
Selenium Protects Mouse Hypothalamic Cells from Glucocorticoid-Induced Endoplasmic Reticulum Stress Vulnerability and Insulin Signaling Impairment
 , and
, and 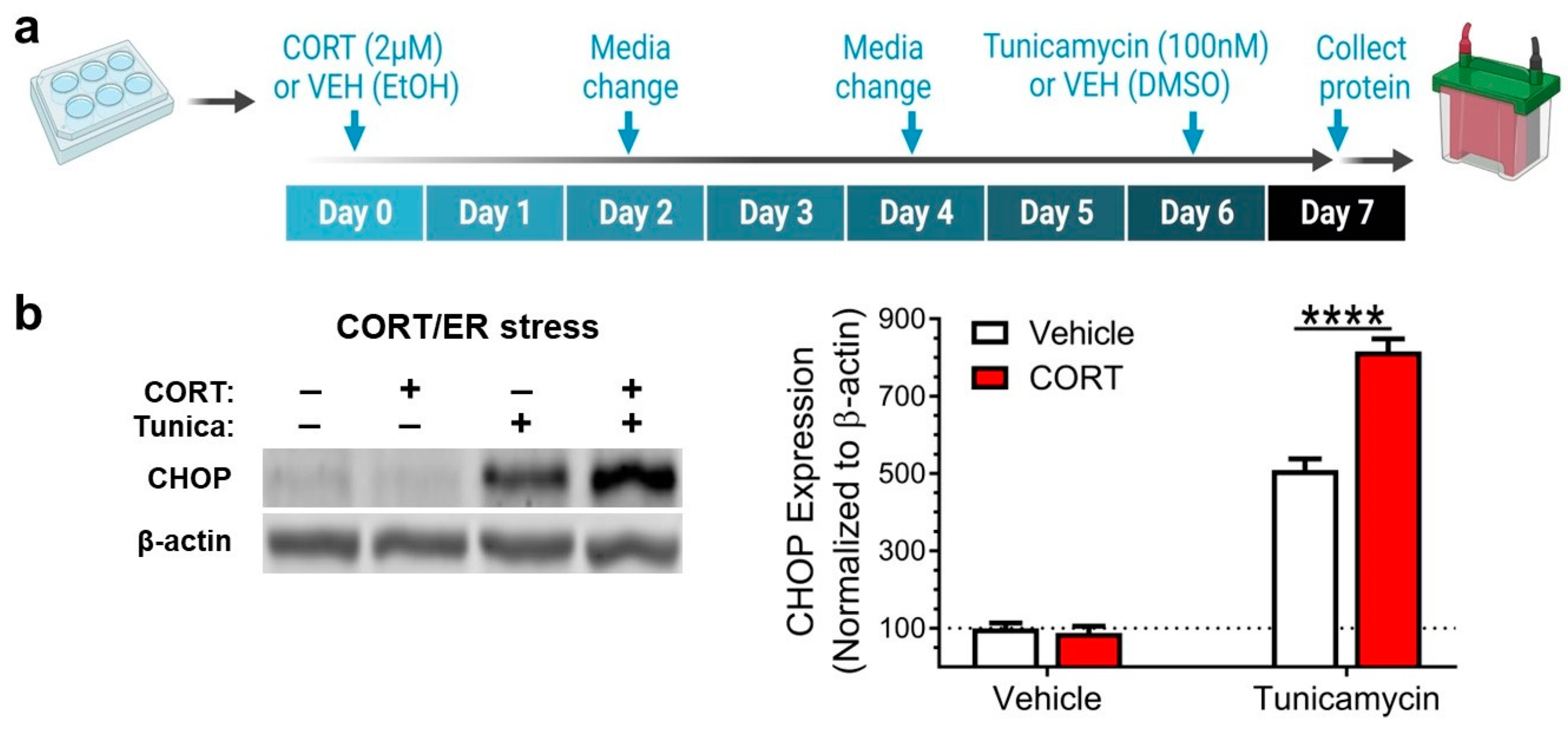{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Chemicals
2.2. Cell Culture
2.3. Western Blotting
2.4. Statistical Analysis
3. Results
3.1. Corticosterone Potentiates Endoplasmic Reticulum Stress Induction
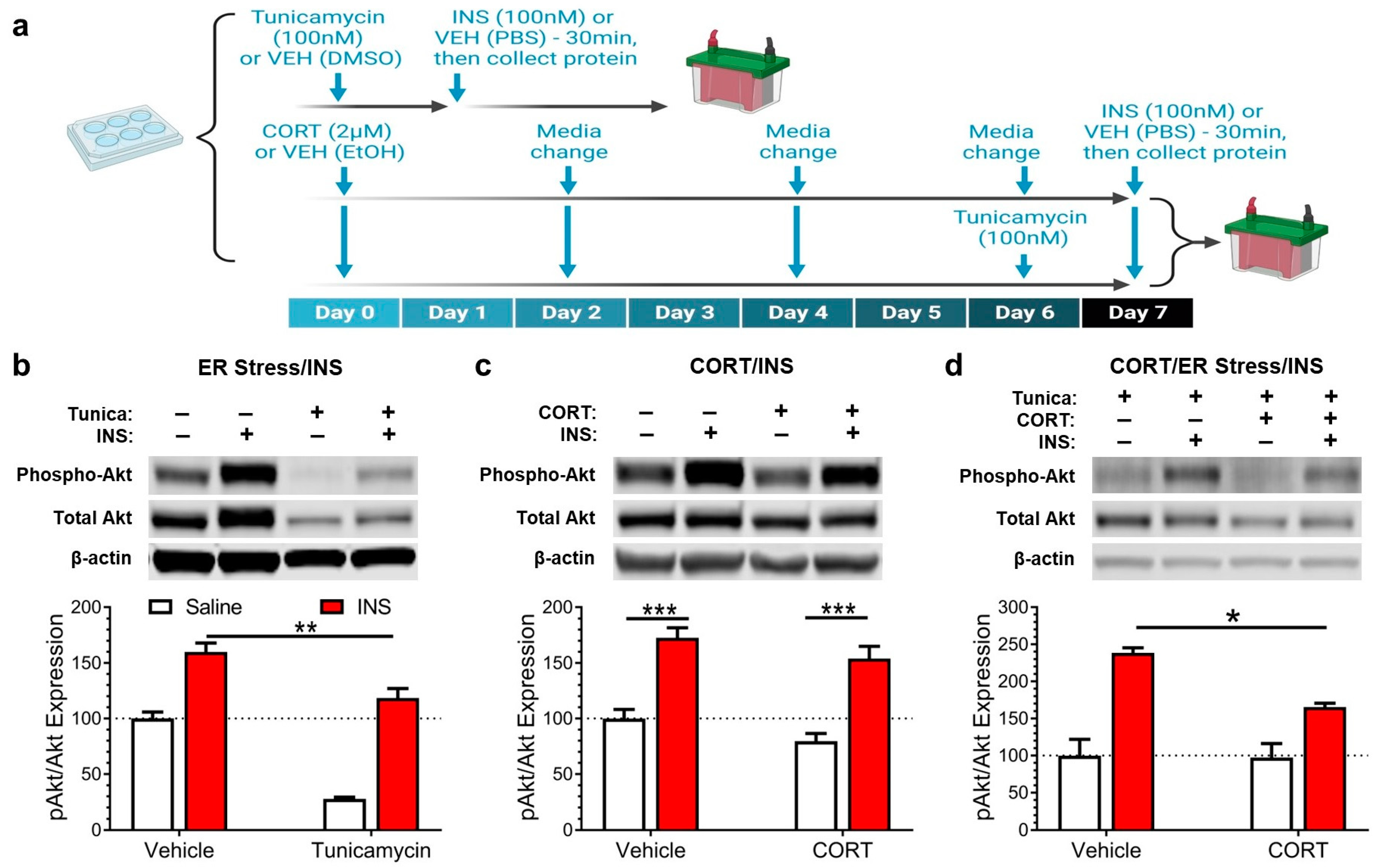
3.2. Corticosterone Exacerbates the Insulin Signaling Impairment Caused by Endoplasmic Reticulum Stress
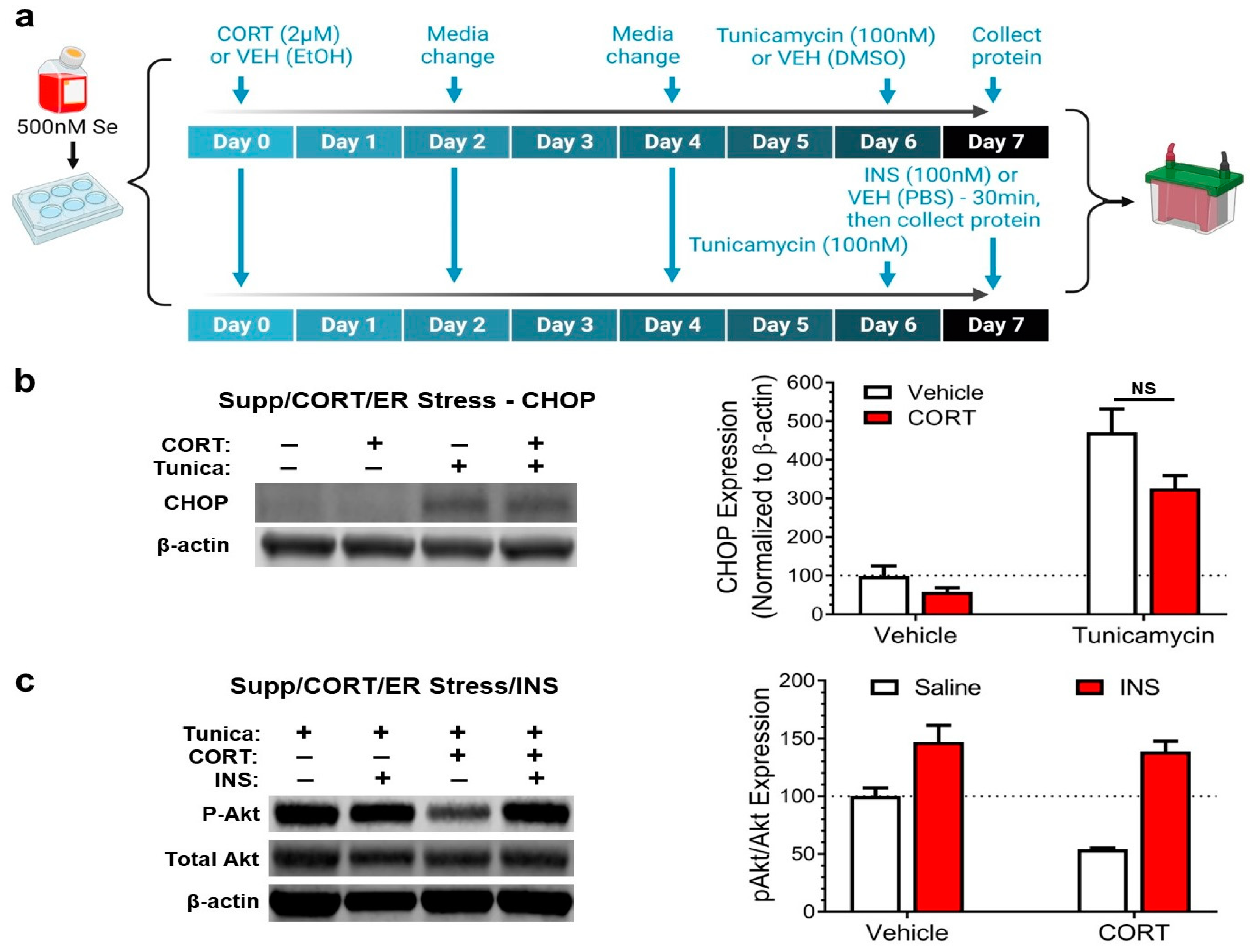
3.3. Selenium Supplementation Prevents Corticosterone from Exacerbating Endoplasmic Reticulum Stress and Impairing Insulin Signaling
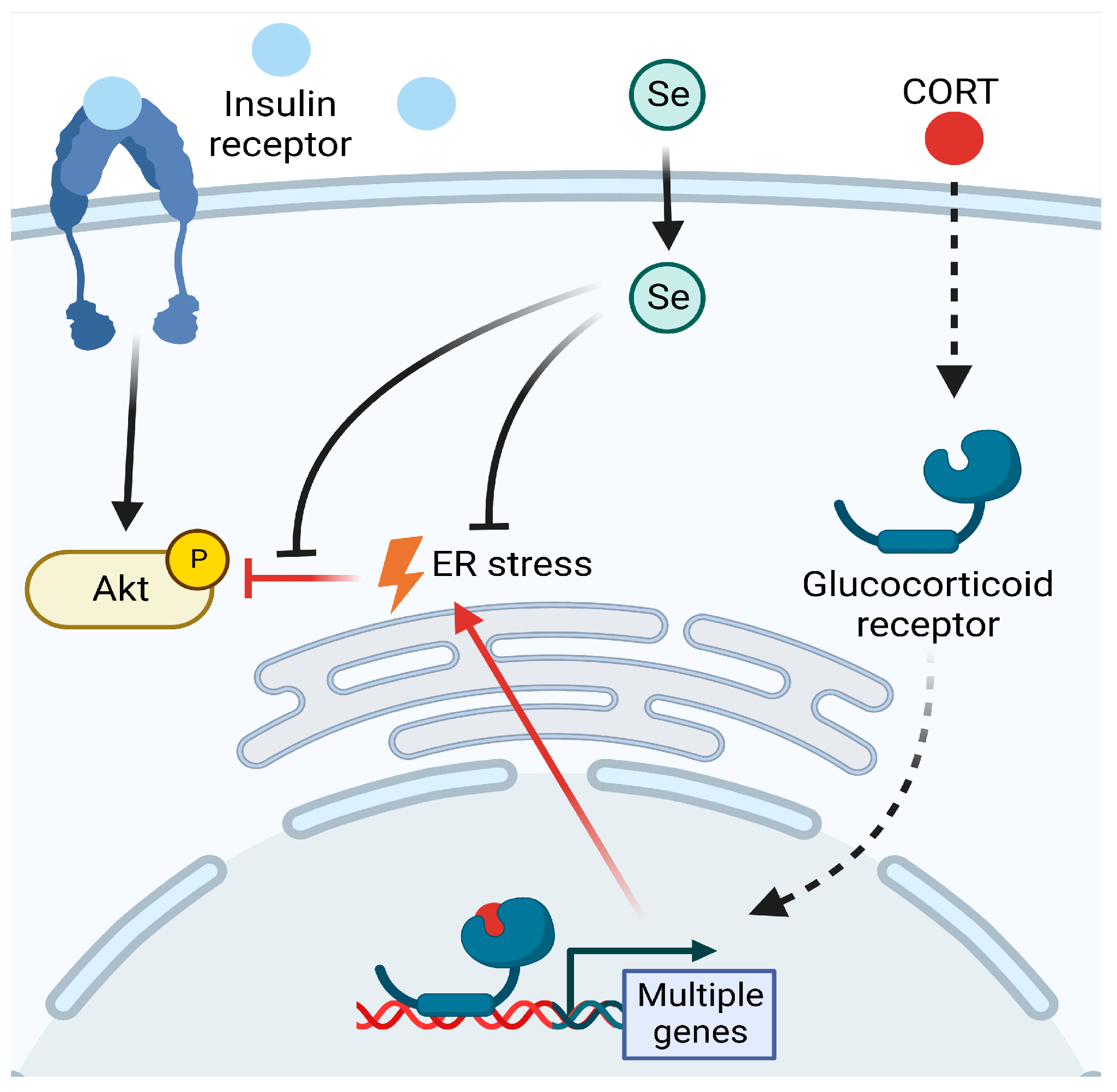
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Leistner, C.; Menke, A. Hypothalamic-pituitary-adrenal axis and stress. Handb. Clin. Neurol. 2020, 175, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Overman, R.A.; Yeh, J.Y.; Deal, C.L. Prevalence of oral glucocorticoid usage in the United States: A general population perspective. Arthritis Care Res. (Hoboken) 2013, 65, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Oray, M.; Abu Samra, K.; Ebrahimiadib, N.; Meese, H.; Foster, C.S. Long-term side effects of glucocorticoids. Expert Opin. Drug Saf. 2016, 15, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Wray, J.R.; Davies, A.; Sefton, C.; Allen, T.J.; Adamson, A.; Chapman, P.; Lam, B.Y.H.; Yeo, G.S.H.; Coll, A.P.; Harno, E.; et al. Global transcriptomic analysis of the arcuate nucleus following chronic glucocorticoid treatment. Mol. Metab. 2019, 26, 5–17. [Google Scholar] [CrossRef]
