

Links between Vitamin K, Ferroptosis and SARS-CoV-2 Infection

 , , ,
, , ,  ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
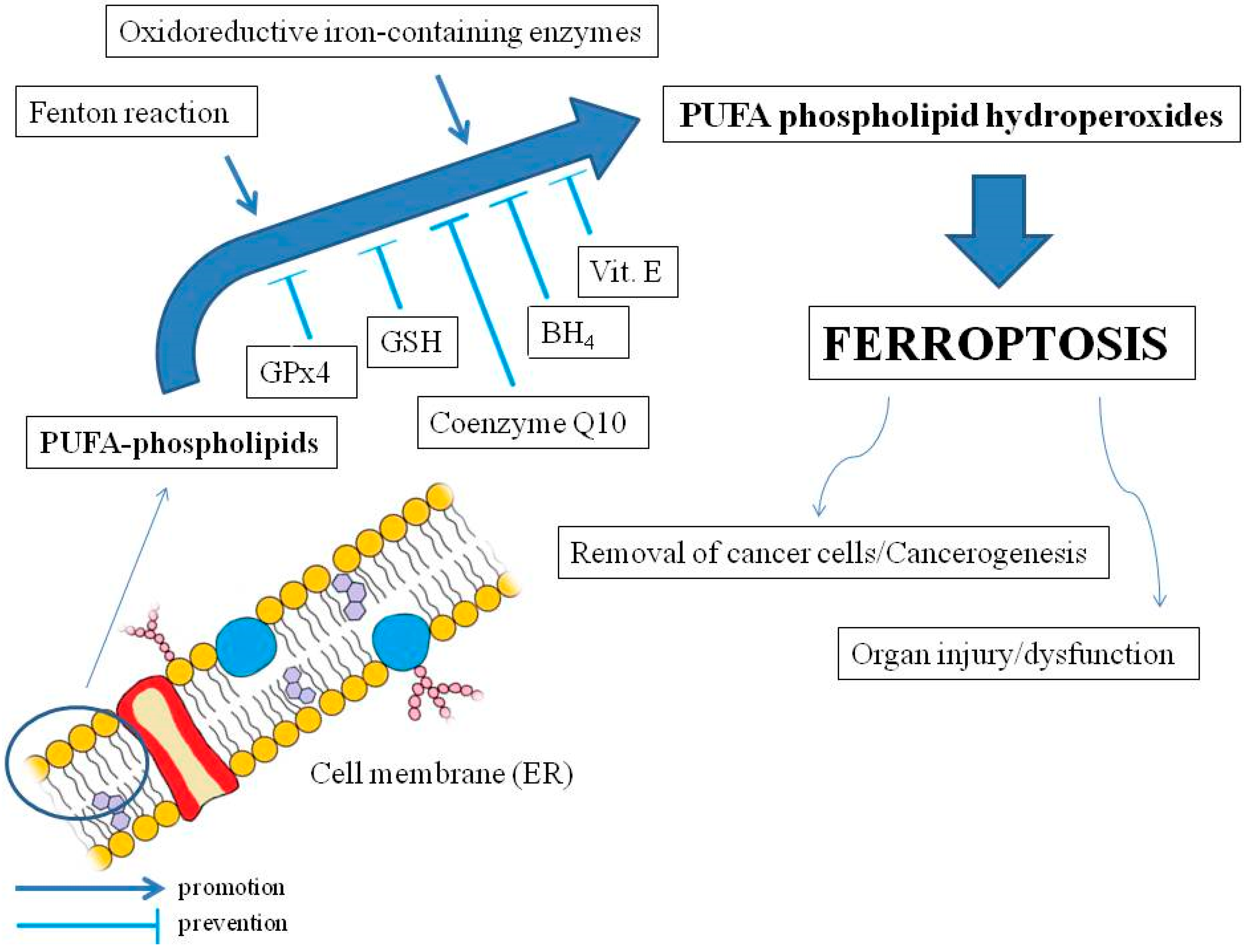
2. Oxidant–Antioxidant Balance and Lipid Peroxidation
3. Ferroptosis Mechanisms and Health Implications
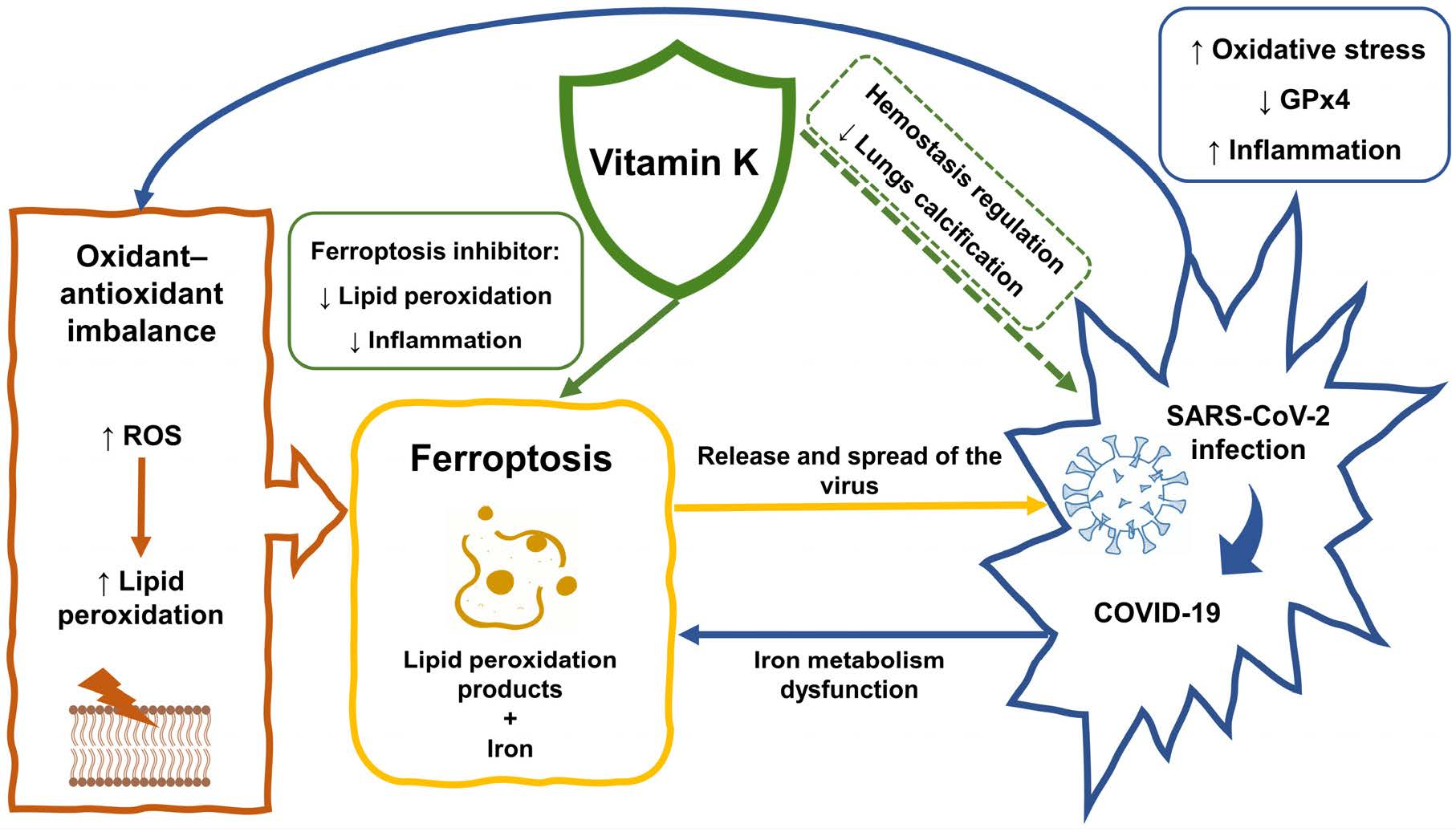
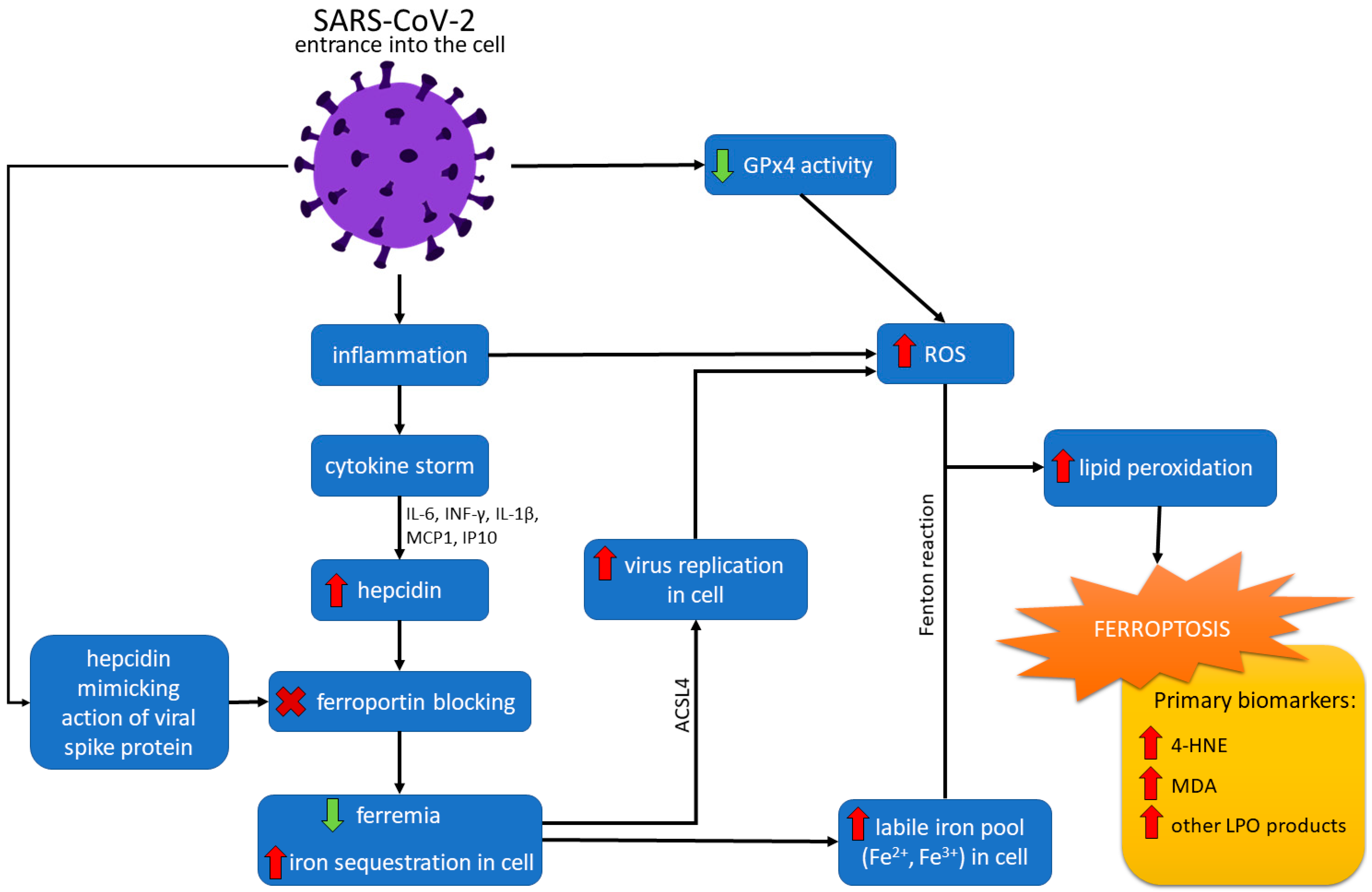
4. Ferroptosis and COVID-19
5. Ferroptosis as a Therapeutic Target in COVID-19
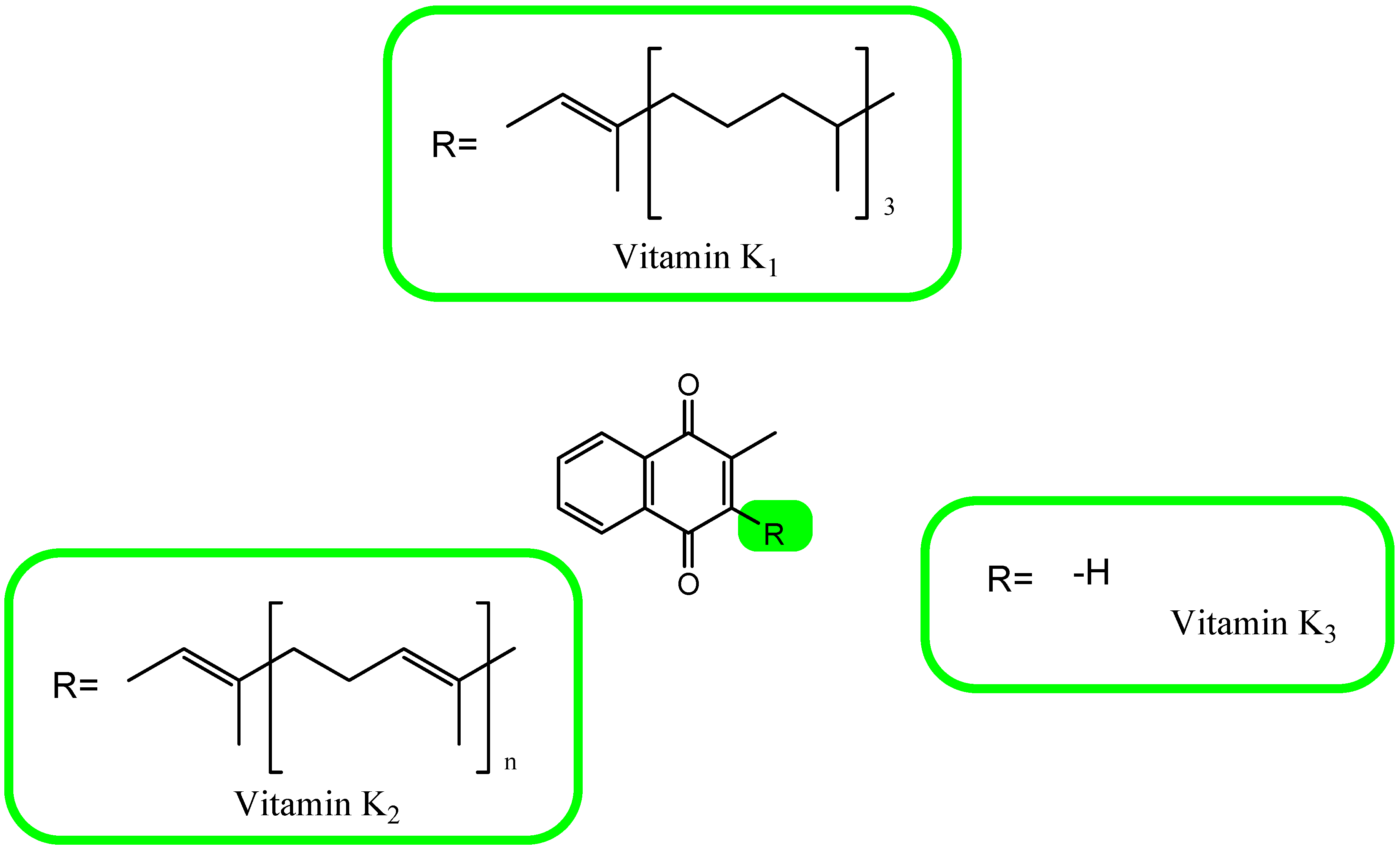
6. A Brief Overview of Vitamin K
7. Vitamin K in COVID-19
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chavda, V.P.; Kapadia, C.; Soni, S.; Prajapati, R.; Chauhan, S.C.; Yallapu, M.M.; Apostolopoulos, V. A Global Picture: Therapeutic Perspectives for COVID-19. Immunotherapy 2022, 14, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Carius, B.M.; Chavez, S.; Liang, S.Y.; Brady, W.J.; Koyfman, A.; Gottlieb, M. Clinical Update on COVID-19 for the Emergency Clinician: Presentation and Evaluation. Am. J. Emerg. Med. 2022, 54, 46–57. [Google Scholar] [CrossRef]
- Alam, M.S.; Czajkowsky, D.M. SARS-CoV-2 Infection and Oxidative Stress: Pathophysiological Insight into Thrombosis and Therapeutic Opportunities. Cytokine Growth Factor Rev. 2022, 63, 44–57. [Google Scholar] [CrossRef]
- Yegiazaryan, A.; Abnousian, A.; Alexander, L.J.; Badaoui, A.; Flaig, B.; Sheren, N.; Aghazarian, A.; Alsaigh, D.; Amin, A.; Mundra, A.; et al. Recent Developments in the Understanding of Immunity, Pathogenesis and Management of COVID-19. Int. J. Mol. Sci. 2022, 23, 9297. [Google Scholar] [CrossRef] [PubMed]
- Wool, G.D.; Miller, J.L. The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiology 2021, 88, 15–27. [Google Scholar] [CrossRef]
- Araki, S.; Shirahata, A. Vitamin K Deficiency Bleeding in Infancy. Nutrients 2020, 12, 780. [Google Scholar] [CrossRef] [Green Version]
- McPherson, C. Vitamin K Deficiency Bleeding: An Ounce of Prevention. Neonatal Netw. 2020, 39, 356–362. [Google Scholar] [CrossRef]
- Ivanova, D.; Zhelev, Z.; Getsov, P.; Nikolova, B.; Aoki, I.; Higashi, T.; Bakalova, R. Vitamin K: Redox-Modulation, Prevention of Mitochondrial Dysfunction and Anticancer Effect. Redox Biol. 2018, 16, 352–358. [Google Scholar] [CrossRef]
- Sengupta, P.; Leisegang, K.; Agarwal, A. The Impact of COVID-19 on the Male Reproductive Tract and Fertility: A Systematic Review. Arab. J. Urol. 2021, 19, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.D.; Shen, Q.; Cintron, S.A.; Hiebert, J.B. Post-COVID-19 Syndrome. Nurs. Res. 2022, 71, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Pomilio, A.B.; Vitale, A.A.; Lazarowski, A.J. COVID-19 and Alzheimer’s Disease: Neuroinflammation, Oxidative Stress, Ferroptosis, and Mechanisms Involved. Curr. Med. Chem. 2022, 29. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Y.; Zhang, K.; Shen, L.; Deng, M. Ferroptosis in COVID-19-Related Liver Injury: A Potential Mechanism and Therapeutic Target. Front. Cell. Infect. Microbiol. 2022, 12, 1058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, C.; Feng, C.; Yan, C.; Yu, Y.; Chen, Z.; Guo, C.; Wang, X. Role of Mitochondrial Reactive Oxygen Species in Homeostasis Regulation. Redox Rep. 2022, 27, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M. Effects of Antioxidant Enzymes in the Molecular Control of Reactive Oxygen Species Toxicology. Toxicology 2000, 153, 83–104. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive Oxygen Species and Mitochondria: A Nexus of Cellular Homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial Composition and Function under the Control of Hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Garza-Lombó, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Redox Homeostasis, Oxidative Stress and Mitophagy. Mitochondrion 2020, 51, 105–117. [Google Scholar] [CrossRef]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolin, I.; Herrera, F.; Martin, V.; Reiter, R.J. Regulation of Antioxidant Enzymes: A Significant Role for Melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Pruchniak, M.P.; Araźna, M.; Demkow, U. Biochemistry of Oxidative Stress. