
Involvement of Intracellular TAGE and the TAGE–RAGE–ROS Axis in the Onset and Progression of NAFLD/NASH

{kind=link}
{kind=link}
Abstract
:1. Introduction
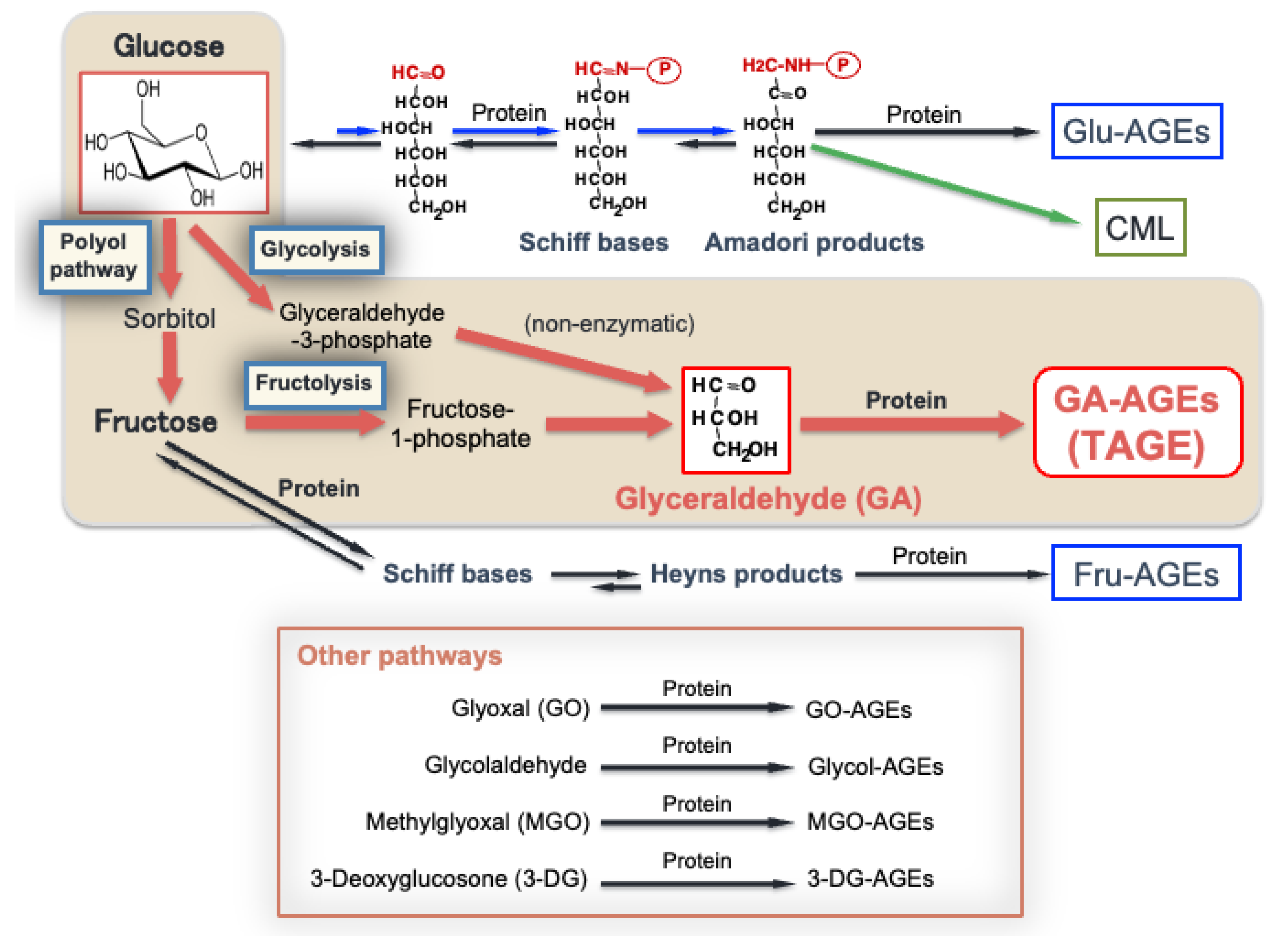
2. Production of AGEs in the Human Body
3. The Intracellular Generation and Accumulation of TAGE in the Liver
4. The TAGE–RAGE Axis
5. Serum TAGE Levels and NASH/Non-B, Non-C (NBNC)-HCC
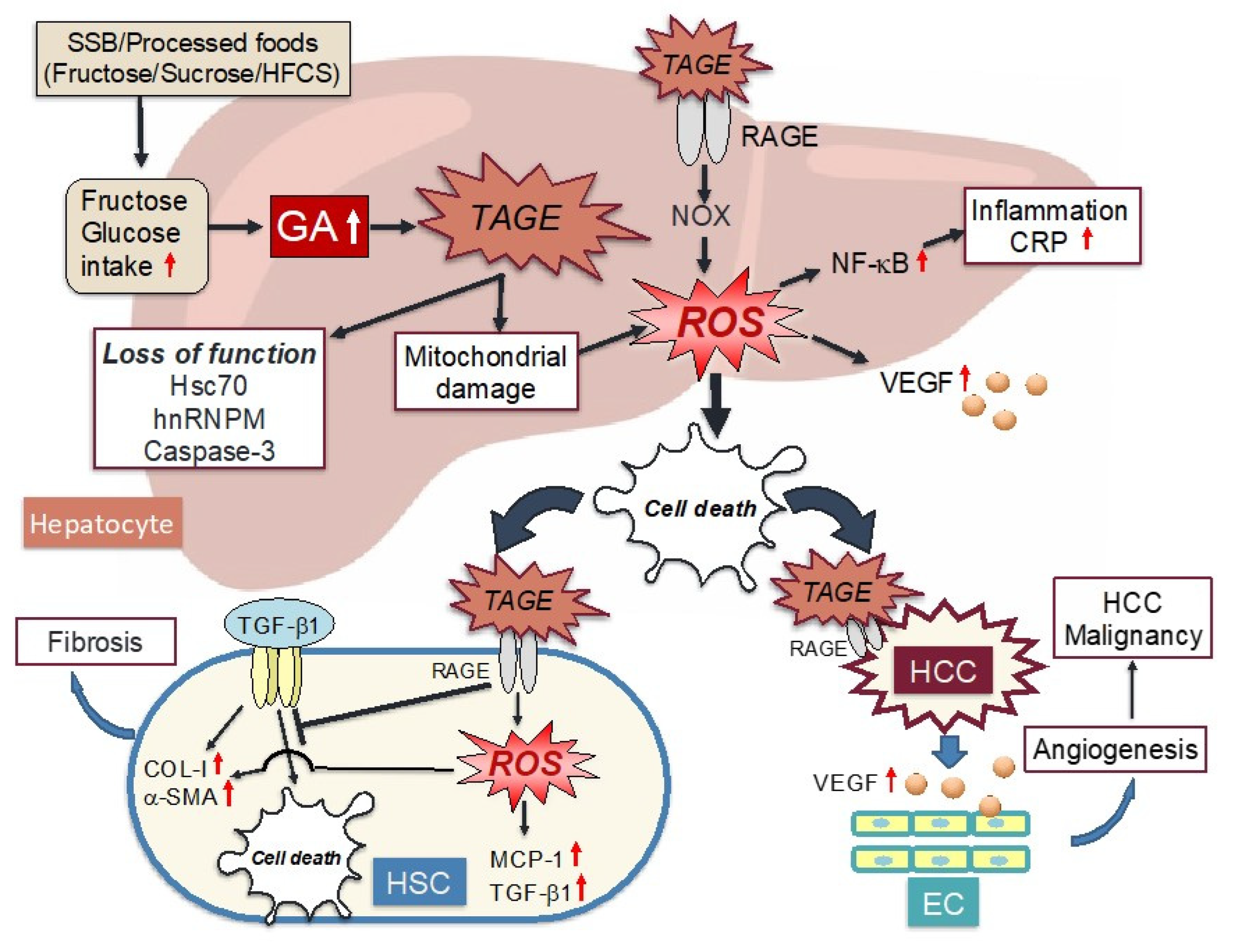
6. Cytotoxicity of TAGE in the Liver
6.1. Intracellular TAGE and Hepatocyte Cell Death
6.2. TAGE-Modified Proteins and Hepatocyte Cell Damage
6.3. Oxidative Stress and Its Response Associated with Intracellular TAGE in Hepatocytes
6.4. Effects of Extracellular TAGE on Hepatocytes
6.5. Effects of Extracellular TAGE on HSC
6.6. Extracellular TAGE and the Malignant Progression of NASH-Related HCC
6.7. Limitations
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end-products |
| CLD | Chronic liver disease |
| CML | Nε-(Carboxymethyl)lysine |
| COL-I | Collagen type I |
| CRP | C-Reactive protein |
| CVD | Cardiovascular disease |
| 3-DG-AGEs | 3-Deoxyglucosone-derived AGEs |
| DHAP | Dihydroxyacetone phosphate |
| DN-HD | Diabetic nephropathy with hemodialysis |
| EC | Endothelial cells |
| ECM | Extracellular matrix |
| Fru | Fructose |
| Fru-AGEs | Fructose-derived AGEs |
| GA | Glyceraldehyde |
| GA-AGEs | Glyceraldehyde-derived AGEs |
| GA-3-P | Glyceraldehyde-3-phosphate |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GLAP | 3-Hydroxy-5-hydroxymethyl-pyridinium compound |
| Glu | Glucose |
| Glu-AGEs | Glucose-derived AGEs |
| Glycol-AGEs | Glycolaldehyde-derived AGEs |
| GO | Glyoxal |
| GO-AGEs | Glyoxal-derived AGEs |
| γ-GTP | γ-Glutamyl transpeptidase |
| HCC | Hepatocellular carcinoma |
| HFCS | High-fructose corn syrup |
| hiPSC | Human induced pluripotent stem cells |
| hiPSC-HLC | HLC differentiated from hiPSC |
| HLC | Hepatocyte-like cells |
| hnRNPM | Heterogeneous nuclear ribonucleoprotein M |
| HO-1 | Hemeoxygenase-1 |
| HOMA-IR | Homeostasis model assessment of IR |
| Hsc70 | Heat shock cognate 70 |
| HSC | Hepatic stellate cells |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IL | Interleukin |
| IR | Insulin resistance |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MG-H1 | Methylglyoxal-derived hydroimidazolone compound |
| MGO | Methylglyoxal |
| MGO-AGEs | Methylglyoxal-derived AGEs |
| MS | Metabolic syndrome |
| NAFL | Nonalcoholic fatty liver |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| NBNC-HCC | Non-B, non-C HCC |
| NF-κB | Nuclear factor kappa B |
| NOX | Nicotinamide adenine dinucleotide phosphate-reduced oxidase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| RAGE | Receptor for AGEs |
| sRAGE | Soluble form of RAGE |
| RasGRP2 | Ras guanyl nucleotide releasing protein 2 |
| ROS | Reactive oxygen species |
| α-SMA | α-smooth muscle actin |
| SSB | Sugar-sweetened beverages |
| TAGE | Toxic AGEs |
| T2DM | Type 2 diabetes mellitus |
| TGF-β1 | Transforming growth factor-β1 |
| TNF-α | Tumor necrosis factor-α |
| VE | Vascular endothelial |
| VEGF | Vascular endothelial growth factor |
References
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Henry, L.; Paik, J.; Younossi, Z.M. Review article: The epidemiologic burden of non-alcoholic fatty liver disease across the world. Aliment. Pharmacol. Ther. 2022, 56, 942–956. [Google Scholar] [CrossRef]