- Cakir, I.; Nillni, E.A. Endoplasmic Reticulum Stress, the Hypothalamus, and Energy Balance. Trends Endocrinol. Metab. 2019, 30, 163–176. [Google Scholar] [CrossRef]
- Spiers, J.G.; Chen, H.J.; Sernia, C.; Lavidis, N.A. Activation of the hypothalamic-pituitary-adrenal stress axis induces cellular oxidative stress. Front. Neurosci. 2014, 8, 456. [Google Scholar] [CrossRef] [Green Version]
- You, J.M.; Yun, S.J.; Nam, K.N.; Kang, C.; Won, R.; Lee, E.H. Mechanism of glucocorticoid-induced oxidative stress in rat hippocampal slice cultures. Can. J. Physiol. Pharmacol. 2009, 87, 440–447. [Google Scholar] [CrossRef]
- Sunena; Mishra, D.N. Stress Etiology of Type 2 Diabetes. Curr. Diabetes Rev. 2022, 18, e240222201413. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Duntas, L.H.; Rayman, M.P. The role of selenium in type-2 diabetes mellitus and its metabolic comorbidities. Redox Biol. 2022, 50, 102236. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Combs, G.F.; Wu, T.-L.; Zeng, H.; Cheng, W.-H. Selenium status and type 2 diabetes risk. Arch. Biochem. Biophys. 2022, 730, 109400. [Google Scholar] [CrossRef]
- Schweizer, U.; Bohleber, S.; Zhao, W.; Fradejas-Villar, N. The Neurobiology of Selenium: Looking Back and to the Future. Front. Neurosci. 2021, 15, 652099. [Google Scholar] [CrossRef] [PubMed]
- Toh, P.; Nicholson, J.L.; Vetter, A.M.; Berry, M.J.; Torres, D.J. Selenium in Bodily Homeostasis: Hypothalamus, Hormones, and Highways of Communication. Int. J. Mol. Sci. 2022, 23, 15445. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.L.; Toh, P.; Alfulaij, N.; Berry, M.J.; Torres, D.J. New insights on selenoproteins and neuronal function. Free. Radic. Biol. Med. 2022, 190, 55–61. [Google Scholar] [CrossRef]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Datta, S.; McEwen, B.S.; Chattarji, S. Corticosterone after acute stress prevents the delayed effects on the amygdala. Neuropsychopharmacology 2020, 45, 2139–2146. [Google Scholar] [CrossRef]
