
Advances of H2S in Regulating Neurodegenerative Diseases by Preserving Mitochondria Function

Abstract
:1. Introduction
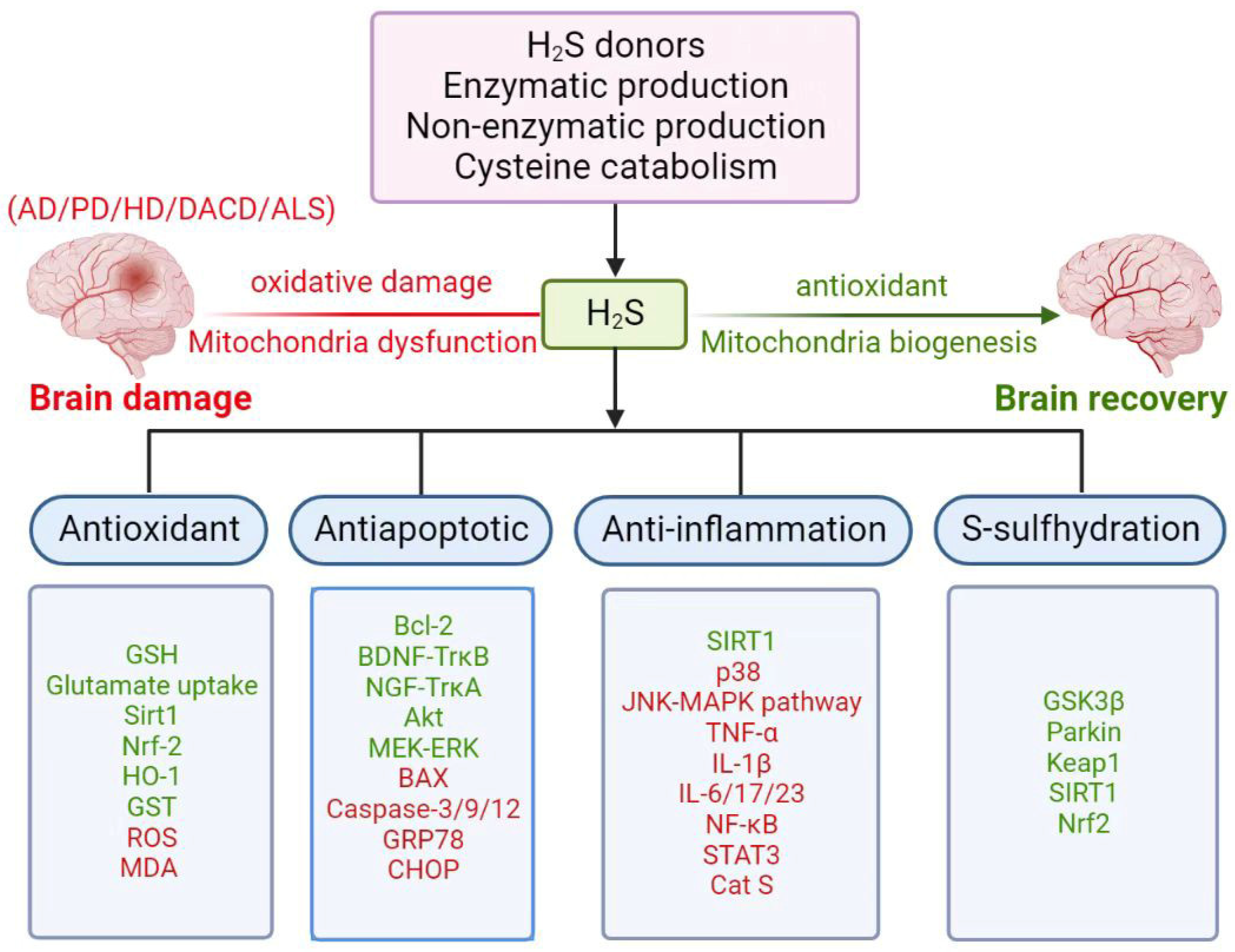
2. Induction of Neurotoxicity
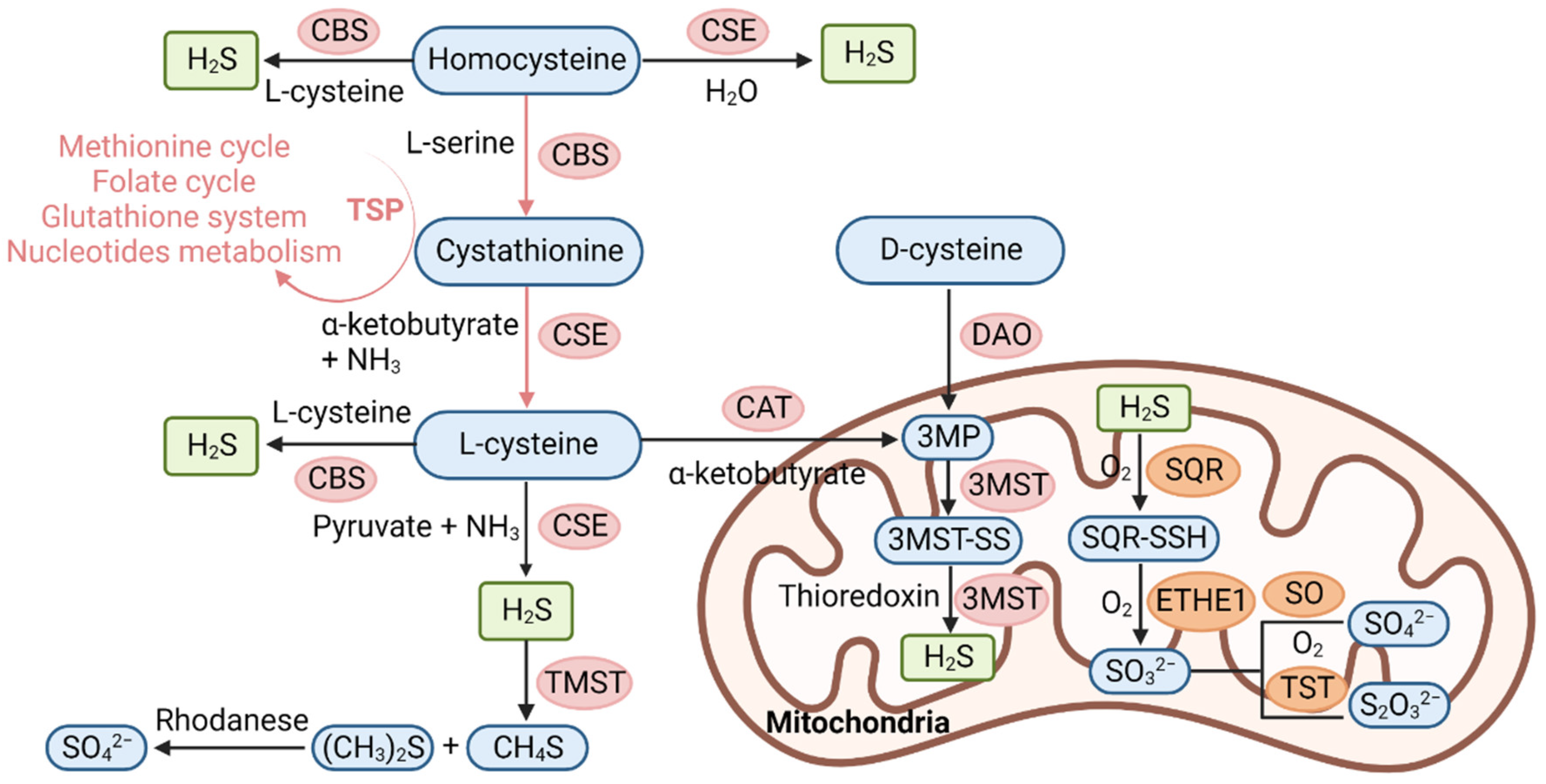
3. Biosynthesis and Metabolism of H2S
3.1. Physiological Function of H2S
3.2. Donors of H2S
4. Involvement of H2S in Neurological Diseases
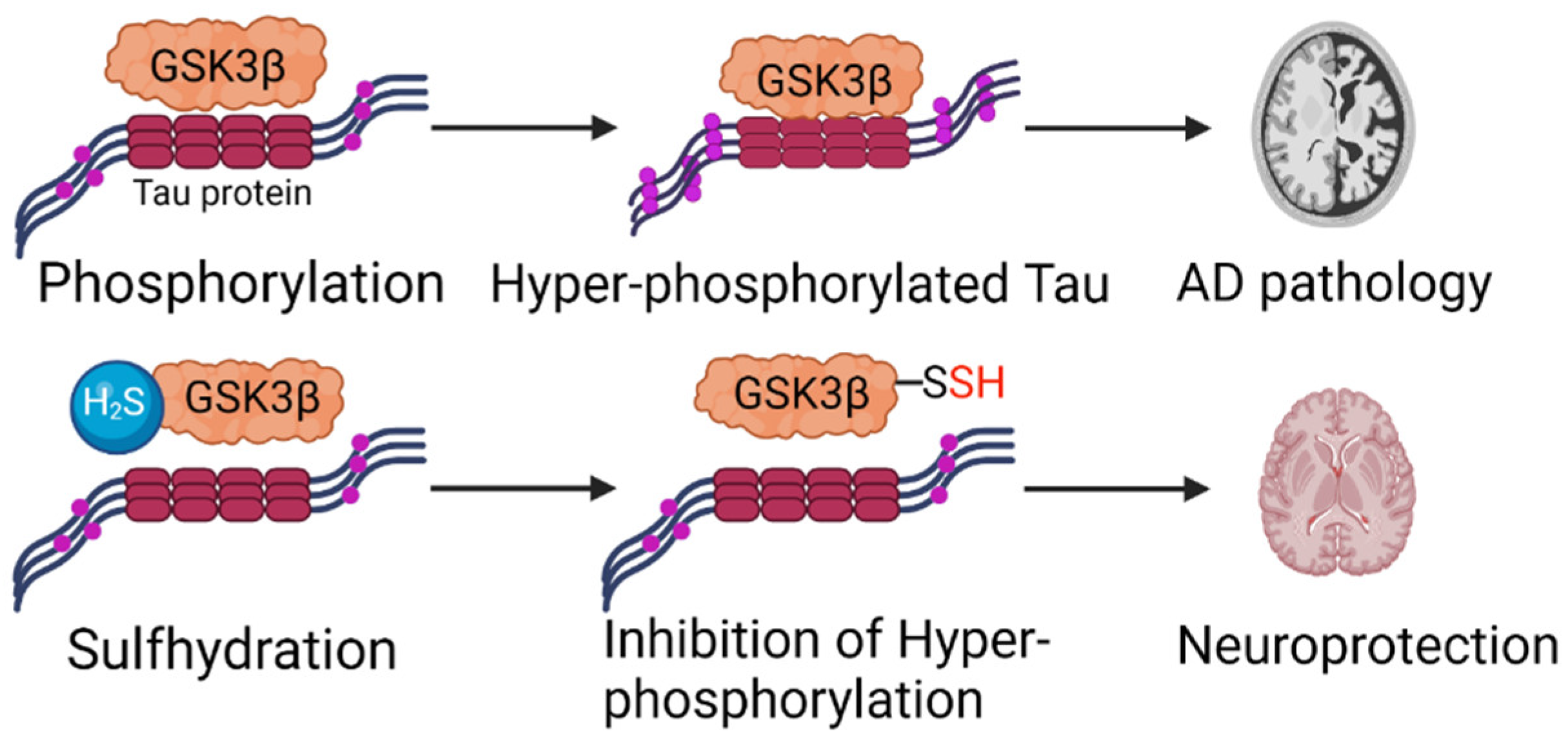
4.1. Alzheimer’s Disease (AD)
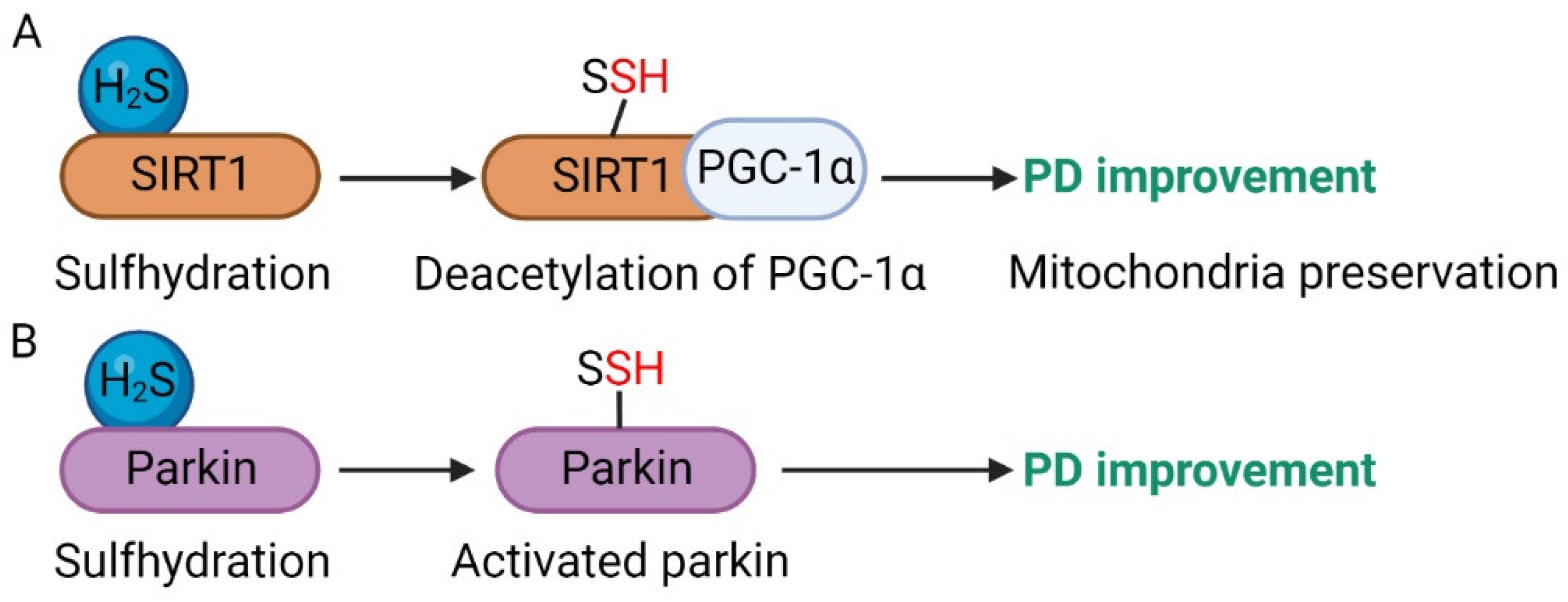
4.2. Parkinson’s Disease (PD)
4.3. Huntington’s Disease
4.4. Diabetes-Associated Cognitive Impairment and Other Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases | Biological Model | Targets | References |
|---|---|---|---|
| AD | PC12 cells | Mitochondrial membrane potential, Redox steady state, Apoptosis pathway | [90,91,98] |
| APP/PS1 neurons | Energy production, Mitochondrial DNA, Redox steady state, APP pathway | [92,102] | |
| HepG2/HEK293 cells | Mitochondrial bioenergetics | [93] | |
| 3xTg-AD mice | Redox steady state, GSK3β | [94,95,105] | |
| APP/PS1 transgenic mice | Nrf2, HO-1, GST | [96] | |
| Homocysteine-induced AD mice | NMDA receptor | [97] | |
| Microglia cells | p38 and JNK-MAPK dependent pathway, STAT3, Cat S | [99,101] | |
| Hippocampus cells | NF-κB pathway | [100] | |
| PD | Rotenone-induced PD mice | Mitochondrial function, JNK-MAPK pathway, anti-inflammatory factors | [82,110] |
| SH-SY5Y cells | Mitochondrial transmembrane potential, Cell apoptotic, Redox steady state | [109] | |
| MPP+-induced PD mouse | SIRT1 expression and sulfhydration, PGC-1α deacetylation | [118,120] | |
| 6-OHDA-induced PD rat | Leptin signaling, Redox steady state | [110] | |
| PD patients | Parkin sulfhydration | [121] | |
| HD | 3-nitropropionic acid (3NP)-induced HD rat | Antioxidative responses, anti-inflammatory and anti-apoptotic signaling | [131] |
| DACD | Streptozotocin (STZ)-induced diabetic rat | GRP78, cleaved caspase-12 and CHOP expression, SIRT1 expression | [142,143] |
| db/db mice | SIRT1 expression, ER stress pathway, anti-apoptotic and anti-inflammatory pathway | [145,147] |
5. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Richardson, J.R.; Wang, Y. Neurotoxicology. Chem. Res. Toxicol. 2021, 34, 1197. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Wallace, K.B.; Starkov, A.A. Mitochondrial targets of drug toxicity. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 353–388. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Hu, L.F.; Hu, G.; Bian, J.S. Hydrogen sulfide protects astrocytes against H2O2-induced neural injury via enhancing glutamate uptake. Free Radic. Biol. Med. 2008, 45, 1705–1713. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Borah, A. Oxidative stress and mitochondrial dysfunction are the underlying events of dopaminergic neurodegeneration in homocysteine rat model of Parkinson’s disease. Neurochem. Int. 2016, 101, 48–55. [Google Scholar] [CrossRef]
- Kimura, H. Hydrogen sulfide: From brain to gut. Antioxid. Redox Signal. 2010, 12, 1111–1123. [Google Scholar] [CrossRef]
- Warenycia, M.W.; Goodwin, L.R.; Benishin, C.G.; Reiffenstein, R.J.; Francom, D.M.; Taylor, J.D.; Dieken, F.P. Acute hydrogen sulfide poisoning. Demonstration of selective uptake of sulfide by the brainstem by measurement of brain sulfide levels. Biochem. Pharmacol. 1989, 38, 973–981. [Google Scholar] [CrossRef]
- Kimura, H. Hydrogen sulfide: Its production, release and functions. Amino Acids 2011, 41, 113–121. [Google Scholar] [CrossRef]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef]
- Kimura, H. Signaling of hydrogen sulfide and polysulfides. Antioxid. Redox Signal. 2015, 22, 347–349. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef] [Green Version]
- Kamat, P.K.; Kalani, A.; Tyagi, N. Role of hydrogen sulfide in brain synaptic remodeling. Methods Enzymol. 2015, 555, 207–229. [Google Scholar]
- Panthi, S.; Manandhar, S.; Gautam, K. Hydrogen sulfide, nitric oxide, and neurodegenerative disorders. Transl. Neurodegener. 2018, 7, 3. [Google Scholar] [CrossRef]
