R.; Thomson, J.P.; Lentini, A.; Nestor, C.E.; Pennings, S. DNA methylation as a genomic marker of exposure to chemical and environmental agents. Curr. Opin. Chem. Biol. 2018, 45, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Dobersch, S.; Romero-Olmedo, A.J.; Barreto, G. Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev. 2015, 34, 229–241. [Google Scholar] [CrossRef]
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef] [Green Version]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Thomson, J.P.; Meehan, R.R. The application of genome-wide 5-hydroxymethylcytosine studies in cancer research. Epigenomics 2017, 9, 77–91. [Google Scholar] [CrossRef]
- Thomson, J.P.; Ottaviano, R.; Unterberger, E.B.; Lempiäinen, H.; Muller, A.; Terranova, R.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; Lyall, M.J.; et al. Loss of Tet1-Associated 5-Hydroxymethylcytosine Is Concomitant with Aberrant Promoter Hypermethylation in Liver Cancer. Cancer Res. 2016, 76, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Ozturk, N.; Singh, I.; Mehta, A.; Braun, T.; Barreto, G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front. Cell Dev. Biol. 2014, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fischle, W.; Cheung, W.; Jacobs, S.; Khorasanizadeh, S.; Allis, C.D. Beyond the double helix: Writing and reading the histone code. Novartis Found. Symp. 2004, 259, 3–17, discussion 17-21, 163–9. [Google Scholar] [PubMed]
- Singh, I.; Ozturk, N.; Cordero, J.; Mehta, A.; Hasan, D.; Cosentino, C.; Sebastian, C.; Krüger, M.; Looso, M.; Carraro, G.; et al. High mobility group protein-mediated transcription requires DNA damage marker gamma-H2AX. Cell Res. 2015, 25, 837–850. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhao, J.; Lv, Y.; Wang, W.; Feng, C.; Zou, W.; Su, L.; Jiao, J. Histone Variants and Histone Modifications in Neurogenesis. Trends Cell Biol. 2020, 30, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Takami, Y. Participation of Histones and Histone-Modifying Enzymes in Cell Functions through Alterations in Chromatin Structure. J. Biochem. 2001, 129, 491–499. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef]
- Stillman, B. Histone Modifications: Insights into Their Influence on Gene Expression. Cell 2018, 175, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The Effects of Cadmium Toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Gao, Y.; Zheng, X.; Liu, C.; Dong, S.; Li, R.; Zhang, G.; Wei, Y.; Qu, H.; Li, Y.; et al. Histone Modifications Regulate Chromatin Compartmentalization by Contributing to a Phase Separation Mechanism. Mol. Cell 2019, 76, 646–659.e6. [Google Scholar] [CrossRef]
- Johnson, M.O.; Siska, P.J.; Contreras, D.C.; Rathmell, J.C. Nutrients and the microenvironment to feed a T cell army. Semin. Immunol. 2016, 28, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Padilla, P.G.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niculescu, M.D.; Zeisel, S.H. Diet, Methyl Donors and DNA Methylation: Interactions between Dietary Folate, Methionine and Choline. J. Nutr. 2002, 132, S2333–S2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twinn, D.; Hjort, L.; Novakovic, B.; Ozanne, S.; Saffery, R. Intrauterine programming of obesity and type 2 diabetes. Diabetologia 2019, 62, 1789–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caudron-Herger, M.; Rippe, K. Nuclear architecture by RNA. Curr. Opin. Genet. Dev. 2012, 22, 179–187. [Google Scholar] [CrossRef]
- Rubio, K.; Castillo-Negrete, R.; Barreto, G. Non-coding RNAs and nuclear architecture during epithelial-mesenchymal transition in lung cancer and idiopathic pulmonary fibrosis. Cell. Signal. 2020, 70, 109593. [Google Scholar] [CrossRef]
- Zhang, W.; Dolan, M.E. Beyond the HapMap Genotypic Data: Prospects of Deep Resequencing Projects. Curr. Bioinform. 2008, 3, 178. [Google Scholar] [CrossRef] [Green Version]
- Vojtech, L.; Woo, S.; Hughes, S.; Levy, C.; Ballweber, L.; Sauteraud, R.P.; Strobl, J.; Westerberg, K.; Gottardo, R.; Tewari, M.; et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res. 2014, 42, 7290–7304. [Google Scholar] [CrossRef] [Green Version]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Park, C.W.; Zeng, Y.; Zhang, X.; Subramanian, S.; Steer, C.J. Mature microRNAs identified in highly purified nuclei from HCT116 colon cancer cells. RNA Biol. 2010, 7, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Leucci, E.; Patella, F.; Waage, J.; Holmstrøm, K.; Lindow, M.; Porse, B.; Kauppinen, S.; Lund, A.H. microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci. Rep. 2013, 3, 2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Politz, J.C.R.; Zhang, F.; Pederson, T. MicroRNA-206 colocalizes with ribosome-rich regions in both the nucleolus and cytoplasm of rat myogenic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 18957–18962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Gutierrez, P.; Politz, J.C.R.; Pederson, T. A mRNA and Cognate MicroRNAs Localize in the Nucleolus. Nucleus 2014, 5, 636–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, K.; Singh, I.; Dobersch, S.; Sarvari, P.; Günther, S.; Cordero, J.; Mehta, A.; Wujak, L.; Cabrera-Fuentes, H.; Chao, C.-M.; et al. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 2229. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, K.-M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Lin, C.; Liu, W.; Zhang, J.; Ohgi, K.A.; Grinstein, J.D.; Dorrestein, P.C.; Rosenfeld, M.G. ncRNA- and Pc2 Methylation-Dependent Gene Relocation between Nuclear Structures Mediates Gene Activation Programs. Cell 2011, 147, 773–788. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Feng, Y.; Zhang, D.; Zhao, S.D.; Hu, Z.; Greshock, J.; Zhang, Y.; Yang, L.; Zhong, X.; Wang, L.-P.; et al. A Functional Genomic Approach Identifies FAL1 as an Oncogenic Long Noncoding RNA that Associates with BMI1 and Represses p21 Expression in Cancer. Cancer Cell 2014, 26, 344–357. [Google Scholar] [CrossRef] [Green Version]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.R.; Taalab, Y.M.; Heinze, S.; Lovaković, B.T.; Pizent, A.; Renieri, E.; Tsatsakis, A.; Farooqi, A.A.; Javorac, D.; Andjelkovic, M.; et al. Toxic-Metal-Induced Alteration in miRNA Expression Profile as a Proposed Mechanism for Disease Development. Cells 2020, 9, 901. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Zamudio, R.; Ha, H.C. Environmental epigenetics in metal exposure. Epigenetics 2011, 6, 820–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezo, Y.J.; Dale, B.; Elder, K. The negative impact of the environment on methylation/epigenetic marking in gametes and embryos: A plea for action to protect the fertility of future generations. Mol. Reprod. Dev. 2019, 86, 1273–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samet, J.M.; Wages, P. Oxidative stress from environmental exposures. Curr. Opin. Toxicol. 2018, 7, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Miguel, V.; Lamas, S.; Espinosa-Diez, C. Role of non-coding-RNAs in response to environmental stressors and consequences on human health. Redox Biol. 2020, 37, 101580. [Google Scholar] [CrossRef]
- Bhargava, A.; Shukla, A.; Bunkar, N.; Shandilya, R.; Lodhi, L.; Kumari, R.; Gupta, P.K.; Rahman, A.; Chaudhury, K.; Tiwari, R.; et al. Exposure to ultrafine particulate matter induces NF-kappabeta mediated epigenetic modifications. Environ. Pollut. 2019, 252 Pt A, 39–50. [Google Scholar] [CrossRef]