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2015; pp. 9–19. [Google Scholar]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Aschner, M.; Nguyen, T.T.; Sinitskii, A.I.; Santamaría, A.; Bornhorst, J.; Ajsuvakova, O.P.; da Rocha, J.B.T.; Skalny, A.V.; Tinkov, A.A. Isolevuglandins (IsoLGs) as Toxic Lipid Peroxidation Byproducts and Their Pathogenetic Role in Human Diseases. Free Radic. Biol. Med. 2021, 162, 266–273. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative Stress, Aging, and Diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Paardekooper, L.M.; van Vroonhoven, E.; ter Beest, M.; van den Bogaart, G. Radical Stress Is More Cytotoxic in the Nucleus than in Other Organelles. Int. J. Mol. Sci. 2019, 20, 4147. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Menezes, L.B.; Segat, B.B.; Tolentino, H.; Pires, D.C.; Mattos, L.M.d.M.; Hottum, H.M.; Pereira, M.D.; Latini, A.; Horn Jr., A.; Fernandes, C. ROS Scavenging of SOD/CAT Mimics Probed by EPR and Reduction of Lipid Peroxidation in S. Cerevisiae and Mouse Liver, under Severe Hydroxyl Radical Stress Condition. J. Inorg. Biochem. 2023, 239, 112062. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Anthonymuthu, T.S.; Kenny, E.M.; Bayır, H. Therapies Targeting Lipid Peroxidation in Traumatic Brain Injury. Brain Res. 2016, 1640, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Woźniak, A.; Górecki, D.; Szpinda, M.; Mila-Kierzenkowska, C.; Woźniak, B. Oxidant-Antioxidant Balance in the Blood of Patients with Chronic Obstructive Pulmonary Disease After Smoking Cessation. Oxid. Med. Cell. Longev. 2013, 2013, 897075. [Google Scholar] [CrossRef] [Green Version]
- Woźniak, B.; Woźniak, A.; Drewa, G.; Mila-Kierzenkowska, C.; Ślusarz, R. The Effect of Treatment on Lipid Peroxidation in Patients with Subarachnoid Haemorrhage. Arch. Med. Sci. 2009, 5, 394–400. [Google Scholar]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid Peroxidation Triggers Neurodegeneration: A Redox Proteomics View into the Alzheimer Disease Brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Girotti, A.W.; Korytowski, W. Cholesterol as a Natural Probe for Free Radical-Mediated Lipid Peroxidation in Biological Membranes and Lipoproteins. J. Chromatogr. B 2016, 1019, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, F.; Bonnefont-Rousselot, D.; Mas, E.; Drai, J.; Thérond, P. Biomarkers of Lipid Peroxidation: Analytical Aspects. Ann. Biol. Clin. (Paris) 2008, 66, 605–620. [Google Scholar] [CrossRef]
- Ramana, K.V.; Srivastava, S.; Singhal, S.S. Lipid Peroxidation Products in Human Health and Disease. Oxid. Med. Cell. Longev. 2013, 2013, 583438. [Google Scholar] [CrossRef] [PubMed]
- Beharier, O.; Kajiwara, K.; Sadovsky, Y. Ferroptosis, Trophoblast Lipotoxic Damage, and Adverse Pregnancy Outcome. Placenta 2021, 108, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated Generation of Lipid Peroxides Enhances Ferroptosis Induced by Erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 583438. [Google Scholar] [CrossRef] [Green Version]
- Ausili, A.; Clemente, J.; Pons-Belda, Ó.D.; de Godos, A.; Corbalán-García, S.; Torrecillas, A.; Teruel, J.A.; Gomez-Fernández, J.C. Interaction of Vitamin K 1 and Vitamin K 2 with Dimyristoylphosphatidylcholine and Their Location in the Membrane. Langmuir 2020, 36, 1062–1073. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The Thioredoxin Antioxidant System. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Jastrząb, A.; Skrzydlewska, E. Thioredoxin-Dependent System. Application of Inhibitors. J. Enzyme Inhib. Med. Chem. 2021, 36, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Suliman, H.B.; Piantadosi, C.A. Extracellular Superoxide Dismutase. Int. J. Biochem. Cell Biol. 2005, 37, 2466–2471. [Google Scholar] [CrossRef]