- Aroca, A.; Gotor, C. Hydrogen Sulfide: A Key Role in Autophagy Regulation from Plants to Mammalians. Antioxidants 2022, 11, 327. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Bhatia, M.; Zhu, Y.Z.; Zhu, Y.C.; Ramnath, R.D.; Wang, Z.J.; Anuar, F.B.; Whiteman, M.; Salto-Tellez, M.; Moore, P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005, 19, 1196–1198. [Google Scholar] [CrossRef]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef]
- Kumar, M.; Ray, R.S.; Sandhir, R. Hydrogen sulfide attenuates homocysteine-induced neurotoxicity by preventing mitochondrial dysfunctions and oxidative damage: In vitro and in vivo studies. Neurochem. Int. 2018, 120, 87–98. [Google Scholar] [CrossRef]
- Grieco, A.J. Homocystinuria: Pathogenetic mechanisms. Am. J. Med. Sci. 1977, 273, 120–132. [Google Scholar] [CrossRef]
- Mudd, S.H.; Skovby, F.; Levy, H.L.; Pettigrew, K.D.; Wilcken, B.; Pyeritz, R.E.; Andria, G.; Boers, G.H.; Bromberg, I.L.; Cerone, R.; et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar]
- Ichinohe, A.; Kanaumi, T.; Takashima, S.; Enokido, Y.; Nagai, Y.; Kimura, H. Cystathionine beta-synthase is enriched in the brains of Down’s patients. Biochem. Biophys. Res. Commun. 2005, 338, 1547–1550. [Google Scholar] [CrossRef]
- Kamath, A.F.; Chauhan, A.K.; Kisucka, J.; Dole, V.S.; Loscalzo, J.; Handy, D.E.; Wagner, D.D. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 2006, 107, 591–593. [Google Scholar] [CrossRef] [Green Version]
- Vitvitsky, V.; Dayal, S.; Stabler, S.; Zhou, Y.; Wang, H.; Lentz, S.R.; Banerjee, R. Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R39–R46. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Dong, Z.; Zhao, Y.; Sai, N.; Wang, X.; Liu, H.; Huang, G.; Zhang, X. Homocysteine induces mitochondrial dysfunction involving the crosstalk between oxidative stress and mitochondrial pSTAT3 in rat ischemic brain. Sci. Rep. 2017, 7, 6932. [Google Scholar] [CrossRef] [Green Version]
- Kamat, P.K.; Kalani, A.; Givvimani, S.; Sathnur, P.B.; Tyagi, S.C.; Tyagi, N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 2013, 252, 302–319. [Google Scholar] [CrossRef] [Green Version]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef] [Green Version]
- Linnebank, M.; Lutz, H.; Jarre, E.; Vielhaber, S.; Noelker, C.; Struys, E.; Jakobs, C.; Klockgether, T.; Evert, B.O.; Kunz, W.S.; et al. Binding of copper is a mechanism of homocysteine toxicity leading to COX deficiency and apoptosis in primary neurons, PC12 and SHSY-5Y cells. Neurobiol. Dis. 2006, 23, 725–730. [Google Scholar] [CrossRef]
- Zieminska, E.; Matyja, E.; Kozlowska, H.; Stafiej, A.; Lazarewicz, J.W. Excitotoxic neuronal injury in acute homocysteine neurotoxicity: Role of calcium and mitochondrial alterations. Neurochem. Int. 2006, 48, 491–497. [Google Scholar] [CrossRef]
- Chiu, K.M.; Lin, T.Y.; Lee, M.Y.; Lu, C.W.; Wang, M.J.; Wang, S.J. Typhaneoside Suppresses Glutamate Release Through Inhibition of Voltage-Dependent Calcium Entry in Rat Cerebrocortical Nerve Terminals. Chem. Res. Toxicol. 2021, 34, 1286–1295. [Google Scholar] [CrossRef]
- Li, Q.Q.; Chen, J.; Hu, P.; Jia, M.; Sun, J.H.; Feng, H.Y.; Qiao, F.C.; Zang, Y.Y.; Shi, Y.Y.; Chen, G.; et al. Enhancing GluN2A-type NMDA receptors impairs long-term synaptic plasticity and learning and memory. Mol. Psychiatry 2022, 27, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Moyano, P.; Sanjuna, J.; Garcia, J.M.; Garcia, J.; Frejo, M.T.; Naval, M.V.; Del Pino, J. Paraquat Treatment Compromises the Clearance of β-Amyloid and Tau Proteins and Induces Primary Hippocampal Neuronal Cell Death through HSP70, P20S, and TFEB Disruption. Chem. Res. Toxicol. 2021, 34, 1240–1244. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Mehrpour, O.; Forouzanfar, F.; Roshanravan, B.; Samarghandian, S. Oxidative stress and mitochondrial dysfunction in organophosphate pesticide-induced neurotoxicity and its amelioration: A review. Environ. Sci. Pollut. Res. Int. 2020, 27, 24799–24814. [Google Scholar] [CrossRef] [PubMed]