- Soberanes, S.; Gonzalez, A.; Urich, D.; Chiarella, S.E.; Radigan, K.A.; Osornio-Vargas, A.; Joseph, J.; Kalyanaraman, B.; Ridge, K.M.; Chandel, N.S.; et al. Particulate matter Air Pollution induces hypermethylation of the p16 promoter Via a mitochondrial ROS-JNK-DNMT1 pathway. Sci. Rep. 2012, 2, 275. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shen, S.; Du, C.; Wang, Y.; Liu, Y.; He, Q. A role for the mitotic proteins Bub3 and BuGZ in transcriptional regulation of catalase-3 expression. PLoS Genet. 2022, 18, e1010254. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Branca, J.J.V.; Pacini, A.; Gulisano, M.; Taddei, N.; Fiorillo, C.; Becatti, M. Cadmium-Induced Cytotoxicity: Effects on Mitochondrial Electron Transport Chain. Front. Cell Dev. Biol. 2020, 8, 604377. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralph, S.J.; Moreno-Sanchez, R.; Neuzil, J.; Rodriguez-Enriquez, S. Inhibitors of succinate: Quinone reductase/Complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm. Res. 2011, 28, 2695–2730. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkstra, G.; Moshage, H.; Van Dullemen, H.M.; De Jager-Krikken, A.; Tiebosch, A.T.M.G.; Kleibeuker, J.H.; Jansen, P.L.M.; van Goor, H. Expression of nitric oxide synthases and formation of nitrotyrosine and reactive oxygen species in inflammatory bowel disease. J. Pathol. 1998, 186, 416–421. [Google Scholar] [CrossRef]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Selvaraju, V.; Baskaran, S.; Agarwal, A.; Henkel, R. Environmental contaminants and male infertility: Effects and mechanisms. Andrologia 2021, 53, e13646. [Google Scholar] [CrossRef]
- Skinner, M.K. Epigenetic transgenerational inheritance. Nat. Rev. Endocrinol. 2016, 12, 68–70. [Google Scholar] [CrossRef] [Green Version]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Plastics Derived Endocrine Disruptors (BPA, DEHP and DBP) Induce Epigenetic Transgenerational Inheritance of Obesity, Reproductive Disease and Sperm Epimutations. PLoS ONE 2013, 8, e55387. [Google Scholar] [CrossRef] [Green Version]
- El Henafy, H.M.A.; Ibrahim, M.A.; Abd El Aziz, S.A.; Gouda, E.M. Oxidative Stress and DNA methylation in male rat pups provoked by the transplacental and translactational exposure to bisphenol A. Environ. Sci. Pollut. Res. Int. 2020, 27, 4513–4519. [Google Scholar] [CrossRef]
- Fan, X.; Hou, T.; Jia, J.; Tang, K.; Wei, X.; Wang, Z. Discrepant dose responses of bisphenol A on oxidative stress and DNA methylation in grass carp ovary cells. Chemosphere 2020, 248, 126110. [Google Scholar] [CrossRef] [PubMed]
- Rattan, S.; Beers, H.K.; Kannan, A.; Ramakrishnan, A.; Brehm, E.; Bagchi, I.; Irudayaraj, J.M.; Flaws, J.A. Prenatal and ancestral exposure to di(2-ethylhexyl) phthalate alters gene expression and DNA methylation in mouse ovaries. Toxicol. Appl. Pharmacol. 2019, 379, 114629. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Cruz, E.Y.; Amador-Martínez, I.; Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Chaverri, J.P. Renal damage induced by cadmium and its possible therapy by mitochondrial transplantation. Chem. Interact. 2022, 361, 109961. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Yao, H.; Rahman, I.; Gupta, I.; Ganguly, S.; Rozanas, C.R.; Stuehr, D.J.; Panda, K.; Ruijters, E.; Haenen, G.; et al. Oxidative Stress and Chromatin Remodeling in Chronic Obstructive Pulmonary Disease and Smoking-Related Diseases. Antioxid. Redox Signal. 2013, 18, 1956–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebratu, Y.A.; Schwalm, K.; Smith, K.R.; Schuyler, M.; Tesfaigzi, Y. Cigarette Smoke Suppresses Bik To Cause Epithelial Cell Hyperplasia and Mucous Cell Metaplasia. Am. J. Respir. Crit. Care Med. 2011, 183, 1531–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Liu, J.; Li, J.; Lv, X.; Yu, L.; Wu, K.; Yang, Y. Metal contamination, bioaccumulation, ROS generation, and epigenotoxicity influences on zebrafish exposed to river water polluted by mining activities. J. Hazard. Mater. 2021, 405, 124150. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Ye, S.; Pan, Y.; Bao, Y.; Chen, H.; Shao, C. Long-term cadmium exposure leads to the enhancement of lymphocyte proliferation via down-regulating p16 by DNA hypermethylation. Mutat. Res. 2013, 757, 125–131. [Google Scholar] [CrossRef]
- Barone, G.; Storelli, A.; Garofalo, R.; Mallamaci, R.; Storelli, M.M. Residual Levels of Mercury, Cadmium, Lead and Arsenic in Some Commercially Key Species from Italian Coasts (Adriatic Sea): Focus on Human Health. Toxics 2022, 10, 223. [Google Scholar] [CrossRef]
- Tellez-Plaza, M.; Tang, W.-Y.; Shang, Y.; Umans, J.G.; Francesconi, K.A.; Goessler, W.; Ledesma, M.; León, M.; Laclaustra, M.; Pollak, J.; et al. Association of Global DNA Methylation and Global DNA Hydroxymethylation with Metals and Other Exposures in Human Blood DNA Samples. Environ. Health Perspect. 2014, 122, 946–954. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yang, X.; Xu, M.; Zhang, J.; Sun, N. Epigenetic marker (LINE-1 promoter) methylation level was associated with occupational lead exposure. Clin. Toxicol. 2013, 51, 225–229. [Google Scholar] [CrossRef]
- Alam, N.; Shapla, U.M.; Shen, H.; Huang, Q. Linking emerging contaminants exposure to adverse health effects: Crosstalk between epigenome and environment. J. Appl. Toxicol. 2021, 41, 878–897. [Google Scholar] [CrossRef] [PubMed]
- El Dine, F.M.M.B.; Abdel Samie Mohamed Hussein, H.; Abdelmawla Ghazala, R. Evaluation of epigenetic changes of liver tissue induced by oral administration of Titanium dioxide nanoparticles and possible protective role of Nigella Sativa oil, in adult male albino rats. Nanomed. J. 2018, 5, 192–198. [Google Scholar]
- Bai, W.; Chen, Y.; Gao, A. Cross talk between poly(ADP-ribose) polymerase 1 methylation and oxidative stress involved in the toxic effect of anatase titanium dioxide nanoparticles. Int. J. Nanomed. 2015, 10, 5561–5569. [Google Scholar] [CrossRef] [Green Version]
- Bellavia, A.; Urch, B.; Speck, M.; Brook, R.D.; Scott, J.A.; Albetti, B.; Behbod, B.; North, M.; Valeri, L.; Bertazzi, P.A.; et al. DNA Hypomethylation, Ambient Particulate Matter, and Increased Blood Pressure: Findings From Controlled Human Exposure Experiments. J. Am. Heart Assoc. 2013, 2, e000212. [Google Scholar] [CrossRef] [Green Version]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Bossis, G.; Hyun, Y.-M.; Park, C.O.; Hyun, J.W. Particulate matter-induced senescence of skin keratinocytes involves oxidative stress-dependent epigenetic modifications. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, G.; Lin, Z.; Wang, Y.; He, H.; Liu, T.; Kamp, D.W. Pro-inflammatory response and oxidative stress induced by specific components in ambient particulate matter in human bronchial epithelial cells. Environ. Toxicol. 2016, 31, 923–936. [Google Scholar] [CrossRef]
- Menezo, Y.J.; Silvestris, E.; Dale, B.; Elder, K. Oxidative stress and alterations in DNA methylation: Two sides of the same coin in reproduction. Reprod. Biomed. Online 2016, 33, 668–683. [Google Scholar] [CrossRef] [Green Version]