- Chan, S.W.; Egan, P.A. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005, 19, 1510–1512. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Mayer, C.M.; Belsham, D.D. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: Rescue of resistance and apoptosis through adenosine 5′ monophosphate-activated protein kinase activation. Endocrinology 2010, 151, 576–585. [Google Scholar] [CrossRef]
- Liu, Z.; Fei, B.; Xie, L.; Liu, J.; Chen, X.; Zhu, W.; Lv, L.; Ma, W.; Gao, Z.; Hou, J.; et al. Glucocorticoids protect HEI-OC1 cells from tunicamycin-induced cell damage via inhibiting endoplasmic reticulum stress. Open Life Sci. 2021, 16, 695–702. [Google Scholar] [CrossRef]
- Hu, D.D.; Mai, J.N.; He, L.Y.; Li, P.Q.; Chen, W.X.; Yan, J.J.; Zhu, W.D.; Deng, L.; Wei, D.; Liu, D.H.; et al. Glucocorticoids Prevent Enterovirus 71 Capsid Protein VP1 Induced Calreticulin Surface Exposure by Alleviating Neuronal ER Stress. Neurotox. Res. 2017, 31, 204–217. [Google Scholar] [CrossRef]
- Guo, Y.; Hao, D.; Hu, H. High doses of dexamethasone induce endoplasmic reticulum stress-mediated apoptosis by promoting calcium ion influx-dependent CHOP expression in osteoblasts. Mol. Biol. Rep. 2021, 48, 7841–7851. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Liu, X.; Gong, F.; Ding, X.; Zhou, X.; Liu, C.; Zhao, F.; Li, X.; Shi, J. Dexamethasone promotes the endoplasmic reticulum stress response of bone marrow mesenchymal stem cells by activating the PERK-Nrf2 signaling pathway. Pharmacol. Res. Perspect. 2021, 9, e00791. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Kim, H.C.; Kim, H.U.; Park, T.; Park, J.; Kim, U.; Kim, M.K.; Jeong, J.H. Asprosin attenuates insulin signaling pathway through PKCdelta-activated ER stress and inflammation in skeletal muscle. J. Cell. Physiol. 2019, 234, 20888–20899. [Google Scholar] [CrossRef] [PubMed]
- Roh, E.; Hwang, H.J.; Kim, J.W.; Hong, S.H.; Kim, J.A.; Lee, Y.B.; Choi, K.M.; Baik, S.H.; Yoo, H.J. Ginsenoside Mc1 improves liver steatosis and insulin resistance by attenuating ER stress. J. Ethnopharmacol. 2020, 259, 112927. [Google Scholar] [CrossRef] [PubMed]
- Park, T.J.; Park, S.Y.; Lee, H.J.; Abd El-Aty, A.M.; Jeong, J.H.; Jung, T.W. alpha-ketoisocaproic acid promotes ER stress through impairment of autophagy, thereby provoking lipid accumulation and insulin resistance in murine preadipocytes. Biochem. Biophys. Res. Commun. 2022, 603, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Torres, D.J.; Berry, M.J.; Pitts, M.W. Hypothalamic redox balance and leptin signaling—Emerging role of selenoproteins. Free. Radic. Biol. Med. 2018, 127, 172–181. [Google Scholar] [CrossRef]
- Cheng, H.; Gang, X.; He, G.; Liu, Y.; Wang, Y.; Zhao, X.; Wang, G. The Molecular Mechanisms Underlying Mitochondria-Associated Endoplasmic Reticulum Membrane-Induced Insulin Resistance. Front. Endocrinol. (Lausanne) 2020, 11, 592129. [Google Scholar] [CrossRef]
- Schomburg, L.; Schweizer, U.; Holtmann, B.; Flohe, L.; Sendtner, M.; Kohrle, J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 2003, 370, 397–402. [Google Scholar] [CrossRef] [Green Version]
- Leiter, O.; Zhuo, Z.; Rust, R.; Wasielewska, J.M.; Gronnert, L.; Kowal, S.; Overall, R.W.; Adusumilli, V.S.; Blackmore, D.G.; Southon, A.; et al. Selenium mediates exercise-induced adult neurogenesis and reverses learning deficits induced by hippocampal injury and aging. Cell Metab. 2022, 34, 408–423.e408. [Google Scholar] [CrossRef]