- Rosa, A.C.; Bruni, N.; Meineri, G.; Corsi, D.; Cavi, N.; Gastaldi, D.; Dosio, F. Strategies to Expand the Therapeutic Potential of Superoxide Dismutase by Exploiting Delivery Approaches. Int. J. Biol. Macromol. 2021, 168, 846–865. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase, a Remarkable Enzyme: Targeting the Oldest Antioxidant Enzyme to Find a New Cancer Treatment Approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Brunelli, L.; Yermilov, V.; Beckman, J.S. Modulation of Catalase Peroxidatic and Catalatic Activity by Nitric Oxide. Free Radic. Biol. Med. 2001, 30, 709–714. [Google Scholar] [CrossRef]
- Böhm, B.; Heinzelmann, S.; Motz, M.; Bauer, G. Extracellular Localization of Catalase Is Associated with the Transformed State of Malignant Cells. Biol. Chem. 2015, 396, 1339–1356. [Google Scholar] [CrossRef] [Green Version]
- Buday, K.; Conrad, M. Emerging Roles for Non-Selenium Containing ER-Resident Glutathione Peroxidases in Cell Signaling and Disease. Biol. Chem. 2021, 402, 271–287. [Google Scholar] [CrossRef]
- Ufer, C.; Wang, C.C. The Roles of Glutathione Peroxidases during Embryo Development. Front. Mol. Neurosci. 2011, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Woźniak, A. Signs of Oxidative Stress after Exercise. Biol. Sport 2003, 20, 93–112. [Google Scholar]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef]
- Girotti, A.W.; Korytowski, W. Intermembrane Translocation of Photodynamically Generated Lipid Hydroperoxides: Broadcasting of Redox Damage. Photochem. Photobiol. 2022, 98, 591–597. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid Peroxidation in Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Li, P.; Jiang, M.; Li, K.; Li, H.; Zhou, Y.; Xiao, X.; Xu, Y.; Krishfield, S.; Lipsky, P.E.; Tsokos, G.C.; et al. Glutathione Peroxidase 4–Regulated Neutrophil Ferroptosis Induces Systemic Autoimmunity. Nat. Immunol. 2021, 22, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R. Ferroptosis Turns 10: Emerging Mechanisms, Physiological Functions, and Therapeutic Applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. IPLA2β-Mediated Lipid Detoxification Controls P53-Driven Ferroptosis Independent of GPX4. Nat. Commun. 2021, 12, 3644. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Ge, C.; Zhang, S.; Mu, H.; Zheng, S.; Tan, Z.; Huang, X.; Xu, C.; Zou, J.; Zhu, Y.; Feng, D.; et al. Emerging Mechanisms and Disease Implications of Ferroptosis: Potential Applications of Natural Products. Front. Cell Dev. Biol. 2022, 9, 3730. [Google Scholar] [CrossRef] [PubMed]
- Schilstra, M.J.; Veldink, G.A.; Vliegenthart, J.F.G. The Dioxygenation Rate In Lipoxygenase Catalysis Is Determined by the Amount of Iron(III) Lipoxygenase in Solution. Biochemistry 1994, 33, 3974–3979. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 Oxidoreductase Contributes to Phospholipid Peroxidation in Ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis Is an Autophagic Cell Death Process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-H.; Wu, J.; Ding, C.-K.C.; Lin, C.-C.; Pan, S.; Bossa, N.; Xu, Y.; Yang, W.-H.; Mathey-Prevot, B.; Chi, J.-T. Kinome Screen of Ferroptosis Reveals a Novel Role of ATM in Regulating Iron Metabolism. Cell Death Differ. 2020, 27, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Frey, A.G.; Palenchar, D.J.; Achar, S.; Bullough, K.Z.; Vashisht, A.; Wohlschlegel, J.A.; Philpott, C.C. A PCBP1–BolA2 Chaperone Complex Delivers Iron for Cytosolic [2Fe–2S] Cluster Assembly. Nat. Chem. Biol. 2019, 15, 872–881. [Google Scholar] [CrossRef]