- Donkena, K.V.; Young, C.Y.F.; Tindall, D.J. Oxidative Stress and DNA Methylation in Prostate Cancer. Obstet. Gynecol. Int. 2010, 2010, 302051. [Google Scholar] [CrossRef] [Green Version]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Wachsman, J.T. DNA methylation and the association between genetic and epigenetic changes: Relation to carcinogenesis. Mutat. Res. 1997, 375, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; A Loeb, L. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G–T and A–C substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Formation of an 8-hydroxyguanine moiety in deoxyribonucleic acid on gamma-irradiation in aqueous solution. Biochemistry 1985, 24, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Turk, P.W.; Laayoun, A.; Smith, S.S.; Weitzman, S.A. DNA adduct 8-hydroxyl-2′-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis 1995, 16, 1253–1255. [Google Scholar] [CrossRef]
- Udomsinprasert, W.; Kitkumthorn, N.; Mutirangura, A.; Chongsrisawat, V.; Poovorawan, Y.; Honsawek, S. Global methylation, oxidative stress and relative telomere length in biliary atresia patients. Sci. Rep. 2016, 6, 26969. [Google Scholar] [CrossRef] [Green Version]
- Valinluck, V.; Tsai, H.H.; Rogstad, D.K.; Burdzy, A.; Bird, A.; Sowers, L.C. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef] [Green Version]
- Weitzman, S.A.; Turk, P.W.; Milkowski, D.H.; Kozlowski, K. Free radical adducts induce alterations in DNA cytosine methylation. Proc. Natl. Acad. Sci. USA 1994, 91, 1261–1264. [Google Scholar] [CrossRef] [Green Version]
- Chia, N.; Wang, L.; Lu, X.; Senut, M.-C.; Brenner, C.A.; Ruden, D.M. Hypothesis: Environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics 2011, 6, 853–856. [Google Scholar] [CrossRef] [Green Version]
- Parry, L.; Clarke, A.R. The Roles of the Methyl-CpG Binding Proteins in Cancer. Genes Cancer 2011, 2, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila, M.A.; Corrales, F.J.; Ruiz, F.; Sánchez-Góngora, E.; Mingorance, J.; Carretero, M.V.; Mato, J.M. Specific interaction of methionine adenosyltransferase with free radicals. Biofactors 1998, 8, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrett, J.T.; Hoover, D.M.; Ludwig, M.L.; Matthews, R.G. The Mechanism of Adenosylmethionine-Dependent Activation of Methionine Synthase: A Rapid Kinetic Analysis of Intermediates in Reductive Methylation of Cob(II)alamin Enzyme. Biochemistry 1998, 37, 12649–12658. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.A.; Durán, C.; Corrales, F.; Pliego, M.M.; Mato, J.M. Modulation of rat liver S-adenosylmethionine synthetase activity by glutathione. J. Biol. Chem. 1992, 267, 17598–17605. [Google Scholar] [CrossRef]
- Cyr, A.R.; Domann, F.E. The Redox Basis of Epigenetic Modifications: From Mechanisms to Functional Consequences. Antioxid. Redox Signal. 2011, 15, 551–589. [Google Scholar] [CrossRef] [Green Version]
- García-Giménez, J.L.; Romá-Mateo, C.; Pérez-Machado, G.; Peiró-Chova, L.; Pallardó, F.V. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 2017, 112, 36–48. [Google Scholar] [CrossRef]
- Ponnaluri, V.C.; Maciejewski, J.P.; Mukherji, M. A mechanistic overview of TET-mediated 5-methylcytosine oxidation. Biochem. Biophys. Res. Commun. 2013, 436, 115–120. [Google Scholar] [CrossRef]
- Dickson, K.M.; Gustafson, C.B.; Young, J.I.; Züchner, S.; Wang, G. Ascorbate-induced generation of 5-hydroxymethylcytosine is unaffected by varying levels of iron and 2-oxoglutarate. Biochem. Biophys. Res. Commun. 2013, 439, 522–527. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef] [Green Version]
- García-Giménez, J.L.; Romá-Mateo, C.; Pallardó, F.V. Oxidative post-translational modifications in histones. Biofactors 2019, 45, 641–650. [Google Scholar] [CrossRef]
- Vrijens, K.; Trippas, A.-J.; Lefebvre, W.; Vanpoucke, C.; Penders, J.; Janssen, B.G.; Nawrot, T.S. Association of Prenatal Exposure to Ambient Air Pollution With Circulating Histone Levels in Maternal Cord Blood. JAMA Netw. Open 2020, 3, e205156. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Dixit, K.; Khan, M.A.; Sharma, Y.; Moinuddin; Alam, K. Physicochemical studies on peroxynitrite-modified H3 histone. Int. J. Biol. Macromol. 2010, 46, 20–26. [Google Scholar] [CrossRef]
- Khan, M.A.; Dixit, K.; Moinuddin; Arif, Z.; Alam, K. Studies on peroxynitrite-modified H1 histone: Implications in systemic lupus erythematosus. Biochimie 2014, 97, 104–113. [Google Scholar] [CrossRef]
- Haqqani, A.S.; Kelly, J.F.; Birnboim, H.C. Selective Nitration of Histone Tyrosine Residues in Vivo in Mutatect Tumors. J. Biol. Chem. 2002, 277, 3614–3621. [Google Scholar] [CrossRef] [Green Version]
- Beisswenger, P.J.; Howell, S.K.; Smith, K.; Szwergold, B.S. Glyceraldehyde-3-phosphate dehydrogenase activity as an independent modifier of methylglyoxal levels in diabetes. Biochim. Biophys. Acta 2003, 1637, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T. 3-Deoxyglucosone: Metabolism, analysis, biological activity, and clinical implication. J. Chromatogr. B Biomed. Sci. Appl. 1999, 731, 23–36. [Google Scholar] [CrossRef]
- Ashraf, J.M.; Ahmad, S.; Rabbani, G.; Jan, A.T.; Lee, E.J.; Khan, R.H.; Choi, I. Physicochemical analysis of structural alteration and advanced glycation end products generation during glycation of H2A histone by 3-deoxyglucosone. IUBMB Life 2014, 66, 686–693. [Google Scholar] [CrossRef]
- Ashraf, J.M.; Rabbani, G.; Ahmad, S.; Hasan, Q.; Khan, R.H.; Alam, K.; Choi, I. Glycation of H1 Histone by 3-Deoxyglucosone: Effects on Protein Structure and Generation of Different Advanced Glycation End Products. PLoS ONE 2015, 10, e0130630. [Google Scholar] [CrossRef] [Green Version]
- Mir, A.R.; Uddin, M.; Alam, K.; Ali, A. Methylglyoxal mediated conformational changes in histone H2A—Generation of carboxyethylated advanced glycation end products. Int. J. Biol. Macromol. 2014, 69, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galligan, J.J.; Rose, K.L.; Beavers, W.N.; Hill, S.; Tallman, K.A.; Tansey, W.P.; Marnett, L.J. Stable Histone Adduction by 4-Oxo-2-nonenal: A Potential Link between Oxidative Stress and Epigenetics. J. Am. Chem. Soc. 2014, 136, 11864–11866. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.; Petroze, R.; Castegna, A.; Ding, Q.; Keller, J.N.; Markesbery, W.R.; Lovell, M.A.; Butterfield, D.A. 4-Hydroxynonenal oxidatively modifies histones: Implications for Alzheimer’s disease. Neurosci. Lett. 2004, 356, 155–158. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Moroni, N.; Serafino, A.; Primavera, A.; Pastore, A.; Pedersen, J.Z.; Petruzzelli, R.; Farrace, M.G.; Pierimarchi, P.; Moroni, G.; et al. Treatment of doxorubicin-resistant MCF7/Dx cells with nitric oxide causes histone glutathionylation and reversal of drug resistance. Biochem. J. 2011, 440, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Giménez, J.L.; Olaso, G.; Hake, S.B.; Bönisch, C.; Wiedemann, S.M.; Markovic, J.; Dasi, F.; Gimeno, A.; Perez-Quilis, C.; Palacios, O.; et al. Histone H3 Glutathionylation in Proliferating Mammalian Cells Destabilizes Nucleosomal Structure. Antioxid. Redox Signal. 2013, 19, 1305–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, R.; Flashman, E.; Mecinović, J.; Kramer, H.B.; Kessler, B.M.; Frapart, Y.M.; Boucher, J.-L.; Clifton, I.J.; McDonough, M.A.; Schofield, C.J. Studies on the Reaction of Nitric Oxide with the Hypoxia-Inducible Factor Prolyl Hydroxylase Domain 2 (EGLN1). J. Mol. Biol. 2011, 410, 268–279. [Google Scholar] [CrossRef]