- Prakash, C.; Soni, M.; Kumar, V. Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: A review. J. Appl. Toxicol. 2016, 36, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Garza-Lombó, C.; Pappa, A.; Panayiotidis, M.I.; Gonsebatt, M.E.; Franco, R. Arsenic-induced neurotoxicity: A mechanistic appraisal. J. Biol. Inorg. Chem. 2019, 24, 1305–1316. [Google Scholar] [CrossRef]
- Kumar, V.; Gill, K.D. Oxidative stress and mitochondrial dysfunction in aluminium neurotoxicity and its amelioration: A review. Neurotoxicology 2014, 41, 154–166. [Google Scholar] [CrossRef]
- Murphy, D.; Patel, H.; Wimalasena, K. Caenorhabditis elegans Model Studies Show MPP(+) Is a Simple Member of a Large Group of Related Potent Dopaminergic Toxins. Chem. Res. Toxicol. 2021, 34, 1275–1285. [Google Scholar] [CrossRef]
- Kabil, O.; Banerjee, R. Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 2010, 285, 21903–21907. [Google Scholar] [CrossRef] [Green Version]
- Qu, K.; Lee, S.W.; Bian, J.S.; Low, C.M.; Wong, P.T. Hydrogen sulfide: Neurochemistry and neurobiology. Neurochem. Int. 2008, 52, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Reiffenstein, R.J.; Hulbert, W.C.; Roth, S.H. Toxicology of hydrogen sulfide. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Burnett, W.W.; King, E.G.; Grace, M.; Hall, W.F. Hydrogen sulfide poisoning: Review of 5 years’ experience. Can. Med. Assoc. J. 1977, 117, 1277–1280. [Google Scholar]
- Beauchamp, R.O., Jr.; Bus, J.S.; Popp, J.A.; Boreiko, C.J.; Andjelkovich, D.A. A critical review of the literature on hydrogen sulfide toxicity. Crit. Rev. Toxicol. 1984, 13, 25–97. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; Gould, D.H. Determination of sulfide in brain tissue and rumen fluid by ion-interaction reversed-phase high-performance liquid chromatography. J. Chromatogr. 1990, 526, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef]
- Kimura, H. Hydrogen sulfide and polysulfides as signaling molecules. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 131–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [Green Version]
- Ishigami, M.; Hiraki, K.; Umemura, K.; Ogasawara, Y.; Ishii, K.; Kimura, H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid. Redox Signal. 2009, 11, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Panthi, S.; Chung, H.J.; Jung, J.; Jeong, N.Y. Physiological Importance of Hydrogen Sulfide: Emerging Potent Neuroprotector and Neuromodulator. Oxidative Med. Cell. Longev. 2016, 2016, 9049782. [Google Scholar] [CrossRef] [Green Version]
- Enokido, Y.; Suzuki, E.; Iwasawa, K.; Namekata, K.; Okazawa, H.; Kimura, H. Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 2005, 19, 1854–1856. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Marutani, E.; Ichinose, F. Emerging pharmacological tools to control hydrogen sulfide signaling in critical illness. Intensive Care Med. Exp. 2020, 8, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caliendo, G.; Cirino, G.; Santagada, V.; Wallace, J.L. Synthesis and biological effects of hydrogen sulfide (H2S): Development of H2S-releasing drugs as pharmaceuticals. J. Med. Chem. 2010, 53, 6275–6286. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picton, R.; Eggo, M.C.; Merrill, G.A.; Langman, M.J.; Singh, S. Mucosal protection against sulphide: Importance of the enzyme rhodanese. Gut 2002, 50, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Donnarumma, E.; Trivedi, R.K.; Lefer, D.J. Protective Actions of H2S in Acute Myocardial Infarction and Heart Failure. Compr. Physiol. 2017, 7, 583–602. [Google Scholar]
- Whiteman, M.; Moore, P.K. Hydrogen sulfide and the vasculature: A novel vasculoprotective entity and regulator of nitric oxide bioavailability? J. Cell. Mol. Med. 2009, 13, 488–507. [Google Scholar] [CrossRef]