- Cao, J.Y.; Poddar, A.; Magtanong, L.; Lumb, J.H.; Mileur, T.R.; Reid, M.A.; Dovey, C.M.; Wang, J.; Locasale, J.W.; Stone, E.; et al. A Genome-Wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep. 2019, 26, 1544–1556.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-Mediated Autophagy Is Involved in the Execution of Ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; De Nicola, G.M. Non-Canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021, 33, 174–189.e7. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Yu, J.; He, W.; Huang, Q.; Zhao, Y.; Liang, B.; Zhang, S.; Wen, Z.; Dong, S.; Rao, J.; et al. Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia 2017, 19, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Van Coillie, S.; Van San, E.; Goetschalckx, I.; Wiernicki, B.; Mukhopadhyay, B.; Tonnus, W.; Choi, S.M.; Roelandt, R.; Dumitrascu, C.; Lamberts, L.; et al. Targeting Ferroptosis Protects against Experimental (Multi)Organ Dysfunction and Death. Nat. Commun. 2022, 13, 1046. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-Mediated Ferroptosis Defence Is a Targetable Vulnerability in Cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Zeitler, L.; Fiore, A.; Meyer, C.; Russier, M.; Zanella, G.; Suppmann, S.; Gargaro, M.; Sidhu, S.S.; Seshagiri, S.; Ohnmacht, C.; et al. Anti-Ferroptotic Mechanism of IL4i1-Mediated Amino Acid Metabolism. eLife 2021, 10, e64806. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, Z.; Zhang, H.; Chen, C.; Zeng, M.; Yunis, J.; Wei, Y.; Wan, Y.; Wang, N.; Zhou, M.; et al. Selenium–GPX4 Axis Protects Follicular Helper T Cells from Ferroptosis. Nat. Immunol. 2021, 22, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Dierge, E.; Debock, E.; Guilbaud, C.; Corbet, C.; Mignolet, E.; Mignard, L.; Bastien, E.; Dessy, C.; Larondelle, Y.; Feron, O. Peroxidation of N-3 and n-6 Polyunsaturated Fatty Acids in the Acidic Tumor Environment Leads to Ferroptosis-Mediated Anticancer Effects. Cell Metab. 2021, 33, 1701–1715.e5. [Google Scholar] [CrossRef]
- Liao, P.; Wang, W.; Wang, W.; Kryczek, I.; Li, X.; Bian, Y.; Sell, A.; Wei, S.; Grove, S.; Johnson, J.K.; et al. CD8+ T Cells and Fatty Acids Orchestrate Tumor Ferroptosis and Immunity via ACSL4. Cancer Cell 2022, 40, 365–378.e6. [Google Scholar] [CrossRef]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-Mediated Ferroptosis Dampens Intratumoral CD8+ T Cell Effector Function and Impairs Their Antitumor Ability. Cell Metab. 2021, 33, 1001–1012.e5. [Google Scholar] [CrossRef]
- Habib, H.M.; Ibrahim, S.; Zaim, A.; Ibrahim, W.H. The Role of Iron in the Pathogenesis of COVID-19 and Possible Treatment with Lactoferrin and Other Iron Chelators. Biomed. Pharmacother. 2021, 136, 111228. [Google Scholar] [CrossRef]
- Edeas, M.; Saleh, J.; Peyssonnaux, C. Iron: Innocent Bystander or Vicious Culprit in COVID-19 Pathogenesis? Int. J. Infect. Dis. 2020, 97, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Suriawinata, E.; Mehta, K.J. Iron and Iron-Related Proteins in COVID-19. Clin. Exp. Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Fratta Pasini, A.M.; Stranieri, C.; Girelli, D.; Busti, F.; Cominacini, L. Is Ferroptosis a Key Component of the Process Leading to Multiorgan Damage in COVID-19? Antioxidants 2021, 10, 1677. [Google Scholar] [CrossRef] [PubMed]
- Cavezzi, A.; Menicagli, R.; Troiani, E.; Corrao, S. COVID-19, Cation Dysmetabolism, Sialic Acid, CD147, ACE2, Viroporins, Hepcidin and Ferroptosis: A Possible Unifying Hypothesis. F1000Research 2022, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Zeinivand, M.; Jamali-Raeufy, N.; Zavvari, F. The Beneficial Role of Hepcidin Peptide Inhibitor in Improved the Symptoms of COVID-19 in Diabetics: Anti-Inflammatory and Potential Therapeutic Effects. J. Diabetes Metab. Disord. 2022, 21, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Čepelak, I.; Dodig, S. Ferroptosis-a Link between Iron Biology, Aging and COVID-19? Rad Hrvat. Akad. Znan. i Umjet. Med. Znan. 2022, 58–59, 58–68. [Google Scholar] [CrossRef]