- Deleve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Makita, Z. Alternative routes for the formation of immunochemically distinct advanced glycation end-products in vivo. Curr. Mol. Med. 2001, 1, 305–315. [Google Scholar] [CrossRef]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bügel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef]
- Takino, J.; Kobayashi, Y.; Takeuchi, M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J. Gastroenterol. 2010, 45, 646–655. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci. Rep. 2017, 7, 14282. [Google Scholar] [CrossRef] [Green Version]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products promote the production of reactive oxygen species in HepG2 cells. Int. J. Mol. Sci. 2020, 21, 4861. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, C.; Sakasai-Sakai, A.; Okimura, R.; Tanaka, H.; Takata, T.; Takeuchi, M.; Matsunaga, T. Accumulation of toxic advanced glycation end-products induces cytotoxicity and inflammation in hepatocyte-like cells differentiated from human induced pluripotent stem cells. Biol. Pharm. Bull. 2021, 44, 1399–1402. [Google Scholar] [CrossRef]
- Koriyama, Y.; Furukawa, A.; Muramatsu, M.; Takino, J.; Takeuchi, M. Glyceraldehyde caused Alzheimer’s disease-like alterations in diagnostic marker levels in SH-SY5Y human neuroblastoma cells. Sci. Rep. 2015, 5, 13313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, T.; Ueda, T.; Sakasai-Sakai, A.; Takeuchi, M. Generation of glyceraldehyde-derived advanced glycation end-products in pancreatic cancer cells and the potential of tumor promotion. World J. Gastroenterol. 2017, 23, 4910–4919. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Sakasai-Sakai, A.; Ueda, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products in cardiomyocytes may cause cardiovascular disease. Sci. Rep. 2019, 9, 2121. [Google Scholar] [CrossRef] [Green Version]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Impact of intracellular toxic advanced glycation end-products (TAGE) on murine myoblast cell death. Diabetol. Metab. Syndr. 2020, 12, 54. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular toxic advanced glycation end-products in 1.4E7 cell line induce death with reduction of microtubule-associated protein 1 light chain 3 and p62. Nutrients 2022, 14, 332. [Google Scholar] [CrossRef]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. The association between accumulation of toxic advanced glycation end-products and cytotoxic effect in MC3T3-E1 cells. Nutrients 2022, 14, 990. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular toxic advanced glycation end-products may induce cell death and suppress cardiac fibroblasts. Metabolites 2022, 12, 615. [Google Scholar] [CrossRef]
- Takeuchi, M. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker for the onset/progression of lifestyle-related diseases. Diagnostics 2016, 6, 23. [Google Scholar] [CrossRef]
- Takeuchi, M. Toxic AGEs (TAGE) theory: A new concept for preventing the development of diseases related to lifestyle. Diabetol. Metab. Syndr. 2020, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.; Koriyama, Y. Effects of toxic AGEs (TAGE) on human health. Cells 2022, 11, 2178. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamagishi, S. Involvement of toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 16, 845–858. [Google Scholar] [CrossRef]