- Hickok, J.R.; Vasudevan, D.; Antholine, W.E.; Thomas, D.D. Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases. J. Biol. Chem. 2013, 288, 16004–16015. [Google Scholar] [CrossRef] [Green Version]
- Hitchler, M.J.; Domann, F.E. Redox regulation of the epigenetic landscape in Cancer: A role for metabolic reprogramming in remodeling the epigenome. Free Radic. Biol. Med. 2012, 53, 2178–2187. [Google Scholar] [CrossRef] [Green Version]
- Chervona, Y.; Costa, M. The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic. Biol. Med. 2012, 53, 1041–1047. [Google Scholar] [CrossRef] [Green Version]
- Arita, A.; Costa, M. Epigenetics in metal carcinogenesis: Nickel, arsenic, chromium and cadmium. Metallomics 2009, 1, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresovich, J.K.; Zhang, Z.; Fang, F.; Zheng, Y.; Sanchez-Guerra, M.; Joyce, B.T.; Zhong, J.; Chervona, Y.; Wang, S.; Chang, D.; et al. Histone 3 modifications and blood pressure in the Beijing Truck Driver Air Pollution Study. Biomarkers 2017, 22, 584–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Liu, H.; Ha, Y.; Luo, X.; Motamedi, M.; Gupta, M.P.; Ma, J.-X.; Tilton, R.G.; Zhang, W. Posttranslational modification of Sirt6 activity by peroxynitrite. Free Radic. Biol. Med. 2015, 79, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Doyle, K.; Fitzpatrick, F. Redox Signaling, Alkylation (Carbonylation) of Conserved Cysteines Inactivates Class I Histone Deacetylases 1, 2, and 3 and Antagonizes Their Transcriptional Repressor Function. J. Biol. Chem. 2010, 285, 17417–17424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, K.S.; Galligan, J.J.; Smathers, R.L.; Roede, J.R.; Shearn, C.T.; Reigan, P.; Petersen, D.R. 4-Hydroxynonenal Inhibits SIRT3 via Thiol-Specific Modification. Chem. Res. Toxicol. 2011, 24, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Moodie, F.M.; Marwick, J.A.; Anderson, C.S.; Szulakowski, P.; Biswas, S.K.; Bauter, M.R.; Kilty, I.; Rahman, I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J. 2004, 18, 1897–1899. [Google Scholar] [CrossRef]
- Adenuga, D.; Yao, H.; March, T.H.; Seagrave, J.; Rahman, I. Histone Deacetylase 2 Is Phosphorylated, Ubiquitinated, and Degraded by Cigarette Smoke. Am. J. Respir. Cell Mol. Biol. 2009, 40, 464–473. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Rahman, I. Role of histone deacetylase 2 in epigenetics and cellular senescence: Implications in lung inflammaging and COPD. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L557–L566. [Google Scholar] [CrossRef]
- Arita, A.; Costa, M. Oxidative stress and the epigenome in human disease. J. Genet. Genome Res. 2014, 1, 2. [Google Scholar] [CrossRef]
- Yang, S.R.; Wright, J.; Bauter, M.; Seweryniak, K.; Kode, A.; Rahman, I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: Implications for chronic inflammation and aging. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L567–L576. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, T.; Goto, A.; Kurokawa, E.; Hara, H.; Adachi, T. Cross Talk Mechanism among EMT, ROS, and Histone Acetylation in Phorbol Ester-Treated Human Breast Cancer MCF-7 Cells. Oxidative Med. Cell. Longev. 2016, 2016, 1284372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agudelo, M.; Gandhi, N.; Saiyed, Z.; Pichili, V.; Thangavel, S.; Khatavkar, P.; Yndart-Arias, A.; Nair, M. Effects of Alcohol on Histone Deacetylase 2 (HDAC2) and the Neuroprotective Role of Trichostatin A (TSA). Alcohol. Clin. Exp. Res. 2011, 35, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-W.; Yao, H.; Caito, S.; Sundar, I.K.; Rahman, I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 2013, 61, 95–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castonguay, Z.; Auger, C.; Thomas, S.C.; Chahma, M.; Appanna, V.D. Nuclear lactate dehydrogenase modulates histone modification in human hepatocytes. Biochem. Biophys. Res. Commun. 2014, 454, 172–177. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Hou, N.; Xiao, L.; Xu, Y.; Xu, H.; Li, F. Dynamic monitoring of oxidative DNA double-strand break and repair in cardiomyocytes. Cardiovasc. Pathol. 2016, 25, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Katsube, T.; Mori, M.; Tsuji, H.; Shiomi, T.; Wang, B.; Liu, Q.; Nenoi, M.; Onoda, M. Most hydrogen peroxide-induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double-strand breaks. J. Biochem. 2014, 156, 85–95. [Google Scholar] [CrossRef]
- Rusnak, F.; Reiter, T. Sensing electrons: Protein phosphatase redox regulation. Trends Biochem. Sci. 2000, 25, 527–529. [Google Scholar] [CrossRef]
- García-Guede, A.; Vera, O.; Ibáñez-De-Caceres, I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants 2020, 9, 468. [Google Scholar] [CrossRef]
- He, J.; Jiang, B.-H. Interplay Between Reactive Oxygen Species and MicroRNAs in Cancer. Curr. Pharmacol. Rep. 2016, 2, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Huang, Z.; Han, J.; Shao, J.; Huang, C. Redox regulation of microRNAs in cancer. Cancer Lett. 2018, 418, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.; Li, Y.; Xu, Y.; Pang, Y.; Shen, L.; Jiang, R.; Zhao, Y.; Yang, X.; Zhang, J.; Zhou, J.; et al. Regulation of miRNA-21 by reactive oxygen species-activated ERK/NF-kappaB in arsenite-induced cell transformation. Free Radic. Biol. Med. 2012, 52, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Barr, I.; Smith, A.T.; Chen, Y.; Senturia, R.; Burstyn, J.N.; Guo, F. Ferric, not ferrous, heme activates RNA-binding protein DGCR8 for primary microRNA processing. Proc. Natl. Acad. Sci. USA 2012, 109, 1919–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaksik, R.; Lalik, A.; Skonieczna, M.; Cieslar-Pobuda, A.; Student, S.; Rzeszowska-Wolny, J. MicroRNAs and reactive oxygen species: Are they in the same regulatory circuit? Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 764–765, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Bunkar, N.; Kumar, R.; Bhargava, A.; Tiwari, R.; Chaudhury, K.; Goryacheva, I.Y.; Mishra, P.K. Air pollution associated epigenetic modifications: Transgenerational inheritance and underlying molecular mechanisms. Sci. Total Environ. 2019, 656, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tao, X.; Yin, L.; Xu, L.; Xu, Y.; Qi, Y.; Han, X.; Song, S.; Zhao, Y.; Lin, Y.; et al. Protective effects of dioscin against cisplatin-induced nephrotoxicity via the microRNA-34a/sirtuin 1 signalling pathway. Br. J. Pharmacol. 2017, 174, 2512–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardim, M.J. microRNAs: Implications for air pollution research. Mutat. Res. 2011, 717, 38–45. [Google Scholar] [CrossRef]
- Zhang, S.; Duan, S.; Xie, Z.; Bao, W.; Xu, B.; Yang, W.; Zhou, L. Epigenetic Therapeutics Targeting NRF2/KEAP1 Signaling in Cancer Oxidative Stress. Front. Pharmacol. 2022, 13, 924817. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Khor, T.O.; Cheung, K.-L.; Li, W.; Wu, T.-Y.; Huang, Y.; Foster, B.A.; Kan, Y.W.; Kong, A.-N. Nrf2 Expression Is Regulated by Epigenetic Mechanisms in Prostate Cancer of TRAMP Mice. PLoS ONE 2010, 5, e8579. [Google Scholar] [CrossRef] [Green Version]