- Watanabe, L.M.; Hashimoto, A.C.; Torres, D.J.; Alfulaij, N.; Peres, R.; Sultana, R.; Maunakea, A.K.; Berry, M.J.; Seale, L.A. Effect of statin treatment in obese selenium-supplemented mice lacking selenocysteine lyase. Mol. Cell. Endocrinol. 2021, 533, 111335. [Google Scholar] [CrossRef]
- Qiao, L.; Men, L.; Yu, S.; Yao, J.; Li, Y.; Wang, M.; Yu, Y.; Wang, N.; Ran, L.; Wu, Y.; et al. Hepatic deficiency of selenoprotein S exacerbates hepatic steatosis and insulin resistance. Cell Death Dis. 2022, 13, 275. [Google Scholar] [CrossRef] [PubMed]
- Tinkov, A.A.; Ajsuvakova, O.P.; Filippini, T.; Zhou, J.C.; Lei, X.G.; Gatiatulina, E.R.; Michalke, B.; Skalnaya, M.G.; Vinceti, M.; Aschner, M.; et al. Selenium and Selenoproteins in Adipose Tissue Physiology and Obesity. Biomolecules 2020, 10, 658. [Google Scholar] [CrossRef] [PubMed]
- Takamura, T. Hepatokine Selenoprotein P-Mediated Reductive Stress Causes Resistance to Intracellular Signal Transduction. Antioxid. Redox Signal. 2020, 33, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.J.; Alfulaij, N.; Berry, M.J. Stress and the Brain: An Emerging Role for Selenium. Front. Neurosci. 2021, 15, 666601. [Google Scholar] [CrossRef]
- Birmann, P.T.; Domingues, M.; Casaril, A.M.; Smaniotto, T.A.; Hartwig, D.; Jacob, R.G.; Savegnago, L. A pyrazole-containing selenium compound modulates neuroendocrine, oxidative stress, and behavioral responses to acute restraint stress in mice. Behav. Brain Res. 2021, 396, 112874. [Google Scholar] [CrossRef]