- Gaiatto, A.C.M.; Bibo, T.A.; de Godoy Moreira, N.; Raimundo, J.R.S.; da Costa Aguiar Alves, B.; Gascón, T.; Carvalho, S.S.; Pereira, E.C.; Fonseca, F.L.A.; da Veiga, G.L. COVID-19 Compromises Iron Homeostasis: Transferrin as a Target of Investigation. J. Trace Elem. Med. Biol. 2023, 76, 127109. [Google Scholar] [CrossRef]
- Yang, M.; Lai, C.L. SARS-CoV-2 Infection: Can Ferroptosis Be a Potential Treatment Target for Multiple Organ Involvement? Cell Death Discov. 2020, 6, 130. [Google Scholar] [CrossRef]
- Ma, T.-L.; Zhou, Y.; Wang, C.; Wang, L.; Chen, J.-X.; Yang, H.-H.; Zhang, C.-Y.; Zhou, Y.; Guan, C.-X. Targeting Ferroptosis for Lung Diseases: Exploring Novel Strategies in Ferroptosis-Associated Mechanisms. Oxid. Med. Cell. Longev. 2021, 2021, 1098970. [Google Scholar] [CrossRef]
- Ehsani, S. COVID-19 and Iron Dysregulation: Distant Sequence Similarity between Hepcidin and the Novel Coronavirus Spike Glycoprotein. Biol. Direct 2020, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Otlu, Ö.; Erdem, M.; Korkmaz, K.; Gunay, Ö.O.; İn, E.; Kıran, T.R.; Karabulut, A.B. Effect of Altered Iron Metabolism on Hyperinflammation and Coagulopathy in Patients with Critical COVID-19:A Retrospective Study. Int. J. Acad. Med. Pharm. 2022, 4, 60–64. [Google Scholar] [CrossRef]
- Claise, C.; Saleh, J.; Rezek, M.; Vaulont, S.; Peyssonnaux, C.; Edeas, M. Low Transferrin Levels Predict Heightened Inflammation in Patients with COVID-19: New Insights. Int. J. Infect. Dis. 2022, 116, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, W.; You, B.; Zhou, C. Advances in Cell Death Mechanisms Involved in Viral Myocarditis. Front. Cardiovasc. Med. 2022, 9, 968752. [Google Scholar] [CrossRef] [PubMed]
- Kung, Y.-A.; Chiang, H.-J.; Li, M.-L.; Gong, Y.-N.; Chiu, H.-P.; Hung, C.-T.; Huang, P.-N.; Huang, S.-Y.; Wang, P.-Y.; Hsu, T.-A.; et al. Acyl-Coenzyme A Synthetase Long-Chain Family Member 4 Is Involved in Viral Replication Organelle Formation and Facilitates Virus Replication via Ferroptosis. MBio 2022, 13, e02717-21. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Shen, W.-B.; Yang, P.; Turan, S. SARS-CoV-2 Infection Induces Activation of Ferroptosis in Human Placenta. Front. Cell Dev. Biol. 2022, 10, 1022747. [Google Scholar] [CrossRef]
- Jacobs, W.; Lammens, M.; Kerckhofs, A.; Voets, E.; Van San, E.; Van Coillie, S.; Peleman, C.; Mergeay, M.; Sirimsi, S.; Matheeussen, V.; et al. Fatal Lymphocytic Cardiac Damage in Coronavirus Disease 2019 (COVID-19): Autopsy Reveals a Ferroptosis Signature. ESC Heart Fail. 2020, 7, 3772–3781. [Google Scholar] [CrossRef]
- Zhang, R.; Sun, C.; Chen, X.; Han, Y.; Zang, W.; Jiang, C.; Wang, J.; Wang, J. COVID-19-Related Brain Injury: The Potential Role of Ferroptosis. J. Inflamm. Res. 2022, 15, 2181–2198. [Google Scholar] [CrossRef]
- Skesters, A.; Kustovs, D.; Lece, A.; Moreino, E.; Petrosina, E.; Rainsford, K.D. Selenium, Selenoprotein P, and Oxidative Stress Levels in SARS-CoV-2 Patients during Illness and Recovery. Inflammopharmacology 2022, 30, 499–503. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Q.; Tang, Y.-D.; Zhai, J.; Hu, W.; Zheng, C. When Ferroptosis Meets Pathogenic Infections. Trends Microbiol. 2022. [Google Scholar] [CrossRef]
- Duan, J.-Y.; Lin, X.; Xu, F.; Shan, S.-K.; Guo, B.; Li, F.-X.-Z.; Wang, Y.; Zheng, M.-H.; Xu, Q.-S.; Lei, L.-M.; et al. Ferroptosis and Its Potential Role in Metabolic Diseases: A Curse or Revitalization? Front. Cell Dev. Biol. 2021, 9, 701788. [Google Scholar] [CrossRef]
- Lin, Z.; Yang, X.; Guan, L.; Qin, L.; Ding, J.; Zhou, L. The Link between Ferroptosis and Airway Inflammatory Diseases: A Novel Target for Treatment. Front. Mol. Biosci. 2022, 9, 878. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Joshua, B.; Jin, N.; Du, S.; Li, C. Ferroptosis in Viral Infection: The Unexplored Possibility. Acta Pharmacol. Sin. 2022, 43, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Dowd, P. The Mechanism of Action of Vitamin K. Annu. Rev. Nutr. 1995, 15, 419–440. [Google Scholar] [CrossRef]