- González-García, P.; Hidalgo-Gutiérrez, A.; Mascaraque, C.; Barriocanal-Casado, E.; Bakkali, M.; Ziosi, M.; Abdihankyzy, U.B.; Sánchez-Hernández, S.; Escames, G.; Prokisch, H.; et al. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum. Mol. Genet. 2020, 29, 3296–3311. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Fu, M.; Módis, K.; Wang, R.; Ju, Y.; Wu, L. Stimulatory effect of CSE-generated H2S on hepatic mitochondrial biogenesis and the underlying mechanisms. Nitric Oxide 2016, 58, 67–76. [Google Scholar] [CrossRef]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.H.; Moore, P.K. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): New insights into the biology of hydrogen sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Feng, H.; Li, S.; Meng, G.; Liu, S.; Tang, X.; Ma, Y.; Han, Y.; Xiao, Y.; Gu, Y.; et al. SIRT3 Mediates the Antioxidant Effect of Hydrogen Sulfide in Endothelial Cells. Antioxid. Redox Signal. 2016, 24, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Yan, Q.; Qian, L.; Jiang, Y.; Chen, X.; Zeng, S.; Xu, Z.; Gong, Z. S-adenosylmethionine administration inhibits levodopa-induced vascular endothelial growth factor-A expression. Aging 2020, 12, 21290–21307. [Google Scholar] [CrossRef] [PubMed]
- Caro, A.A.; Adlong, L.W.; Crocker, S.J.; Gardner, M.W.; Luikart, E.F.; Gron, L.U. Effect of garlic-derived organosulfur compounds on mitochondrial function and integrity in isolated mouse liver mitochondria. Toxicol. Lett. 2012, 214, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Liu, H.M.; Wei, X.; Gong, X.; Lu, Z.Y.; Huang, Z.H. Diallyl trisulfide attenuates hyperglycemia-induced endothelial apoptosis by inhibition of Drp1-mediated mitochondrial fission. Acta Diabetol. 2019, 56, 1177–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polhemus, D.J.; Li, Z.; Pattillo, C.B.; Gojon, G., Sr.; Gojon, G., Jr.; Giordano, T.; Krum, H. A novel hydrogen sulfide prodrug, SG1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc. Ther. 2015, 33, 216–226. [Google Scholar] [CrossRef]
- Polhemus, D.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef] [Green Version]
- Saif, J.; Ahmad, S.; Rezai, H.; Litvinova, K.; Sparatore, A.; Alzahrani, F.A.; Wang, K.; Ahmed, A. Hydrogen sulfide releasing molecule MZe786 inhibits soluble Flt-1 and prevents preeclampsia in a refined RUPP mouse model. Redox Biol. 2021, 38, 101814. [Google Scholar] [CrossRef]
- Rezai, H.; Ahmad, S.; Alzahrani, F.A.; Sanchez-Aranguren, L.; Dias, I.H.; Agrawal, S.; Sparatore, A.; Wang, K.; Ahmed, A. MZe786, a hydrogen sulfide-releasing aspirin prevents preeclampsia in heme oxygenase-1 haplodeficient pregnancy under high soluble flt-1 environment. Redox Biol. 2021, 38, 101768. [Google Scholar] [CrossRef]
- Sanchez-Aranguren, L.C.; Rezai, H.; Ahmad, S.; Alzahrani, F.A.; Sparatore, A.; Wang, K.; Ahmed, A. MZe786 Rescues Cardiac Mitochondrial Activity in High sFlt-1 and Low HO-1 Environment. Antioxidants 2020, 9, 598. [Google Scholar] [CrossRef] [PubMed]
- De Koning, M.L.Y.; Assa, S.; Maagdenberg, C.G.; van Veldhuisen, D.J.; Pasch, A.; van Goor, H.; Lipsic, E.; van der Harst, P. Safety and Tolerability of Sodium Thiosulfate in Patients with an Acute Coronary Syndrome Undergoing Coronary Angiography: A Dose-Escalation Safety Pilot Study (SAFE-ACS). J. Interv. Cardiol. 2020, 2020, 6014915. [Google Scholar] [CrossRef]
- Szczesny, B.; Módis, K.; Yanagi, K.; Coletta, C.; Le Trionnaire, S.; Perry, A.; Wood, M.E.; Whiteman, M.; Szabo, C. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 2014, 41, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Gerő, D.; Torregrossa, R.; Perry, A.; Waters, A.; Le-Trionnaire, S.; Whatmore, J.L.; Wood, M.; Whiteman, M. The novel mitochondria-targeted hydrogen sulfide (H2S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol. Res. 2016, 113 Pt A, 186–198. [Google Scholar] [CrossRef]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int. J. Med. Sci. 2019, 16, 1386–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabassum, R.; Jeong, N.Y.; Jung, J. Protective effect of hydrogen sulfide on oxidative stress-induced neurodegenerative diseases. Neural Regen. Res. 2020, 15, 232–241. [Google Scholar] [PubMed]
- Tabassum, R.; Jeong, N.Y.; Jung, J. Therapeutic importance of hydrogen sulfide in age-associated neurodegenerative diseases. Neural Regen. Res. 2020, 15, 653–662. [Google Scholar]