- Casaril, A.M.; Lourenco, D.A.; Domingues, M.; Smaniotto, T.A.; Birmann, P.T.; Vieira, B.; Sonego, M.S.; Seixas, F.K.; Collares, T.; Lenardao, E.J.; et al. Anhedonic- and anxiogenic-like behaviors and neurochemical alterations are abolished by a single administration of a selenium-containing compound in chronically stressed mice. Compr. Psychoneuroendocrinol. 2021, 6, 100054. [Google Scholar] [CrossRef]
- Beytut, E.; Yilmaz, S.; Aksakal, M.; Polat, S. The possible protective effects of vitamin E and selenium administration in oxidative stress caused by high doses of glucocorticoid administration in the brain of rats. J. Trace Elem. Med. Biol. 2018, 45, 131–135. [Google Scholar] [CrossRef]
- Tanaka, H.; Makino, Y.; Okamoto, K.; Iida, T.; Yan, K.; Yoshikawa, N. Redox regulation of the glucocorticoid receptor. Antioxid. Redox Signal. 1999, 1, 403–423. [Google Scholar] [CrossRef]
- Tashima, Y.; Terui, M.; Itoh, H.; Mizunuma, H.; Kobayashi, R.; Marumo, F. Effect of selenite on glucocorticoid receptor. J. Biochem. 1989, 105, 358–361. [Google Scholar] [CrossRef]
- Rossier, M.F.; Lenglet, S.; Vetterli, L.; Python, M.; Maturana, A. Corticosteroids and redox potential modulate spontaneous contractions in isolated rat ventricular cardiomyocytes. Hypertension 2008, 52, 721–728. [Google Scholar] [CrossRef]
- Psarra, A.M.; Hermann, S.; Panayotou, G.; Spyrou, G. Interaction of mitochondrial thioredoxin with glucocorticoid receptor and NF-kappaB modulates glucocorticoid receptor and NF-kappaB signalling in HEK-293 cells. Biochem. J. 2009, 422, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Rock, C.; Moos, P.J. Selenoprotein P regulation by the glucocorticoid receptor. Biometals 2009, 22, 995–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, B.C.; Jung, N.K.; Park, C.Y.; Oh, I.J.; Choi, Y.D.; Park, J.I.; Lee, S.W. Epigenetic