- Barve, A.; Khor, T.O.; Nair, S.; Reuhl, K.; Suh, N.; Reddy, B.; Newmark, H.; Kong, A.N. Gamma-tocopherol-enriched mixed tocopherol diet inhibits prostate carcinogenesis in TRAMP mice. Int. J. Cancer. 2009, 124, 1693–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbano, R.; Muscarella, L.A.; Pasculli, B.; Valori, V.M.; Fontana, A.; Coco, M.; la Torre, A.; Balsamo, T.; Poeta, M.L.; Marangi, G.F.; et al. Aberrant Keap1 methylation in breast cancer and association with clinicopathological features. Epigenetics 2013, 8, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Yu, S.; Zhang, C.; Kong, A.-N.T. Epigenetic regulation of Keap1-Nrf2 signaling. Free Radic. Biol. Med. 2015, 88 Pt B, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Motta, V.; Angelici, L.; Nordio, F.; Bollati, V.; Fossati, S.; Frascati, F.; Tinaglia, V.; Bertazzi, P.A.; Battaglia, C.; Baccarelli, A.A. Integrative Analysis of miRNA and Inflammatory Gene Expression After Acute Particulate Matter Exposure. Toxicol. Sci. 2013, 132, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fossati, S.; Baccarelli, A.; Zanobetti, A.; Hoxha, M.; Vokonas, P.S.; Wright, R.; Schwartz, J. Ambient Particulate Air Pollution and MicroRNAs in Elderly Men. Epidemiology 2014, 25, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Izzotti, A.; Calin, G.A.; Arrigo, P.; Steele, V.E.; Croce, C.M.; De Flora, S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009, 23, 806–812. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Luo, F.; Xu, Y.; Wang, B.; Zhao, Y.; Xu, W.; Shi, L.; Lu, X.; Liu, Q. Epithelial-mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol. Appl. Pharmacol. 2015, 282, 9–19. [Google Scholar] [CrossRef]
- Rubin, H. Dynamics of cell transformation in culture and its significance for tumor development in animals. Proc. Natl. Acad. Sci. USA 2017, 114, 12237–12242. [Google Scholar] [CrossRef] [Green Version]
- Ou, K.; Hamo, D.; Schulze, A.; Roemhild, A.; Kaiser, D.; Gasparoni, G.; Salhab, A.; Zarrinrad, G.; Amini, L.; Schlickeiser, S.; et al. Strong Expansion of Human Regulatory T Cells for Adoptive Cell Therapy Results in Epigenetic Changes Which May Impact Their Survival and Function. Front. Cell Dev. Biol. 2021, 9, 751590. [Google Scholar] [CrossRef]
- Cox, M.C.; Deng, C.; Naler, L.B.; Lu, C.; Verbridge, S.S. Effects of Culture Condition on Epigenomic Profiles of Brain Tumor Cells. ACS Biomater. Sci. Eng. 2019, 5, 1544–1552. [Google Scholar] [CrossRef]
- Weissbein, U.; Plotnik, O.; Vershkov, D.; Benvenisty, N. Culture-induced recurrent epigenetic aberrations in human pluripotent stem cells. PLoS Genet. 2017, 13, e1006979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazor, K.L.; Altun, G.; Lynch, C.; Tran, H.; Harness, J.V.; Slavin, I.; Garitaonandia, I.; Müller, F.-J.; Wang, Y.-C.; Boscolo, F.S.; et al. Recurrent Variations in DNA Methylation in Human Pluripotent Stem Cells and Their Differentiated Derivatives. Cell Stem Cell 2012, 10, 620–634. [Google Scholar] [CrossRef] [Green Version]
- Mallon, B.S.; Chenoweth, J.G.; Johnson, K.R.; Hamilton, R.S.; Tesar, P.J.; Yavatkar, A.S.; Tyson, L.J.; Park, K.; Chen, K.G.; Fann, Y.C.; et al. StemCellDB: The Human Pluripotent Stem Cell Database at the National Institutes of Health. Stem Cell Res. 2013, 10, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference Maps of Human ES and iPS Cell Variation Enable High-Throughput Characterization of Pluripotent Cell Lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Allegrucci, C.; Wu, Y.-Z.; Thurston, A.; Denning, C.N.; Priddle, H.; Mummery, C.L.; Oostwaard, D.W.-V.; Andrews, P.W.; Stojkovic, M.; Smith, N.; et al. Restriction landmark genome scanning identifies culture-induced DNA methylation instability in the human embryonic stem cell epigenome. Hum. Mol. Genet. 2007, 16, 1253–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, S.; Benvenisty, N. Epigenetic aberrations in human pluripotent stem cells. EMBO J. 2019, 38, e101033. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Huebner, A.J.; Clement, K.; Walsh, R.M.; Savol, A.; Lin, K.; Gu, H.; Di Stefano, B.; Brumbaugh, J.; Kim, S.Y.; et al. Prolonged Mek1/2 suppression impairs the developmental potential of embryonic stem cells. Nature 2017, 548, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Park, J.; Bang, Y.-J.; Kim, T.-Y. Gene silencing of TSPYL5 mediated by aberrant promoter methylation in gastric cancers. Lab. Investig. 2008, 88, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-Y.; Zhong, S.; Fields, C.R.; Kim, J.H.; Robertson, K.D. Epigenomic Profiling Reveals Novel and Frequent Targets of Aberrant DNA Methylation-Mediated Silencing in Malignant Glioma. Cancer Res 2006, 66, 7490–7501. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tang, C.B.; Kilian, K.A. Matrix Mechanics Influence Fibroblast–Myofibroblast Transition by Directing the Localization of Histone Deacetylase 4. Cell. Mol. Bioeng. 2017, 10, 405–415. [Google Scholar] [CrossRef]
- Plachot, C.; Lelièvre, S.A. DNA methylation control of tissue polarity and cellular differentiation in the mammary epithelium. Exp. Cell Res. 2004, 298, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Chandramouly, G.; Abad, P.C.; Knowles, D.W.; Lelievre, S. The control of tissue architecture over nuclear organization is crucial for epithelial cell fate. J. Cell Sci. 2007, 120 Pt 9, 1596–1606. [Google Scholar] [CrossRef] [Green Version]
- Chhetri, A.; Rispoli, J.V.; Lelièvre, S.A. 3D Cell Culture for the Study of Microenvironment-Mediated Mechanostimuli to the Cell Nucleus: An Important Step for Cancer Research. Front. Mol. Biosci. 2021, 8, 628386. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Xiao, P.; Wang, X.; Yang, X.; Xu, H.; Lu, K.; Lu, S.; Liang, X. Melatonin prevents deterioration in quality by preserving epigenetic modifications of porcine oocytes after prolonged culture. Aging 2018, 10, 3897–3909. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dal-Pra, S.; Mirotsou, M.; Jayawardena, T.M.; Hodgkinson, C.P.; Bursac, N.; Dzau, V.J. Tissue-engineered 3-dimensional (3D) microenvironment enhances the direct reprogramming of fibroblasts into cardiomyocytes by microRNAs. Sci. Rep. 2016, 6, 38815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Shen, Q.; Yang, X.; Qiu, Y.; Zhang, W. The Role of Extracellular Vesicles: An Epigenetic View of the Cancer Microenvironment. BioMed Res. Int. 2015, 2015, 649161. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, C.; Melo, S.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of Double-stranded Genomic DNA Spanning All Chromosomes with Mutated KRAS and p53 DNA in the Serum Exosomes of Patients with Pancreatic Cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; You, Y.; Li, Q.; Zeng, C.; Fu, F.; Guo, A.; Zhang, H.; Zou, P.; Zhong, Z.; Wang, H.; et al. BCR-ABL1–positive microvesicles transform normal hematopoietic transplants through genomic instability: Implications for donor cell leukemia. Leukemia 2014, 28, 1666–1675. [Google Scholar] [CrossRef]
- Schiera, G.; DI Liegro, C.M.; Saladino, P.; Pitti, R.; Savettieri, G.; Proia, P.; DI Liegro, I. Oligodendroglioma cells synthesize the differentiation-specific linker histone H1° and release it into the extracellular environment through shed vesicles. Int. J. Oncol. 2013, 43, 1771–1776. [Google Scholar] [CrossRef] [Green Version]