- Kumar, M.; Sandhir, R. Hydrogen Sulfide in Physiological and Pathological Mechanisms in Brain. CNS Neurol. Disord. Drug Targets 2018, 17, 654–670. [Google Scholar] [CrossRef]
- Hu, L.F.; Wong, P.T.; Moore, P.K.; Bian, J.S. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. J. Neurochem. 2007, 100, 1121–1128. [Google Scholar] [CrossRef]
- Lee, M.; Schwab, C.; Yu, S.; McGeer, E.; McGeer, P.L. Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide. Neurobiol. Aging 2009, 30, 1523–1534. [Google Scholar] [CrossRef]
- Aschner, M.; Skalny, A.V.; Ke, T.; da Rocha, J.B.; Paoliello, M.M.; Santamaria, A.; Bornhorst, J.; Rongzhu, L.; Svistunov, A.A.; Djordevic, A.B.; et al. Hydrogen sulfide (H2S) signaling as a protective mechanism against endogenous and exogenous neurotoxicants. Curr. Neuropharmacol. 2022, 20, 1908–1924. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.F.; Lu, M.; Wu, Z.Y.; Wong, P.T.; Bian, J.S. Hydrogen sulfide inhibits rotenone-induced apoptosis via preservation of mitochondrial function. Mol. Pharmacol. 2009, 75, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis. 2013, 36, 197–209. [Google Scholar] [CrossRef]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Eto, K.; Asada, T.; Arima, K.; Makifuchi, T.; Kimura, H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 293, 1485–1488. [Google Scholar] [CrossRef]
- Liu, X.Q.; Liu, X.Q.; Jiang, P.; Huang, H.; Yan, Y. Plasma levels of endogenous hydrogen sulfide and homocysteine in patients with Alzheimer’s disease and vascular dementia and the significance thereof. Zhonghua Yi Xue Za Zhi 2008, 88, 2246–2249. [Google Scholar]
- Peng, S.Y.; Wu, X.; Lu, T.; Cui, G.; Chen, G. Research progress of hydrogen sulfide in Alzheimer’s disease from laboratory to hospital: A narrative review. Med. Gas Res. 2020, 10, 125–129. [Google Scholar] [CrossRef]
- McCarty, M.F.; O’Keefe, J.H.; DiNicolantonio, J.J. A diet rich in taurine, cysteine, folate, B(12) and betaine may lessen risk for Alzheimer’s disease by boosting brain synthesis of hydrogen sulfide. Med. Hypotheses 2019, 132, 109356. [Google Scholar] [CrossRef]
- Tang, X.Q.; Shen, X.T.; Huang, Y.E.; Ren, Y.K.; Chen, R.Q.; Hu, B.; He, J.Q.; Yin, W.L.; Xu, J.H.; Jiang, Z.S. Hydrogen sulfide antagonizes homocysteine-induced neurotoxicity in PC12 cells. Neurosci. Res. 2010, 68, 241–249. [Google Scholar] [CrossRef]
- Tang, X.Q.; Ren, Y.K.; Zhou, C.F.; Yang, C.T.; Gu, H.F.; He, J.Q.; Chen, R.Q.; Zhuang, Y.Y.; Fang, H.R.; Wang, C.Y. Hydrogen sulfide prevents formaldehyde-induced neurotoxicity to PC12 cells by attenuation of mitochondrial dysfunction and pro-apoptotic potential. Neurochem. Int. 2012, 61, 16–24. [Google Scholar] [CrossRef]
- Zhao, F.L.; Fang, F.; Qiao, P.F.; Yan, N.; Gao, D.; Yan, Y. AP39, a Mitochondria-Targeted Hydrogen Sulfide Donor, Supports Cellular Bioenergetics and Protects against Alzheimer’s Disease by Preserving Mitochondrial Function in APP/PS1 Mice and Neurons. Oxidative Med. Cell. Longev. 2016, 2016, 8360738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Módis, K.; Ju, Y.; Ahmad, A.; Untereiner, A.A.; Altaany, Z.; Wu, L.; Szabo, C.; Wang, R. S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol. Res. 2016, 113 Pt A, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.J.; Li, X.; Tang, X.Q. Therapeutic benefits of H₂S in Alzheimer’s disease. J. Clin. Neurosci. 2014, 21, 1665–1669. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, Y.; Liu, H.; Yin, C.; Li, X.; Gong, Q. Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: A novel mechanism mediated by the activation of Nrf2. Pharmacol. Biochem. Behav. 2016, 150–151, 207–216. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kyles, P.; Kalani, A.; Tyagi, N. Hydrogen Sulfide Ameliorates Homocysteine-Induced Alzheimer’s Disease-Like Pathology, Blood-Brain Barrier Disruption, and Synaptic Disorder. Mol. Neurobiol. 2016, 53, 2451–2467. [Google Scholar] [CrossRef]