- Booth, S.L. Roles for Vitamin K Beyond Coagulation. Annu. Rev. Nutr. 2009, 29, 89–110. [Google Scholar] [CrossRef]
- Ferland, G. The Discovery of Vitamin K and Its Clinical Applications. Ann. Nutr. Metab. 2012, 61, 213–218. [Google Scholar] [CrossRef]
- Beulens, J.W.J.; Booth, S.L.; van den Heuvel, E.G.H.M.; Stoecklin, E.; Baka, A.; Vermeer, C. The Role of Menaquinones (Vitamin K2) in Human Health. Br. J. Nutr. 2013, 110, 1357–1368. [Google Scholar] [CrossRef] [Green Version]
- El Asmar, M.; Naoum, J.; Arbid, E. Vitamin K Dependent Proteins and the Role of Vitamin K2 in the Modulation of Vascular Calcification: A Review. Oman Med. J. 2014, 29, 172–177. [Google Scholar] [CrossRef]
- Mladěnka, P.; Macáková, K.; Kujovská Krčmová, L.; Javorská, L.; Mrštná, K.; Carazo, A.; Protti, M.; Remião, F.; Nováková, L. Vitamin K–Sources, Physiological Role, Kinetics, Deficiency, Detection, Therapeutic Use, and Toxicity. Nutr. Rev. 2022, 80, 677–698. [Google Scholar] [CrossRef]
- Fusaro, M.; Gallieni, M.; Rizzo, M.A.; Stucchi, A.; Delanaye, P.; Cavalier, E.; Moysés, R.M.A.; Jorgetti, V.; Iervasi, G.; Giannini, S.; et al. Vitamin K Plasma Levels Determination in Human Health. Clin. Chem. Lab. Med. 2017, 55, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Kaźmierczak-Barańska, J.; Karwowski, B.T. Vitamin K Contribution to DNA Damage—Advantage or Disadvantage? A Human Health Response. Nutrients 2022, 14, 4219. [Google Scholar] [CrossRef] [PubMed]
- Thane, C.W.; Bolton-Smith, C.; Coward, W.A. Comparative Dietary Intake and Sources of Phylloquinone (Vitamin K1) among British Adults in 1986–7 and 2000–1. Br. J. Nutr. 2006, 96, 1105–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcone, T.D.; Kim, S.S.W.; Cortazzo, M.H. Vitamin K: Fracture Prevention and Beyond. PM&R 2011, 3, S82–S87. [Google Scholar] [CrossRef]
- Hodges, S.J.; Pilkington, M.J.; Shearer, M.J.; Bitensky, L.; Chayen, J. Age-Related Changes in the Circulating Levels of Congeners of Vitamin K2, Menaquinone-7 and Menaquinone-8. Clin. Sci. 1990, 78, 63–66. [Google Scholar] [CrossRef]
- Harrington, D.J.; Booth, S.L.; Card, D.J.; Shearer, M.J. Excretion of the Urinary 5C- and 7C-Aglycone Metabolites of Vitamin K by Young Adults Responds to Changes in Dietary Phylloquinone and Dihydrophylloquinone Intakes. J. Nutr. 2007, 137, 1763–1768. [Google Scholar] [CrossRef] [Green Version]
- Patracchini, P.; Aiello, V.; Palazzi, P.; Calzolari, E.; Bernardi, F. Sublocalization of the Human Protein C Gene on Chromosome 2q13-Q14. Hum. Genet. 1989, 81, 191–192. [Google Scholar] [CrossRef]
- Greer, F.R. Vitamin K the Basics—What’s New? Early Hum. Dev. 2010, 86, 43–47. [Google Scholar] [CrossRef]
- Dahlbäck, B. Protein S and C4b-Binding Protein: Components Involved in the Regulation of the Protein C Anticoagulant System. Thromb. Haemost. 1991, 66, 49–61. [Google Scholar] [CrossRef]
- Sofi, F.; Cesari, F.; Fedi, S.; Abbate, R.; Gensini, G.F. Protein Z: “Light and Shade” of a New Thrombotic Factor. Clin. Lab. 2004, 50, 647–652. [Google Scholar]
- Mark, M.R.; Chen, J.; Hammonds, R.G.; Sadick, M.; Godowsk, P.J. Characterization of Gas6, a Member of the Superfamily of G Domain-Containing Proteins, as a Ligand for Rse and Axl. J. Biol. Chem. 1996, 271, 9785–9789. [Google Scholar] [CrossRef] [Green Version]