- Takeuchi, M.; Takino, J.; Yamagishi, S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: A novel therapeutic strategy. Curr. Drug Targets 2010, 11, 1468–1482. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Takino, J.; Sakasai-Sakai, A.; Takata, T.; Ueda, T.; Tsutsumi, M.; Hyogo, H.; Yamagishi, S. Involvement of the TAGE-RAGE system in non-alcoholic steatohepatitis: Novel treatment strategies. World J. Hepatol. 2014, 6, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Sakasai-Sakai, A.; Takata, T.; Tsutsumi, M. Toxic AGE (TAGE) theory for the pathophysiology of the onset/progression of NAFLD and ALD. Nutrients 2017, 9, 634. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular toxic AGEs (TAGE) triggers numerous types of cell damage. Biomolecules 2021, 11, 387. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Shirai, H.; Kawakami, M.; Furuno, S.; Kobayashi, Y. Assessment of total sugar and glucose concentrations in commonly consumed beverages in Japan. Nutr. Food Technol. 2015, 1, 2. [Google Scholar] [CrossRef]
- Takeuchi, M.; Takino, J.; Furuno, S.; Shirai, H.; Kawakami, M.; Muramatsu, M.; Kobayashi, Y.; Yamagishi, S. Assessment of the concentrations of various advanced glycation end-products in beverages and foods that are commonly consumed in Japan. PLoS ONE 2015, 10, e0118652. [Google Scholar] [CrossRef] [Green Version]
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: Systematic review, meta-analysis, and estimation of population attributable fraction. Br. J. Sports Med. 2016, 50, 496–504. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Hoffmann, G.; Lampousi, A.M.; Knüppel, S.; Iqbal, K.; Schwedhelm, C.; Bechthold, A.; Schlesinger, S.; Boeing, H. Food groups and risk of type 2 diabetes mellitus: A systematic review and meta-analysis of prospective studies. Eur. J. Epidemiol. 2017, 32, 363–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, V.S. Sugar sweetened beverages and cardiometabolic health. Curr. Opin. Cardiol. 2017, 32, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H.; Luukkonen, P.K.; Hodson, L.; Moore, J.B. Dietary carbohydrates and fats in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 770–786. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Baker, S.S.; Liu, W.; Desai, S.; Alkhouri, R.; Kozielski, R.; Mastrandrea, L.; Sarfraz, A.; Cai, W.; Vlassara, H.; et al. Effect of dietary advanced glycation end products on mouse liver. PLoS ONE 2012, 7, e35143. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Takeuchi, M.; Makita, Z.; Yanagisawa, K.; Kameda, Y.; Koike, T. Detection of noncarboxymethyllysine and carboxymethyllysine advanced glycation end products (AGE) in serum of diabetic patients. Mol. Med. 1999, 5, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Makita, Z.; Bucala, R.; Suzuki, T.; Koike, T.; Kameda, Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol. Med. 2000, 6, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Yanase, Y.; Matsuura, N.; Yamagishi, S.; Kameda, Y.; Bucala, R.; Makita, Z. Immunological detection of a novel advanced glycation end-product. Mol. Med. 2001, 7, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Iwaki, M.; Takino, J.; Shirai, H.; Kawakami, M.; Bucala, R.; Yamagishi, S. Immunological detection of fructose-derived advanced glycation end-products. Lab. Invest 2010, 90, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Bucala, R.; Suzuki, T.; Ohkubo, T.; Yamazaki, M.; Koike, T.; Kameda, Y.; Makita, Z. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J. Neuropathol. Exp. Neurol. 2000, 59, 1094–1105. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Yamagishi, S. TAGE (toxic AGEs) hypothesis in various chronic diseases. Med. Hypotheses 2004, 63, 449–452. [Google Scholar] [CrossRef]