- Tang, X.Q.; Chen, R.Q.; Ren, Y.K.; Soldato, P.D.; Sparatore, A.; Zhuang, Y.Y.; Fang, H.R.; Wang, C.Y. ACS6, a Hydrogen sulfide-donating derivative of sildenafil, inhibits homocysteine-induced apoptosis by preservation of mitochondrial function. Med. Gas Res. 2011, 1, 20. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Y.; Bian, J.S. Hydrogen sulfide protects amyloid-β induced cell toxicity in microglia. J. Alzheimers Dis. 2010, 22, 1189–1200. [Google Scholar] [CrossRef]
- Fan, H.; Guo, Y.; Liang, X.; Yuan, Y.; Qi, X.; Wang, M.; Ma, J.; Zhou, H. Hydrogen sulfide protects against amyloid beta-peptide induced neuronal injury via attenuating inflammatory responses in a rat model. J. Biomed. Res. 2013, 27, 296–304. [Google Scholar]
- Cao, L.; Cao, X.; Zhou, Y.; Nagpure, B.V.; Wu, Z.Y.; Hu, L.F.; Yang, Y.; Sethi, G.; Moore, P.K.; Bian, J.S. Hydrogen sulfide inhibits ATP-induced neuroinflammation and Aβ(1-42) synthesis by suppressing the activation of STAT3 and cathepsin S. Brain Behav. Immun. 2018, 73, 603–614. [Google Scholar] [CrossRef] [PubMed]
- He, X.L.; Yan, N.; Zhang, H.; Qi, Y.W.; Zhu, L.J.; Liu, M.J.; Yan, Y. Hydrogen sulfide improves spatial memory impairment and decreases production of Aβ in APP/PS1 transgenic mice. Neurochem. Int. 2014, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.L.; Qiao, P.F.; Yan, N.; Gao, D.; Liu, M.J.; Yan, Y. Hydrogen Sulfide Selectively Inhibits γ-Secretase Activity and Decreases Mitochondrial Aβ Production in Neurons from APP/PS1 Transgenic Mice. Neurochem. Res. 2016, 41, 1145–1159. [Google Scholar] [CrossRef]
- Vandini, E.; Ottani, A.; Zaffe, D.; Calevro, A.; Canalini, F.; Cavallini, G.M.; Rossi, R.; Guarini, S.; Giuliani, D. Mechanisms of Hydrogen Sulfide against the Progression of Severe Alzheimer’s Disease in Transgenic Mice at Different Ages. Pharmacology 2019, 103, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3β and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236; discussion 222. [Google Scholar] [CrossRef]
- Shefa, U.; Kim, M.S.; Jeong, N.Y.; Jung, J. Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxidative Med. Cell. Longev. 2018, 2018, 1873962. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, J.; Wang, H. Hydrogen sulfide alleviates oxidative stress injury and reduces apoptosis induced by MPP(+) in Parkinson’s disease cell model. Mol. Cell Biochem. 2020, 472, 231–240. [Google Scholar] [CrossRef]
- Hu, L.F.; Lu, M.; Tiong, C.X.; Dawe, G.S.; Hu, G.; Bian, J.S. Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell 2010, 9, 135–146. [Google Scholar] [CrossRef]
- Jiang, W.; Zou, W.; Hu, M.; Tian, Q.; Xiao, F.; Li, M.; Zhang, P.; Chen, Y.J.; Jiang, J.M. Hydrogen sulphide attenuates neuronal apoptosis of substantia nigra by re-establishing autophagic flux via promoting leptin signalling in a 6-hydroxydopamine rat model of Parkinson’s disease. Clin. Exp. Pharmacol. Physiol. 2022, 49, 122–133. [Google Scholar] [CrossRef]
- Yuan, Y.Q.; Wang, Y.L.; Yuan, B.S.; Yuan, X.; Hou, X.O.; Bian, J.S.; Liu, C.F.; Hu, L.F. Impaired CBS-H2S signaling axis contributes to MPTP-induced neurodegeneration in a mouse model of Parkinson’s disease. Brain Behav. Immun. 2018, 67, 77–90. [Google Scholar] [CrossRef]
- Tang, X.Q.; Fan, L.L.; Li, Y.J.; Shen, X.T.; Zhuan, Y.Y.; He, J.Q.; Xu, J.H.; Hu, B.; Li, Y.J. Inhibition of hydrogen sulfide generation contributes to 1-methy-4-phenylpyridinium ion-induced neurotoxicity. Neurotox. Res. 2011, 19, 403–411. [Google Scholar] [CrossRef]
- Hou, X.; Yuan, Y.; Sheng, Y.; Yuan, B.; Wang, Y.; Zheng, J.; Liu, C.F.; Zhang, X.; Hu, L.F. GYY4137, an H2S Slow-Releasing Donor, Prevents Nitrative Stress and α-Synuclein Nitration in an MPTP Mouse Model of Parkinson’s Disease. Front. Pharmacol. 2017, 8, 741. [Google Scholar] [CrossRef] [Green Version]
- Murros, K.E. Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease. Cells 2022, 11, 978. [Google Scholar] [CrossRef]
- Kida, K.; Yamada, M.; Tokuda, K.; Marutani, E.; Kakinohana, M.; Kaneki, M.; Ichinose, F. Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson’s disease. Antioxid. Redox Signal. 2011, 15, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakmak, Y.O. Rotorua, hydrogen sulphide and Parkinson’s disease-A possible beneficial link? N. Z. Med. J. 2017, 130, 123–125. [Google Scholar] [PubMed]