- Zlatanova, J.; Doenecke, D. Histone H1 zero: A major player in cell differentiation? FASEB J. 1994, 8, 1260–1268. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Cheng, P.; Fan, Y.; Yu, B.; Nikitina, E.; Sirotkin, A.; Shurin, M.; Oyama, T.; Adachi, Y.; Nadaf, S.; et al. H1(0) histone and differentiation of dendritic cells. A molecular target for tumor-derived factors. J. Leukoc. Biol. 2002, 72, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Jiang, T.; Kang, X. Circulating microRNAs in cancer: Origin, function and application. J. Exp. Clin. Cancer Res. 2012, 31, 38–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom. 2013, 14, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, N.J.; Zhou, H.; Elashoff, D.; Henson, B.S.; Kastratovic, D.A.; Abemayor, E.; Wong, D.T. Salivary microRNA: Discovery, Characterization, and Clinical Utility for Oral Cancer Detection. Clin. Cancer Res. 2009, 15, 5473–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating MicroRNA-208b and MicroRNA-499 Reflect Myocardial Damage in Cardiovascular Disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Lodes, M.J.; Caraballo, M.; Suciu, D.; Munro, S.; Kumar, A.; Anderson, B. Detection of Cancer with Serum miRNAs on an Oligonucleotide Microarray. PLoS ONE 2009, 4, e6229. [Google Scholar] [CrossRef]
- Stepkowski, S.M.; Chen, H.; Daloze, P.; Kahan, B.D. Rapamycin, a potent immunosuppressive drug for vascularized heart, kidney, and small bowel transplantation in the rat. Transplantation 1991, 51, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Etheridge, A.; Lee, I.; Hood, L.; Galas, D.; Wang, K. Extracellular microRNA: A new source of biomarkers. Mutat. Res. 2011, 717, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Shen, N.; Pang, J.; Xie, D.; Deng, B.; Molina, J.R.; Yang, P.; Liu, S. Restoration of miR-101 suppresses lung tumorigenesis through inhibition of DNMT3a-dependent DNA methylation. Cell Death Dis. 2014, 5, e1413. [Google Scholar] [CrossRef] [Green Version]
- Kremer, E.A.; Gaur, N.; Lee, M.A.; Engmann, O.; Bohacek, J.; Mansuy, I.M. Interplay between TETs and microRNAs in the adult brain for memory formation. Sci. Rep. 2018, 8, 1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; You, R.; Yuan, X.; Yang, T.; Samuel, E.L.; Marcano, D.C.; Sikkema, W.K.; Tour, J.M.; Rodriguez, A.; Kheradmand, F.; et al. The microRNA miR-22 inhibits the histone deacetylase HDAC4 to promote T(H)17 cell-dependent emphysema. Nat. Immunol. 2015, 16, 1185–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorico, A.; Mercapide, J.; Rappa, G. Prominin-1 (CD133) and Metastatic Melanoma: Current Knowledge and Therapeutic Perspectives. Adv. Exp. Med. Biol. 2012, 777, 197–211. [Google Scholar] [CrossRef]
- Rappa, G.; Mercapide, J.; Anzanello, F.; Le, T.T.; Johlfs, M.G.; Fiscus, R.R.; Wilsch-Bräuninger, M.; Corbeil, D.; Lorico, A. Wnt interaction and extracellular release of prominin-1/CD133 in human malignant melanoma cells. Exp. Cell Res. 2013, 319, 810–819. [Google Scholar] [CrossRef] [Green Version]
- Rappa, G.; Fodstad, O.; Lorico, A. The Stem Cell-Associated Antigen CD133 (Prominin-1) Is a Molecular Therapeutic Target for Metastatic Melanoma. Stem Cells 2008, 26, 3008–3017. [Google Scholar] [CrossRef] [Green Version]
- Rappa, G.; Mercapide, J.; Anzanello, F.; Pope, R.M.; Lorico, A. Biochemical and biological characterization of exosomes containing prominin-1/CD133. Mol. Cancer 2013, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, K.; Inoue, K.; Fujiwara, A.; Hatakeyama, K.; Kanto, K.; Watanabe, Y.; Muramatsu, K.; Fukuda, Y.; Ogura, S.-I.; Yamaguchi, K.; et al. Let-7 MicroRNA Family Is Selectively Secreted into the Extracellular Environment via Exosomes in a Metastatic Gastric Cancer Cell Line. PLoS ONE 2010, 5, e13247. [Google Scholar] [CrossRef] [Green Version]
- Taverna, S.; Amodeo, V.; Saieva, L.; Russo, A.; Giallombardo, M.; De Leo, G.; Alessandro, R. Exosomal shuttling of miR-126 in endothelial cells modulates adhesive and migratory abilities of chronic myelogenous leukemia cells. Mol. Cancer 2014, 13, 169. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Tapparo, M.; Collino, F.; Vitillo, L.; Damasco, C.; Deregibus, M.C.; Tetta, C.; Bussolati, B.; Camussi, G. Microvesicles Released from Human Renal Cancer Stem Cells Stimulate Angiogenesis and Formation of Lung Premetastatic Niche. Cancer Res. 2011, 71, 5346–5356. [Google Scholar] [CrossRef] [Green Version]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.-U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef] [PubMed]
- Aguilo, F.; Zhou, M.M.; Walsh, M.J. Long noncoding RNA, polycomb, and the ghosts haunting INK4b-ARF-INK4a expression. Cancer Res. 2011, 71, 5365–5369. [Google Scholar] [CrossRef] [Green Version]
- Jovicic, A.; Zaldivar Jolissaint, J.F.; Moser, R.; Silva Santos, M.D.; Luthi-Carter, R. MicroRNA-22 (miR-22) Overexpression Is Neuroprotective via General Anti-Apoptotic Effects and May also Target Specific Huntington’s Disease-Related Mechanisms. PLoS ONE 2013, 8, e54222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, C.; Fang, Q.Q.; Chen, W.Z.; Jiang, J.X.; Jiang, Z.; Ye, J.; Zhang, T.; Yang, L.; Meng, F.B.; Xia, W.J.; et al. Breast cancer-derived exosomes transmit lncRNA SNHG16 to induce CD73+gammadelta1 Treg cells. Signal Transduct. Target. Ther. 2020, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.P.; et al. Extracellular vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nature 2019, 21, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.-J.; Lin, X.-J.; Tang, X.-Y.; Zheng, T.-T.; Lin, Y.-Y.; Hua, K.-Q. Exosomal Metastasis-Associated Lung Adenocarcinoma Transcript 1 Promotes Angiogenesis and Predicts Poor Prognosis in Epithelial Ovarian Cancer. Int. J. Biol. Sci. 2018, 14, 1960–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmati, M.; Gyukity-Sebestyen, E.; Dobra, G.; Janovak, L.; Dekany, I.; Saydam, O.; Hunyadi-Gulyas, E.; Nagy, I.; Farkas, A.; Pankotai, T.; et al. Small extracellular vesicles convey the stress-induced adaptive responses of melanoma cells. Sci. Rep. 2019, 9, 15329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci. 2011, 46, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; Patel, T.; Freedman, J.E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 958–966. [Google Scholar] [CrossRef]
- Yarana, C.; Carroll, D.; Chen, J.; Chaiswing, L.; Zhao, Y.; Noel, T.; Alstott, M.; Bae, Y.; Dressler, E.V.; Moscow, J.A.; et al. Extracellular Vesicles Released by Cardiomyocytes in a Doxorubicin-Induced Cardiac Injury Mouse Model Contain Protein Biomarkers of Early Cardiac Injury. Clin. Cancer Res. 2018, 24, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Sultan, S.A.; T, R.; Chen, X. Biotechnological applications of S-adenosyl-methionine-dependent methyltransferases for natural products biosynthesis and diversification. Bioresour. Bioprocess. 2021, 8, 72. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.G. SAM/SAH Analogs as Versatile Tools for SAM-Dependent Methyltransferases. ACS Chem. Biol. 2015, 11, 583–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Gong, Y.; Meng, H.; Li, C.; Xue, L. Symphony of epigenetic and metabolic regulation—Interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin. Epigenetics 2020, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Xu, Z.; Xie, J.; Zhang, J.; Wang, X.; Peng, C.; Li, H.; Chen, H.; Shen, B.; Deng, X. Epigenetic silencing of LncRNA LINC00261 promotes c-myc-mediated aerobic glycolysis by regulating miR-222-3p/HIPK2/ERK axis and sequestering IGF2BP1. Oncogene 2021, 40, 277–291. [Google Scholar] [CrossRef]