- Tahara, N.; Yamagishi, S.; Takeuchi, M.; Honda, A.; Tahara, A.; Nitta, Y.; Kodama, N.; Mizoguchi, M.; Kaida, H.; Ishibashi, M.; et al. Positive association between serum level of glyceraldehyde-derived advanced glycation end products and vascular inflammation evaluated by [18F] fluorodeoxyglucose positron emission tomography. Diabetes Care 2012, 35, 2618–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, T.; Hyase, F. Isolation and identification of the 3-hydroxy-5-hydroxymethyl-pyridinium compound as a novel advanced glycation end product on glyceraldehyde-related Maillard reaction. Biosci. Biotechnol. Biochem. 2003, 67, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Tessier, F.J.; Monnier, V.M.; Sayre, L.M.; Kornfield, J.A. Triosidines: Novel Maillard reaction products and cross-links from the reaction of triose sugars with lysine and arginine residues. Biochem. J. 2003, 369, 705–719. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Watanabe, H.; Hayase, F. Isolation and identification of 5-methyl-imidazolin-4-one derivative as glyceraldehyde-derived advanced glycation end product. Biosci. Biotechnol. Biochem. 2006, 70, 1496–1498. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Kikuchi, S.; Sasaki, N.; Suzuki, T.; Watai, T.; Iwaki, M.; Bucala, R.; Yamagishi, S. Involvement of advanced glycation end-products (AGEs) in Alzheimer’s disease. Curr. Alzheimer Res. 2004, 1, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Litwinowicz, K.; Waszczuk, E.; Gamian, A. Advanced glycation end-products in common non-infectious liver diseases: Systematic review and meta-analysis. Nutrients 2021, 13, 3370. [Google Scholar] [CrossRef] [PubMed]
- Twarda-Clapa, A.; Olczak, A.; Białkowska, A.M.; Koziołkiewicz, M. Advanced glycation end-products (AGEs): Formation, chemistry, classification, receptors, and diseases related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. Alternative routes for the formation of glyceraldehyde-derived AGEs (TAGE) in vivo. Med. Hypotheses 2004, 63, 453–455. [Google Scholar] [CrossRef]
- Oates, P.J. Polyol pathway and diabetic peripheral neuropathy. Int. Rev. Neurobiol. 2002, 50, 325–392. [Google Scholar] [CrossRef]
- Bais, R.; James, H.M.; Rofe, A.M.; Conyers, R.A. The purification and properties of human liver ketohexokinase. A role for ketohexokinase and fructose-bisphosphate aldolase in the metabolic production of oxalate from xylitol. Biochem. J. 1985, 230, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Schalkwijk, C.G.; Wouters, K. Immunometabolism and the modulation of immune responses and host defense: A role for methylglyoxal? Biochim. Biophys. Acta–Mol. Basis Dis. 2022, 1868, 166425. [Google Scholar] [CrossRef]
- Malinská, H.; Hüttl, M.; Miklánková, D.; Trnovská, J.; Zapletalová, I.; Poruba, M.; Marková, I. Ovariectomy-induced hepatic lipid and cytochrome P450 dysmetabolism precedes serum dyslipidemia. Int. J. Mol. Sci. 2021, 22, 4527. [Google Scholar] [CrossRef]