- Li, J.; Li, M.; Wang, C.; Zhang, S.; Gao, Q.; Wang, L.; Ma, L. NaSH increases SIRT1 activity and autophagy flux through sulfhydration to protect SH-SY5Y cells induced by MPP+. Cell Cycle 2020, 19, 2216–2225. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef] [Green Version]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 2014, 509, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.D.; Snyder, S.H. Neurodegeneration in Huntington’s disease involves loss of cystathionine γ-lyase. Cell Cycle 2014, 13, 2491–2493. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.J.; Gray, L.J.; Finkelstein, D.I.; Crouch, P.J.; Pow, D.; Pang, T.Y.; Li, S.; Smith, Z.M.; Francis, P.S.; Renoir, T.; et al. N-acetylcysteine modulates glutamatergic dysfunction and depressive behavior in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 2923–2933. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D. Cysteine metabolism and hydrogen sulfide signaling in Huntington’s disease. Free Radic. Biol. Med. 2022, 186, 93–98. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170.e1113. [Google Scholar] [CrossRef] [PubMed]
- Boutell, J.M.; Wood, J.D.; Harper, P.S.; Jones, A.L. Huntingtin interacts with cystathionine beta-synthase. Hum. Mol. Genet. 1998, 7, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, R.A.; Mansour, S.M. Sodium hydrogen sulfide upregulates cystathionine β-synthase and protects striatum against 3-nitropropionic acid-induced neurotoxicity in rats. J. Pharm. Pharmacol. 2021, 73, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Dubinsky, R.; Gray, C. CYTE-I-HD: Phase I dose finding and tolerability study of cysteamine (Cystagon) in Huntington’s disease. Mov. Disord. 2006, 21, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.L.; Yi, R.; Dong, Q.; Yao, L.F.; Liu, B. The relationship between diabetes-related cognitive dysfunction and leukoaraiosis. Acta Neurol. Belg. 2021, 121, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Liu, Z.; Chen, Y.; Xu, Y.; Qin, J.; Guo, S.; Huang, J.; Tao, J. The prevalence of mild cognitive impairment in type 2 diabetes mellitus patients: A systematic review and meta-analysis. Acta Diabetol. 2021, 58, 671–685. [Google Scholar] [CrossRef]
- Kong, F.J.; Ma, L.L.; Guo, J.J.; Xu, L.H.; Li, Y.; Qu, S. Endoplasmic reticulum stress/autophagy pathway is involved in diabetes-induced neuronal apoptosis and cognitive decline in mice. Clin. Sci. 2018, 132, 111–125. [Google Scholar] [CrossRef]
- Wu, X.L.; Deng, M.Z.; Gao, Z.J.; Dang, Y.Y.; Li, Y.C.; Li, C.W. Neferine alleviates memory and cognitive dysfunction in diabetic mice through modulation of the NLRP3 inflammasome pathway and alleviation of endoplasmic-reticulum stress. Int. Immunopharmacol. 2020, 84, 106559. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Meng, X.; Zhai, Y.; Xie, W.; Wang, R.; Sun, G.; Sun, X. Gastrodin Ameliorates Cognitive Dysfunction in Diabetes Rat Model via the Suppression of Endoplasmic Reticulum Stress and NLRP3 Inflammasome Activation. Front. Pharmacol. 2018, 9, 1346. [Google Scholar] [CrossRef]
- Trujillo-Estrada, L.; Nguyen, C.; da Cunha, C.; Cai, L.; Forner, S.; Martini, A.C.; Ager, R.R.; Prieto, G.A.; Cotman, C.W.; Baglietto-Vargas, D. Tau underlies synaptic and cognitive deficits for type 1, but not type 2 diabetes mouse models. Aging Cell 2019, 18, e12919. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.J.; Lin, C.C.; Yeh, C.M.; Chien, M.E.; Tsao, M.C.; Tseng, P.; Huang, C.W.; Hsu, K.S. Repeated transcranial direct current stimulation improves cognitive dysfunction and synaptic plasticity deficit in the prefrontal cortex of streptozotocin-induced diabetic rats. Brain Stimul. 2017, 10, 1079–1087. [Google Scholar] [CrossRef]
- Szabo, C. Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signal. 2012, 17, 68–80. [Google Scholar] [CrossRef]