- Halliwell, B. Commentary Oxidative Stress, Nutrition and Health. Experimental Strategies for Optimization of Nutritional Antioxidant Intake in Humans. Free Radic. Res. 1996, 25, 57–74. [Google Scholar] [CrossRef]
- Thompson, L.P.; Al-Hasan, Y. Impact of Oxidative Stress in Fetal Programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Dandona, P.; Ghanim, H.; Chaudhuri, A.; Dhindsa, S.; Kim, S.S. Macronutrient intake induces oxidative and inflammatory stress: Potential relevance to atherosclerosis and insulin resistance. Exp. Mol. Med. 2010, 42, 245–253. [Google Scholar] [CrossRef]
- Mishra, P.; Beura, S.; Ghosh, R.; Modak, R. Nutritional Epigenetics: How Metabolism Epigenetically Controls Cellular Physiology, Gene Expression and Disease. Subcell. Biochem. 2022, 100, 239–267. [Google Scholar] [CrossRef]
- Bhotla, H.K.; Meyyazhagan, A.; Pappusamy, M.; Park, S.; Arumugam, V.A.; Pushparaj, K.; Rengasamy, K.R.; Liu, W.; Balasubramanian, B. Dietary nutrients and their control of the redox bioenergetic networks as therapeutics in redox dysfunctions sustained pathologies. Pharmacol. Res. 2021, 170, 105709. [Google Scholar] [CrossRef]
- Lee, H.-S. Impact of Maternal Diet on the Epigenome during In Utero Life and the Developmental Programming of Diseases in Childhood and Adulthood. Nutrients 2015, 7, 9492–9507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, J.A.; Taylor, P.D.; Poston, L.; Bae, S.; Gilbert, R.D.; Ducsay, C.A.; Zhang, L. Experimental models of developmental programming: Consequences of exposure to an energy rich diet during development. J. Physiol. 2005, 565 Pt 1, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Simmons, R.A. Maternal Antioxidant Supplementation Prevents Adiposity in the Offspring of Western Diet–Fed Rats. Diabetes 2010, 59, 3058–3065. [Google Scholar] [CrossRef] [Green Version]
- Ceriello, A.; Esposito, K.; La Sala, L.; Pujadas, G.; De Nigris, V.; Testa, R.; Bucciarelli, L.; Rondinelli, M.; Genovese, S. The protective effect of the Mediterranean diet on endothelial resistance to GLP-1 in type 2 diabetes: A preliminary report. Cardiovasc. Diabetol. 2014, 13, 140. [Google Scholar] [CrossRef]
- Gómez-Virgilio, L.; Silva-Lucero, M.-D.; Flores-Morelos, D.-S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.-M.; Zacapala-Gómez, A.-E.; Luna-Muñoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. [Google Scholar] [CrossRef]
- Ryter, S.W.; Bhatia, D.; Choi, M.E. Autophagy: A Lysosome-Dependent Process with Implications in Cellular Redox Homeostasis and Human Disease. Antioxid. Redox Signal. 2019, 30, 138–159. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Mallamaci, R.; Budriesi, R.; Clodoveo, M.L.; Biotti, G.; Micucci, M.; Ragusa, A.; Curci, F.; Muraglia, M.; Corbo, F.; Franchini, C. Olive Tree in Circular Economy as a Source of Secondary Metabolites Active for Human and Animal Health Beyond Oxidative Stress and Inflammation. Molecules 2021, 26, 1072. [Google Scholar] [CrossRef]
- Fernandes, J.; Fialho, M.; Santos, R.; Peixoto-Plácido, C.; Madeira, T.; Sousa-Santos, N.; Virgolino, A.; Santos, O.; Carneiro, A.V. Is olive oil good for you? A systematic review and meta-analysis on anti-inflammatory benefits from regular dietary intake. Nutrition 2020, 69, 110559. [Google Scholar] [CrossRef]
- Recinella, L.; Chiavaroli, A.; Orlando, G.; Menghini, L.; Ferrante, C.; Di Cesare Mannelli, L.; Ghelardini, C.; Brunetti, L.; Leone, S. Protective Effects Induced by Two Polyphenolic Liquid Complexes from Olive (Olea europaea, mainly Cultivar Coratina) pressing juice in rat isolated tissues challenged with LPS. Molecules 2019, 24, 3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantano, D.; Luccarini, I.; Nardiello, P.; Servili, M.; Stefani, M.; Casamenti, F. Oleuropein aglycone and polyphenols from olive mill waste water ameliorate cognitive deficits and neuropathology. Br. J. Clin. Pharmacol. 2017, 83, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Interactions between reactive oxygen species and autophagy: Special issue: Death mechanisms in cellular homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119041. [Google Scholar] [CrossRef] [PubMed]
- Janda, E.; Lascala, A.; Carresi, C.; Parafati, M.; Aprigliano, S.; Russo, V.; Savoia, C.; Ziviani, E.; Musolino, V.; Morani, F.; et al. Parkinsonian toxin-induced oxidative stress inhibits basal autophagy in astrocytes via NQO2/quinone oxidoreductase 2: Implications for neuroprotection. Autophagy 2015, 11, 1063–1080. [Google Scholar] [CrossRef] [Green Version]
- Vidoni, C.; Castiglioni, A.; Seca, C.; Secomandi, E.; Melone, M.A.; Isidoro, C. Dopamine exacerbates mutant Huntingtin toxicity via oxidative-mediated inhibition of autophagy in SH-SY5Y neuroblastoma cells: Beneficial effects of anti-oxidant therapeutics. Neurochem. Int. 2016, 101, 132–143. [Google Scholar] [CrossRef]
- Shi, Y.; Shen, H.-M.; Gopalakrishnan, V.; Gordon, N. Epigenetic Regulation of Autophagy Beyond the Cytoplasm: A Review. Front. Cell Dev. Biol. 2021, 9, 675599. [Google Scholar] [CrossRef]
- Serzan, M.T.; Atkins, M.B. Current and emerging therapies for first line treatment of metastatic clear cell renal cell carcinoma. J. Cancer Metastasis Treat. 2021, 7, 39. [Google Scholar] [CrossRef]
- Cătană, C.-S.; Atanasov, A.G.; Berindan-Neagoe, I. Natural products with anti-aging potential: Affected targets and molecular mechanisms. Biotechnol. Adv. 2018, 36, 1649–1656. [Google Scholar] [CrossRef]
- Esposito, A.; Ferraresi, A.; Salwa, A.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. Resveratrol Contrasts IL-6 Pro-Growth Effects and Promotes Autophagy-Mediated Cancer Cell Dormancy in 3D Ovarian Cancer: Role of miR-1305 and of Its Target ARH-I. Cancers 2022, 14, 2142. [Google Scholar] [CrossRef]
- Ferraresi, A.; Esposito, A.; Girone, C.; Vallino, L.; Salwa, A.; Ghezzi, I.; Thongchot, S.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. Resveratrol Contrasts LPA-Induced Ovarian Cancer Cell Migration and Platinum Resistance by Rescuing Hedgehog-Mediated Autophagy. Cells 2021, 10, 3213. [Google Scholar] [CrossRef]
- Fan, G.; Li, F.; Wang, P.; Jin, X.; Liu, R. Natural-Product-Mediated Autophagy in the Treatment of Various Liver Diseases. Int. J. Mol. Sci. 2022, 23, 15109. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Ferraresi, A.; Secomandi, E.; Vallino, L.; Dhanasekaran, D.N.; Isidoro, C. Epigenetic targeting of autophagy for cancer prevention and treatment by natural compounds. Semin. Cancer Biol. 2020, 66, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lopez, O.; Milagro, F.I.; Riezu-Boj, J.I.; Martinez, J.A. Epigenetic signatures underlying inflammation: An interplay of nutrition, physical activity, metabolic diseases, and environmental factors for personalized nutrition. Inflamm. Res. 2020, 70, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.; Mishra, S.R.; Behera, B.P.; Mahapatra, K.K.; Panigrahi, D.P.; Bhol, C.S.; Praharaj, P.P.; Sethi, G.; Patra, S.K.; Bhutia, S.K. Autophagy-modulating phytochemicals in cancer therapeutics: Current evidences and future perspectives. Semin. Cancer Biol. 2022, 80, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Fareed, M.M.; Shityakov, S.; Ullah, S.; Qasmi, M. The Role of Vitamins in DNA Methylation as Dietary Supplements or Neutraceuticals: A systematic review. Curr. Mol. Med. 2022. [Google Scholar] [CrossRef]