- Lai, S.W.T.; Lopez Gonzalez, E.J.; Zoukari, T.; Ki, P.; Shuck, S.C. Methylglyoxal and its adducts: Induction, repair, and association with disease. Chem. Res. Toxicol. 2022, 35, 1720–1746. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Sakasai-Sakai, A.; Takino, J.; Takeuchi, M. Evidence for toxic advanced glycation end-products generated in the normal rat liver. Nutrients 2019, 11, 1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Takata, T.; Nakazawa, Y.; Nakamura, Y.; Guo, X.; Yamada, S.; Ishigaki, Y.; Takeuchi, M.; Miyazawa, K. Potential of an interorgan network mediated by toxic advanced glycation end-products in a rat model. Nutrients 2021, 13, 80. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Vianna, M.; Gerlach, M.; Brett, J.; Ryan, J.; Kao, J.; Esposito, C.; Hegarty, H.; Hurley, W.; Clauss, M.; et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem. 1992, 267, 14987–14997. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kato, I.; Doi, T.; Yonekura, H.; Ohashi, S.; Takeuchi, M.; Watanabe, T.; Yamagishi, S.; Sakurai, S.; Takasawa, S.; et al. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J. Clin. Invest 2001, 108, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Leerach, N.; Harashima, A.; Munesue, S.; Kimura, K.; Oshima, Y.; Goto, H.; Yamamoto, H.; Higashida, H.; Yamamoto, Y. Glycation reaction and the role of the receptor for advanced glycation end-products in immunity and social behavior. Glycoconj. J. 2021, 38, 303–310. [Google Scholar] [CrossRef]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. The diverse ligand repertoire of the receptor for advanced glycation endproducts and pathways to the complications of diabetes. Vascul. Pharmacol. 2012, 57, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorci, G.; Riuzzi, F.; Giambanco, I.; Donato, R. RAGE in tissue homeostasis, repair and regeneration. Biochim. Biophys. Acta 2013, 1833, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, R.; Yamagishi, S. AGE-RAGE system and carcinogenesis. Curr. Pharm. Des. 2008, 14, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Yamagishi, S. Advanced glycation end products (AGEs) and their involvement in liver disease. Curr. Pharm. Des. 2008, 14, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Navarra, T.; De Simone, P.; Del Turco, S.; Gastaldelli, A.; Filipponi, F. What is the role of the receptor for advanced glycation end products-ligand axis in liver injury? Liver Transplant. 2011, 17, 633–640. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.; Abedin, M.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Yonekura, H.; Watanabe, T.; Sakurai, S.; Li, H.; Harashima, A.; Myint, K.M.; Osawa, M.; Takeuchi, A.; Takeuchi, M.; et al. Short-chain aldehyde-derived ligands for RAGE and their actions on endothelial cells. Diabetes Res. Clin. Pract. 2007, 77 (Suppl. S1), S30–S40. [Google Scholar] [CrossRef]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef] [Green Version]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J. Gastroenterol. 2012, 18, 1781–1788. [Google Scholar] [CrossRef]
- Takino, J.; Nagamine, K.; Hori, T.; Sakasai-Sakai, A.; Takeuchi, M. Contribution of the toxic advanced glycation end-products-receptor axis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2459–2469. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, Q.; Wu, J.; Zhou, X.; Weng, J.; Xu, J.; Wang, W.; Huang, Q.; Guo, X. Role of Src in vascular hyperpermeability induced by advanced glycation end products. Sci. Rep. 2015, 5, 14090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takino, J.; Sato, T.; Kanetaka, T.; Okihara, K.; Nagamine, K.; Takeuchi, M.; Hori, T. RasGRP2 inhibits glyceraldehyde-derived toxic advanced glycation end-products from inducing permeability in vascular endothelial cells. Sci. Rep. 2021, 11, 2959. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes Metab. Res. Rev. 2001, 17, 436–443. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am. J. Physiol. Cell Physiol. 2008, 294, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Hyogo, H.; Yamagishi, S.; Iwamoto, K.; Arihiro, K.; Takeuchi, M.; Sato, T.; Ochi, H.; Nonaka, M.; Nabeshima, Y.; Inoue, M.; et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2007, 22, 1112–1119. [Google Scholar] [CrossRef]