- Gheibi, S.; Samsonov, A.P.; Gheibi, S.; Vazquez, A.B.; Kashfi, K. Regulation of carbohydrate metabolism by nitric oxide and hydrogen sulfide: Implications in diabetes. Biochem. Pharmacol. 2020, 176, 113819. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Yuan, J.; Tang, Z.J.; Wei, H.J.; Zhu, W.W.; Zhang, P.; Gu, H.F.; Wang, C.Y.; Tang, X.Q. Hydrogen sulfide ameliorates cognitive dysfunction in streptozotocin-induced diabetic rats: Involving suppression in hippocampal endoplasmic reticulum stress. Oncotarget 2017, 8, 64203–64216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Chen, Z.; Kang, X.; Wu, L.; Jiang, J.M.; Liu, S.M.; Wei, H.J.; Chen, Y.J.; Zou, W.; Wang, C.Y.; et al. SIRT1 Mediates H(2)S-Ameliorated Diabetes-Associated Cognitive Dysfunction in Rats: Possible Involvement of Inhibiting Hippocampal Endoplasmic Reticulum Stress and Synaptic Dysfunction. Neurochem. Res. 2021, 46, 611–623. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, L.; Lu, B.; Wen, J.; Wang, M.; Zhang, S.; Li, Q.; Shu, F.; Lu, F.; Liu, N.; et al. Hydrogen sulphide reduced the accumulation of lipid droplets in cardiac tissues of db/db mice via Hrd1 S-sulfhydration. J. Cell. Mol. Med. 2021, 25, 9154–9167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.; Gong, D.; Yang, Y.; Liu, X.; Chen, Z. Inhibition of the SIRT1 signaling pathway exacerbates endoplasmic reticulum stress induced by renal ischemia/reperfusion injury in type 1 diabetic rats. Mol. Med. Rep. 2020, 21, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Chen, Q.; Tian, X.; Qian, N.; Chai, P.; Liu, B.; Hu, J.; Blackstone, C.; Zhu, D.; Teng, J.; et al. DNA damage triggers tubular endoplasmic reticulum extension to promote apoptosis by facilitating ER-mitochondria signaling. Cell Res. 2018, 28, 833–854. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Zhong, D.; Ma, P.; Li, G.; Hua, W.; Sun, Y.; Liu, N.; Zhang, L.; Zhang, W. Exogenous Hydrogen Sulfide Ameliorates Diabetes-Associated Cognitive Decline by Regulating the Mitochondria-Mediated Apoptotic Pathway and IL-23/IL-17 Expression in db/db Mice. Cell. Physiol. Biochem. 2017, 41, 1838–1850. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Davoli, A.; Greco, V.; Spalloni, A.; Guatteo, E.; Neri, C.; Rizzo, G.R.; Cordella, A.; Romigi, A.; Cortese, C.; Bernardini, S.; et al. Evidence of hydrogen sulfide involvement in amyotrophic lateral sclerosis. Ann. Neurol. 2015, 77, 697–709. [Google Scholar] [CrossRef]
- Greco, V.; Spalloni, A.; Corasolla Carregari, V.; Pieroni, L.; Persichilli, S.; Mercuri, N.B.; Urbani, A.; Longone, P. Proteomics and Toxicity Analysis of Spinal-Cord Primary Cultures upon Hydrogen Sulfide Treatment. Antioxidants 2018, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Spalloni, A.; Greco, V.; Ciriminna, G.; Corasolla Carregari, V.; Marini, F.; Pieroni, L.; Mercuri, N.B.; Urbani, A.; Longone, P. Impact of Pharmacological Inhibition of Hydrogen Sulphide Production in the SOD1G93A-ALS Mouse Model. Int. J. Mol. Sci. 2019, 20, 2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, P.; Shrestha, B.; Kaskas, A.M.; Green, J.; Alexander, J.S.; Pattillo, C.B. Hydrogen sulfide: Therapeutic or injurious in ischemic stroke? Pathophysiology 2019, 26, 1–10. [Google Scholar] [CrossRef]
- Chan, S.J.; Wong, P.T. Hydrogen sulfide in stroke: Protective or deleterious? Neurochem. Int. 2017, 105, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Wang, Z.; Chen, G. The role of hydrogen sulfide in stroke. Med. Gas Res. 2016, 6, 79–84. [Google Scholar] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Wang, Q. Advances of H2S in Regulating Neurodegenerative Diseases by Preserving Mitochondria Function. Antioxidants 2023, 12, 652. https://doi.org/10.3390/antiox12030652
Zhou L, Wang Q. Advances of H2S in Regulating Neurodegenerative Diseases by Preserving Mitochondria Function. Antioxidants. 2023; 12(3):652. https://doi.org/10.3390/antiox12030652
Chicago/Turabian StyleZhou, Lina, and Qiang Wang. 2023. "Advances of H2S in Regulating Neurodegenerative Diseases by Preserving Mitochondria Function" Antioxidants 12, no. 3: 652. https://doi.org/10.3390/antiox12030652