- Viegas, C.S.B.; Cavaco, S.; Neves, P.L.; Ferreira, A.; João, A.; Williamson, M.K.; Price, P.A.; Cancela, M.L.; Simes, D.C. Gla-Rich Protein Is a Novel Vitamin K-Dependent Protein Present in Serum That Accumulates at Sites of Pathological Calcifications. Am. J. Pathol. 2009, 175, 2288–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsaki, Y.; Shirakawa, H.; Hiwatashi, K.; Furukawa, Y.; Mizutani, T.; Komai, M. Vitamin K Suppresses Lipopolysaccharide-Induced Inflammation in the Rat. Biosci. Biotechnol. Biochem. 2006, 70, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Shea, M.K.; Dallal, G.E.; Dawson-Hughes, B.; Ordovas, J.M.; O’Donnell, C.J.; Gundberg, C.M.; Peterson, J.W.; Booth, S.L. Vitamin K, Circulating Cytokines, and Bone Mineral Density in Older Men and Women. Am. J. Clin. Nutr. 2008, 88, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Reddi, K.; Henderson, B.; Meghji, S.; Wilson, M.; Poole, S.; Hopper, C.; Harris, M.; Hodges, S.J. Interleukin 6 Production by Lipopolysaccharide-Stimulated Human Fibroblasts Is Potently Inhibited by Naphthoquinone (Vitamin K) Compounds. Cytokine 1995, 7, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Akbari, S.; Rasouli-Ghahroudi, A.A. Vitamin K and Bone Metabolism: A Review of the Latest Evidence in Preclinical Studies. BioMed Res. Int. 2018, 2018, 4629383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishima, E.; Ito, J.; Wu, Z.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; et al. A Non-Canonical Vitamin K Cycle Is a Potent Ferroptosis Suppressor. Nature 2022, 608, 778–783. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. Vitamin K: A New Guardian against Ferroptosis. Mol. Cell 2022, 82, 3760–3762. [Google Scholar] [CrossRef]
- Kolbrink, B.; von Samson-Himmelstjerna, F.A.; Messtorff, M.L.; Riebeling, T.; Nische, R.; Schmitz, J.; Bräsen, J.H.; Kunzendorf, U.; Krautwald, S. Vitamin K1 Inhibits Ferroptosis and Counteracts a Detrimental Effect of Phenprocoumon in Experimental Acute Kidney Injury. Cell. Mol. Life Sci. 2022, 79, 387. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbiny, M.; Atef, H.; Helal, G.M.; Al-Serwi, R.H.; Elkattawy, H.A.; Shaker, G.A.; Said, E.; Abulfaraj, M.; Albalawi, M.A.; Elsherbiny, N.M. Vitamin K2 (MK-7) Intercepts Keap-1/Nrf-2/HO-1 Pathway and Hinders Inflammatory/Apoptotic Signaling and Liver Aging in Naturally Aging Rat. Antioxidants 2022, 11, 2150. [Google Scholar] [CrossRef]
- Lv, J.; Hou, B.; Song, J.; Xu, Y.; Xie, S. The Relationship Between Ferroptosis and Diseases. J. Multidiscip. Healthc. 2022, 15, 2261–2275. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Amirah, S.R.; Harmonis, J.A.; Ikram, E.H.K. Role of Nuclear Factor Erythroid 2 (Nrf2) in the Recovery of Long COVID-19 Using Natural Antioxidants: A Systematic Review. Antioxidants 2022, 11, 1551. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, D.; Sperhake, J.-P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.-D.; Sacco, C.; Bertuzzi, A.; et al. Venous and Arterial Thromboembolic Complications in COVID-19 Patients Admitted to an Academic Hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef]
- Dofferhoff, A.S.M.; Piscaer, I.; Schurgers, L.J.; Visser, M.P.J.; van den Ouweland, J.M.W.; de Jong, P.A.; Gosens, R.; Hackeng, T.M.; van Daal, H.; Lux, P.; et al. Reduced Vitamin K Status as a Potentially Modifiable Risk Factor of Severe Coronavirus Disease 2019. Clin. Infect. Dis. 2021, 73, e4039–e4046. [Google Scholar] [CrossRef] [PubMed]
- Sobczyk, M.K.; Gaunt, T.R. The Effect of Circulating Zinc, Selenium, Copper and Vitamin K1 on COVID-19 Outcomes: A Mendelian Randomization Study. Nutrients 2022, 14, 233. [Google Scholar] [CrossRef]
- Janssen, R.; Visser, M.P.J.; Dofferhoff, A.S.M.; Vermeer, C.; Janssens, W.; Walk, J. Vitamin K Metabolism as the Potential Missing Link between Lung Damage and Thromboembolism in Coronavirus Disease 2019. Br. J. Nutr. 2021, 126, 191–198. [Google Scholar] [CrossRef]
- Linneberg, A.; Kampmann, F.B.; Israelsen, S.B.; Andersen, L.R.; Jørgensen, H.L.; Sandholt, H.; Jørgensen, N.R.; Thysen, S.M.; Benfield, T. The Association of Low Vitamin K Status with Mortality in a Cohort of 138 Hospitalized Patients with COVID-19. Nutrients 2021, 13, 1985. [Google Scholar] [CrossRef]