- Kimura, Y.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Ishitobi, T.; Nabeshima, Y.; Arihiro, K.; Chayama, K. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: Clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J. Gastroenterol. 2010, 45, 750–757. [Google Scholar] [CrossRef]
- Kan, H.; Yamagishi, S.; Ojima, A.; Fukami, K.; Ueda, S.; Takeuchi, M.; Hyogo, H.; Aikata, H.; Chayama, K. Elevation of serum levels of advanced glycation end products in patients with non-B or non-C hepatocellular carcinoma. J. Clin. Lab. Anal. 2015, 29, 480–484. [Google Scholar] [CrossRef]
- Yamagishi, S.; Adachi, H.; Nakamura, K.; Matsui, T.; Jinnouchi, Y.; Takenaka, K.; Takeuchi, M.; Enomoto, M.; Furuki, K.; Hino, A.; et al. Positive association between serum levels of advanced glycation end products and the soluble form of receptor for advanced glycation end products in nondiabetic subjects. Metabolism. 2006, 55, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Yamagishi, S.; Tahara, A.; Ishibashi, M.; Hayabuchi, N.; Takeuchi, M.; Imaizumi, T. Adiponectin is inversely associated with ratio of serum levels of AGEs to sRAGE and vascular inflammation. Int. J. Cardiol. 2012, 158, 461–462. [Google Scholar] [CrossRef]
- Kajikawa, M.; Nakashima, A.; Fujimura, N.; Maruhashi, T.; Iwamoto, Y.; Iwamoto, A.; Matsumoto, T.; Oda, N.; Hidaka, T.; Kihara, Y.; et al. Ratio of serum levels of AGEs to soluble form of RAGE is a predictor of endothelial function. Diabetes Care 2015, 38, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Yamagishi, S.; Adachi, H.; Matsui, T.; Kurita-Nakamura, Y.; Takeuchi, M.; Inoue, H.; Imaizumi, T. Serum levels of soluble form of receptor for advanced glycation end products (sRAGE) are positively associated with circulating AGEs and soluble form of VCAM-1 in patients with type 2 diabetes. Microvasc. Res. 2008, 76, 52–56. [Google Scholar] [CrossRef]
- Nakamura, K.; Yamagishi, S.; Adachi, H.; Matsui, T.; Kurita-Nakamura, Y.; Takeuchi, M.; Inoue, H.; Imaizumi, T. Circulating advanced glycation end products (AGEs) and soluble form of receptor for AGEs (sRAGE) are independent determinants of serum monocyte chemoattractant protein-1 (MCP-1) levels in patients with type 2 diabetes. Diabetes. Metab. Res. Rev. 2008, 24, 109–114. [Google Scholar] [CrossRef]
- Takino, J.; Nagamine, K.; Takeuchi, M.; Hori, T. In vitro identification of nonalcoholic fatty liver disease-related protein hnRNPM. World J. Gastroenterol. 2015, 21, 1784–1793. [Google Scholar] [CrossRef]
- Takino, J.; Nagamine, K.; Suzuki, M.; Sakasai-Sakai, A.; Takeuchi, M.; Hori, T. Gene expression changes associated with the loss of heterogeneous nuclear ribonucleoprotein M function. Am. J. Mol. Biol. 2017, 7, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.; Takeuchi, M. The relevance of toxic AGEs (TAGE) cytotoxicity to NASH pathogenesis: A mini-review. Nutrients 2019, 11, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaie, L.; Iorga, A.; Dara, L. Cell death in liver diseases: A review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef] [PubMed]