- Pehlivan, F.E. Vitamin C: An Epigenetic Regulator. In Vitamin C—An Update on Current Uses and Functions; IntechOpen Limited: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, M.H. Attention in infancy and the prediction of cognitive capacities in childhood. Semin. Perinatol. 1989, 13, 450–457. [Google Scholar]
- Guz, J.; Oliński, R. The role of vitamin C in epigenetic regulation. Postep. Hig. Med. Dosw. 2017, 71, 747–760. [Google Scholar] [CrossRef]
- Camarena, V.; Wang, G. The epigenetic role of vitamin C in health and disease. Cell. Mol. Life Sci. 2016, 73, 1645–1658. [Google Scholar] [CrossRef] [Green Version]
- Brabson, J.P.; Leesang, T.; Mohammad, S.; Cimmino, L. Epigenetic Regulation of Genomic Stability by Vitamin C. Front. Genet. 2021, 12, 675780. [Google Scholar] [CrossRef] [PubMed]
- Hore, T.A.; von Meyenn, F.; Ravichandran, M.; Bachman, M.; Ficz, G.; Oxley, D.; Santos, F.; Balasubramanian, S.; Jurkowski, T.P.; Reik, W. Retinol and ascorbate drive erasure of epigenetic memory and enhance reprogramming to naïve pluripotency by complementary mechanisms. Proc. Natl. Acad. Sci. USA 2016, 113, 12202–12207. [Google Scholar] [CrossRef] [Green Version]
- Urvalek, A.; Laursen, K.B.; Gudas, L.J. The Roles of Retinoic Acid and Retinoic Acid Receptors in Inducing Epigenetic Changes. Subcell. Biochem. 2014, 70, 129–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angrisano, T.; Sacchetti, S.; Natale, F.; Cerrato, A.; Pero, R.; Keller, S.; Peluso, S.; Perillo, B.; Avvedimento, V.E.; Fusco, A.; et al. Chromatin and DNA methylation dynamics during retinoic acid-induced RET gene transcriptional activation in neuroblastoma cells. Nucleic Acids Res. 2011, 39, 1993–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uekawa, A.; Katsushima, K.; Ogata, A.; Kawata, T.; Maeda, N.; Kobayashi, K.-I.; Maekawa, A.; Tadokoro, T.; Yamamoto, Y. Change of epigenetic control of cystathionine beta-synthase gene expression through dietary vitamin B12 is not recovered by methionine supplementation. J. Nutrigenet. Nutr. 2009, 2, 29–36. [Google Scholar]
- Mahajan, A.; Sapehia, D.; Thakur, S.; Mohanraj, P.S.; Bagga, R.; Kaur, J. Effect of imbalance in folate and vitamin B12 in maternal/parental diet on global methylation and regulatory miRNAs. Sci. Rep. 2019, 9, 17602. [Google Scholar] [CrossRef] [Green Version]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and Human Health: Lessons from Vitamin D Receptor Null Mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef] [Green Version]
- Karlic, H.; Varga, F. Impact of vitamin D metabolism on clinical epigenetics. Clin. Epigenetics 2011, 2, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, X.; Shi, H.; Su, S.; Harshfield, G.A.; Gutin, B.; Snieder, H.; Dong, Y. A Genome-Wide Methylation Study of Severe Vitamin D Deficiency in African American Adolescents. J. Pediatr. 2013, 162, 1004–1009.e1. [Google Scholar] [CrossRef] [Green Version]
- Bikle, D.D. Vitamin D regulation of and by long non coding RNAs. Mol. Cell. Endocrinol. 2021, 532, 111317. [Google Scholar] [CrossRef]
- Luna, R.C.; dos Santos Nunes, M.K.; Monteiro, M.G.; da Silva, C.S.; do Nascimento, R.A.; Lima, R.P.; Pimenta, F.C.; de Oliveira, N.F.; Persuhn, D.C.; de Almeida, A.T.; et al. α-Tocopherol influences glycaemic control and miR-9-3 DNA methylation in overweight and obese women under an energy-restricted diet: A randomized, double-blind, exploratory, controlled clinical trial. Nutr. Metab. 2018, 15, 49. [Google Scholar] [CrossRef] [Green Version]
- Shankar, S.; Kumar, D.; Srivastava, R.K. Epigenetic modifications by dietary phytochemicals: Implications for personalized nutrition. Pharmacol. Ther. 2013, 138, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.-D.; Guo, Z.S. Epigenetic drugs for cancer treatment and prevention: Mechanisms of action. Biomol. Concepts 2010, 1, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Cione, E.; La Torre, C.; Cannataro, R.; Caroleo, M.C.; Plastina, P.; Gallelli, L. Quercetin, Epigallocatechin Gallate, Curcumin, and Resveratrol: From Dietary Sources to Human MicroRNA Modulation. Molecules 2019, 25, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallino, L.; Ferraresi, A.; Vidoni, C.; Secomandi, E.; Esposito, A.; Dhanasekaran, D.N.; Isidoro, C. Modulation of non-coding RNAs by resveratrol in ovarian cancer cells: In silico analysis and literature review of the anti-cancer pathways involved. J. Tradit. Complement. Med. 2020, 10, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Hatami, M.; Abdolahi, M.; Soveyd, N.; Djalali, M.; Togha, M.; Honarvar, N.M. Molecular Mechanisms of Curcumin in Neuroinflammatory Disorders: A Mini Review of Current Evidences. Endocr. Metab. Immune Disord.-Drug Targets 2019, 19, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Kalea, A.Z.; Drosatos, K.; Buxton, J.L. Nutriepigenetics and cardiovascular disease. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 252–259. [Google Scholar] [CrossRef]
- Pham, T.X.; Lee, J. Dietary Regulation of Histone Acetylases and Deacetylases for the Prevention of Metabolic Diseases. Nutrients 2012, 4, 1868–1886. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Abdelsalam, S.A.; Renu, K.; Veeraraghavan, V.; Ben Ammar, R.; Ahmed, E.A. Polyphenols as Potent Epigenetics Agents for Cancer. Int. J. Mol. Sci. 2022, 23, 11712. [Google Scholar] [CrossRef]
- Ferraresi, A.; Phadngam, S.; Morani, F.; Galetto, A.; Alabiso, O.; Chiorino, G.; Isidoro, C. Resveratrol inhibits IL-6-induced ovarian cancer cell migration through epigenetic up-regulation of autophagy. Mol. Carcinog. 2017, 56, 1164–1181. [Google Scholar] [CrossRef]
- Carlos-Reyes, Á.; Lopez-Gonzalez, J.S.; Meneses-Flores, M.; Gallardo-Rincón, D.; Ruíz-García, E.; Marchat, L.; La Vega, H.A.-D.; De La Cruz, O.N.H.; López-Camarillo, C. Dietary Compounds as Epigenetic Modulating Agents in Cancer. Front. Genet. 2019, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Zam, W.; Khadour, A. Impact of Phytochemicals and Dietary Patterns on Epigenome and Cancer. Nutr. Cancer 2017, 69, 184–200. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubio, K.; Hernández-Cruz, E.Y.; Rogel-Ayala, D.G.; Sarvari, P.; Isidoro, C.; Barreto, G.; Pedraza-Chaverri, J. Nutriepigenomics in Environmental-Associated Oxidative Stress. Antioxidants 2023, 12, 771. https://doi.org/10.3390/antiox12030771
Rubio K, Hernández-Cruz EY, Rogel-Ayala DG, Sarvari P, Isidoro C, Barreto G, Pedraza-Chaverri J. Nutriepigenomics in Environmental-Associated Oxidative Stress. Antioxidants. 2023; 12(3):771. https://doi.org/10.3390/antiox12030771
Chicago/Turabian StyleRubio, Karla, Estefani Y. Hernández-Cruz, Diana G. Rogel-Ayala, Pouya Sarvari, Ciro Isidoro, Guillermo Barreto, and José Pedraza-Chaverri. 2023. "Nutriepigenomics in Environmental-Associated Oxidative Stress" Antioxidants 12, no. 3: 771. https://doi.org/10.3390/antiox12030771