- Jespersen, T.; Møllehave, L.T.; Thuesen, B.H.; Skaaby, T.; Rossing, P.; Toft, U.; Jørgensen, N.R.; Corfixen, B.L.; Jakobsen, J.; Frimodt-Møller, M.; et al. Uncarboxylated Matrix Gla-Protein: A Biomarker of Vitamin K Status and Cardiovascular Risk. Clin. Biochem. 2020, 83, 49–56. [Google Scholar] [CrossRef]
- Piscaer, I.; van den Ouweland, J.M.W.; Vermeersch, K.; Reynaert, N.L.; Franssen, F.M.E.; Keene, S.; Wouters, E.F.M.; Janssens, W.; Vermeer, C.; Janssen, R. Low Vitamin K Status Is Associated with Increased Elastin Degradation in Chronic Obstructive Pulmonary Disease. J. Clin. Med. 2019, 8, 1116. [Google Scholar] [CrossRef] [Green Version]
- Desai, A.P.; Dirajlal-Fargo, S.; Durieux, J.C.; Tribout, H.; Labbato, D.; McComsey, G.A. Vitamin K & D Deficiencies Are Independently Associated With COVID-19 Disease Severity. Open Forum Infect. Dis. 2021, 8, 1–8. [Google Scholar] [CrossRef]
- Visser, M.P.J.; Walk, J.; Vermeer, C.; Bílková, S.; Janssen, R.; Mayer, O. Enhanced Vitamin K Expenditure as a Major Contributor to Vitamin K Deficiency in COVID-19. Int. J. Infect. Dis. 2022, 125, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, E.; Ialongo, C.; Labriola, R.; Ferraguti, G.; Lucarelli, M.; Angeloni, A. Vitamin K Deficiency and COVID-19. Scand. J. Clin. Lab. Investig. 2020, 80, 525–527. [Google Scholar] [CrossRef]
- Ferland, G. Vitamin K and Brain Function. Semin. Thromb. Hemost. 2013, 39, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nishiumi, S.; Nishida, M.; Mizushina, Y.; Kobayashi, K.; Masuda, A.; Fujita, T.; Morita, Y.; Mizuno, S.; Kutsumi, H.; et al. Vitamin K3 Attenuates Lipopolysaccharide-Induced Acute Lung Injury through Inhibition of Nuclear Factor-ΚB Activation. Clin. Exp. Immunol. 2010, 160, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Shea, M.K.; Booth, S.L.; Massaro, J.M.; Jacques, P.F.; D’Agostino, R.B.; Dawson-Hughes, B.; Ordovas, J.M.; O’Donnell, C.J.; Kathiresan, S.; Keaney, J.F.; et al. Vitamin K and Vitamin D Status: Associations with Inflammatory Markers in the Framingham Offspring Study. Am. J. Epidemiol. 2007, 167, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, H.; Ishizawa, H.; Nakamura, Y.; Tadokoro, H.; Tanaka, S.; Onda, K.; Sugiyama, K.; Hirano, T. Effects of Vitamin K3 and K5 on Proliferation, Cytokine Production, and Regulatory T Cell-Frequency in Human Peripheral-Blood Mononuclear Cells. Life Sci. 2014, 99, 61–68. [Google Scholar] [CrossRef]
- Ménager, P.; Brière, O.; Gautier, J.; Riou, J.; Sacco, G.; Brangier, A.; Annweiler, C. Regular Use of VKA Prior to COVID-19 Associated with Lower 7-Day Survival in Hospitalized Frail Elderly COVID-19 Patients: The GERIA-COVID Cohort Study. Nutrients 2020, 13, 39. [Google Scholar] [CrossRef]
- Norooznezhad, A.H.; Mansouri, K. Endothelial Cell Dysfunction, Coagulation, and Angiogenesis in Coronavirus Disease 2019 (COVID-19). Microvasc. Res. 2021, 137, 104188. [Google Scholar] [CrossRef]
- Veenith, T.; Martin, H.; Le Breuilly, M.; Whitehouse, T.; Gao-Smith, F.; Duggal, N.; Lord, J.M.; Mian, R.; Sarphie, D.; Moss, P. High Generation of Reactive Oxygen Species from Neutrophils in Patients with Severe COVID-19. Sci. Rep. 2022, 12, 10484. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuszkiewicz, J.; Sutkowy, P.; Wróblewski, M.; Pawłowska, M.; Wesołowski, R.; Wróblewska, J.; Woźniak, A. Links between Vitamin K, Ferroptosis and SARS-CoV-2 Infection. Antioxidants 2023, 12, 733. https://doi.org/10.3390/antiox12030733
Nuszkiewicz J, Sutkowy P, Wróblewski M, Pawłowska M, Wesołowski R, Wróblewska J, Woźniak A. Links between Vitamin K, Ferroptosis and SARS-CoV-2 Infection. Antioxidants. 2023; 12(3):733. https://doi.org/10.3390/antiox12030733
Chicago/Turabian StyleNuszkiewicz, Jarosław, Paweł Sutkowy, Marcin Wróblewski, Marta Pawłowska, Roland Wesołowski, Joanna Wróblewska, and Alina Woźniak. 2023. "Links between Vitamin K, Ferroptosis and SARS-CoV-2 Infection" Antioxidants 12, no. 3: 733. https://doi.org/10.3390/antiox12030733