- Stricher, F.; Macri, C.; Ruff, M.; Muller, S. HSPA8/HSC70 chaperone protein: Structure, function, and chemical targeting. Autophagy 2013, 9, 1937–1954. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [Green Version]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative stress in non-alcoholic fatty liver disease. An updated mini review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of NRF2: A review of their potential for clinical development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.J.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, L.; Madeddu, R.; Palio, E.; Arena, N.; Malaguarnera, M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J. Hepatol. 2005, 42, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Jarukamjorn, K.; Jearapong, N.; Pimson, C.; Chatuphonprasert, W. A high-fat, high-fructose diet induces antioxidant imbalance and increases the risk and progression of nonalcoholic fatty liver disease in mice. Scientifica 2016, 2016, 5029414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Liu, J.; Pang, X.; Wang, S.; Wu, D.; Zhang, X.; Feng, L. Angiotensin II induces C-reactive protein expression via AT1-ROS-MAPK-NF-κB signal pathway in hepatocytes. Cell. Physiol. Biochem. 2013, 32, 569–580. [Google Scholar] [CrossRef]
- Yoshida, T.; Yamagishi, S.; Nakamura, K.; Matsui, T.; Imaizumi, T.; Takeuchi, M.; Ueno, T.; Sata, M. Pigment epithelium- derived factor (PEDF) inhibits advanced glycation end product (AGE)-induced C-reactive protein expression in hepatoma cells by suppressing Rac-1 activation. FEBS Lett. 2006, 580, 2788–2796. [Google Scholar] [CrossRef] [Green Version]
- Videla, L.A.; Tapia, G.; Rodrigo, R.; Pettinelli, P.; Haim, D.; Santibãez, C.; Araya, A.V.; Smok, G.; Csendes, A.; Gutierrez, L.; et al. Liver NF-κB and AP-1 DNA binding in obese patients. Obesity 2009, 17, 973–979. [Google Scholar] [CrossRef]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology 2015, 149, 468–480. [Google Scholar] [CrossRef] [Green Version]
- Nasiri-Ansari, N.; Androutsakos, T.; Flessa, C.M.; Kyrou, I.; Siasos, G.; Randeva, H.; Kassi, E.; Papavassiliou, A.G. Endothelial cell dysfunction and nonalcoholic fatty liver disease (NAFLD): A concise review. Cells 2022, 11, 2511. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Fukami, K.; Okuda, S.; Imaizumi, T. Olmesartan blocks advanced glycation end products (AGEs)-induced angiogenesis in vitro by suppressing receptor for AGEs (RAGE) expression. Microvasc. Res. 2008, 75, 130–134. [Google Scholar] [CrossRef]
- Niiya, Y.; Abumiya, T.; Yamagishi, S.; Takino, J.; Takeuchi, M. Advanced glycation end products increase permeability of brain microvascular endothelial cells through reactive oxygen species-induced vascular endothelial growth factor expression. J. Stroke Cerebrovasc. Dis. 2012, 21, 293–298. [Google Scholar] [CrossRef]
- Inagaki, Y.; Yamagishi, S.; Okamoto, T.; Takeuchi, M.; Amano, S. Pigment epithelium-derived factor prevents advanced glycation end products-induced monocyte chemoattractant protein-1 production in microvascular endothelial cells by suppressing intracellular reactive oxygen species generation. Diabetologia 2003, 46, 284–287. [Google Scholar] [CrossRef] [Green Version]