and Glucocorticoid Receptor-Mediated Regulation of Glutathione Peroxidase 3 in Lung Cancer Cells. Mol. Cells 2016, 39, 631–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.Y.; Kim, K.H. Dexamethasone-induced selenoprotein S degradation is required for adipogenesis. J. Lipid Res. 2013, 54, 2069–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Takahashi, T.; Sumitani, K.; Takatsu, H.; Urano, S. Glucocorticoid Generates ROS to Induce Oxidative Injury in the Hippocampus, Leading to Impairment of Cognitive Function of Rats. J. Clin. Biochem. Nutr. 2010, 47, 224–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, K.J.; Hanato, A.N.; Hui, K.W.; Pitts, M.W.; Seale, L.A.; Nicholson, J.L.; Toh, P.; Kim, J.K.; Berry, M.J.; Torres, D.J. Selenium Protects Mouse Hypothalamic Cells from Glucocorticoid-Induced Endoplasmic Reticulum Stress Vulnerability and Insulin Signaling Impairment. Antioxidants 2023, 12, 526. https://doi.org/10.3390/antiox12020526
An KJ, Hanato AN, Hui KW, Pitts MW, Seale LA, Nicholson JL, Toh P, Kim JK, Berry MJ, Torres DJ. Selenium Protects Mouse Hypothalamic Cells from Glucocorticoid-Induced Endoplasmic Reticulum Stress Vulnerability and Insulin Signaling Impairment. Antioxidants. 2023; 12(2):526. https://doi.org/10.3390/antiox12020526
Chicago/Turabian StyleAn, Katlyn J., Ashley N. Hanato, Katherine W. Hui, Matthew W. Pitts, Lucia A. Seale, Jessica L. Nicholson, Pamela Toh, Jun Kyoung Kim, Marla J. Berry, and Daniel J. Torres. 2023. "Selenium Protects Mouse Hypothalamic Cells from Glucocorticoid-Induced Endoplasmic Reticulum Stress Vulnerability and Insulin Signaling Impairment" Antioxidants 12, no. 2: 526. https://doi.org/10.3390/antiox12020526