- Niiya, Y.; Abumiya, T.; Shichinohe, H.; Kuroda, S.; Kikuchi, S.; Ieko, M.; Yamagishi, S.; Takeuchi, M.; Sato, T.; Iwasaki, Y. Susceptibility of brain microvascular endothelial cells to advanced glycation end products-induced tissue factor upregulation is associated with intracellular reactive oxygen species. Brain Res. 2006, 1108, 179–187. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Olmesartan blocks inflammatory reactions in endothelial cells evoked by advanced glycation end products by suppressing generation of reactive oxygen species. Ophthalmic Res. 2008, 40, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Baiocchini, A.; Montaldo, C.; Conigliaro, A.; Grimaldi, A.; Correani, V.; Mura, F.; Ciccosanti, F.; Rotiroti, N.; Brenna, A.; Montalbano, M.; et al. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PLoS ONE 2016, 11, e0151736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Iwamoto, K.; Kanno, K.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Tazuma, S.; Chayama, K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol. 2008, 43, 298–304. [Google Scholar] [CrossRef]
- Takino, J.; Sato, T.; Nagamine, K.; Sakasai-Sakai, A.; Takeuchi, M.; Hori, T. Suppression of hepatic stellate cell death by toxic advanced glycation end-products. Biol. Pharm. Bull. 2021, 44, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Yano, H.; Iemura, A.; Ogasawara, S.; Haramaki, M.; Kojiro, M. Expression of vascular endothelial growth factor in human hepatocellular carcinoma. Hepatology 1998, 28, 68–77. [Google Scholar] [CrossRef]
- Sato, T.; Wu, X.; Shimogaito, N.; Takino, J.; Yamagishi, S.; Takeuchi, M. Effects of high-AGE beverage on RAGE and VEGF expressions in the liver and kidneys. Eur. J. Nutr. 2009, 48, 6–11. [Google Scholar] [CrossRef]
- Okamoto, T.; Yamagishi, S.; Inagaki, Y.; Amano, S.; Koga, K.; Abe, R.; Takeuchi, M.; Ohno, S.; Yoshimura, A.; Makita, Z. Angiogenesis induced by advanced glycation end products and its prevention by cerivastatin. FASEB J. 2002, 16, 1928–1930. [Google Scholar] [CrossRef]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Yoshida, T.; Takeuchi, M.; Imaizumi, T. Pigment epithelium-derived factor (PEDF) blocks advanced glycation end product (AGE)-induced angiogenesis in vitro. Horm. Metab. Res. 2007, 39, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Ueda, S.; Fukami, K.; Yamagishi, S. Advanced glycation end products potentiate citrated plasma-evoked oxidative and inflammatory reactions in endothelial cells by up-regulating protease-activated receptor-1 expression. Cardiovasc. Diabetol. 2014, 13, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’Nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxid. Med. Cell. Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef] [Green Version]
- Remigante, A.; Spinelli, S.; Trichilo, V.; Loddo, S.; Sarikas, A.; Pusch, M.; Dossena, S.; Marino, A.; Morabito, R. d-Galactose induced early aging in human erythrocytes: Role of band 3 protein. J. Cell. Physiol. 2022, 237, 1586–1596. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakasai-Sakai, A.; Takeda, K.; Takeuchi, M. Involvement of Intracellular TAGE and the TAGE–RAGE–ROS Axis in the Onset and Progression of NAFLD/NASH. Antioxidants 2023, 12, 748. https://doi.org/10.3390/antiox12030748
Sakasai-Sakai A, Takeda K, Takeuchi M. Involvement of Intracellular TAGE and the TAGE–RAGE–ROS Axis in the Onset and Progression of NAFLD/NASH. Antioxidants. 2023; 12(3):748. https://doi.org/10.3390/antiox12030748
Chicago/Turabian StyleSakasai-Sakai, Akiko, Kenji Takeda, and Masayoshi Takeuchi. 2023. "Involvement of Intracellular TAGE and the TAGE–RAGE–ROS Axis in the Onset and Progression of NAFLD/NASH" Antioxidants 12, no. 3: 748. https://doi.org/10.3390/antiox12030748