
Cardiac Hepatopathy: New Perspectives on Old Problems through a Prism of Endogenous Metabolic Regulations by Hepatokines
 ,
,
Abstract
:1. Introduction
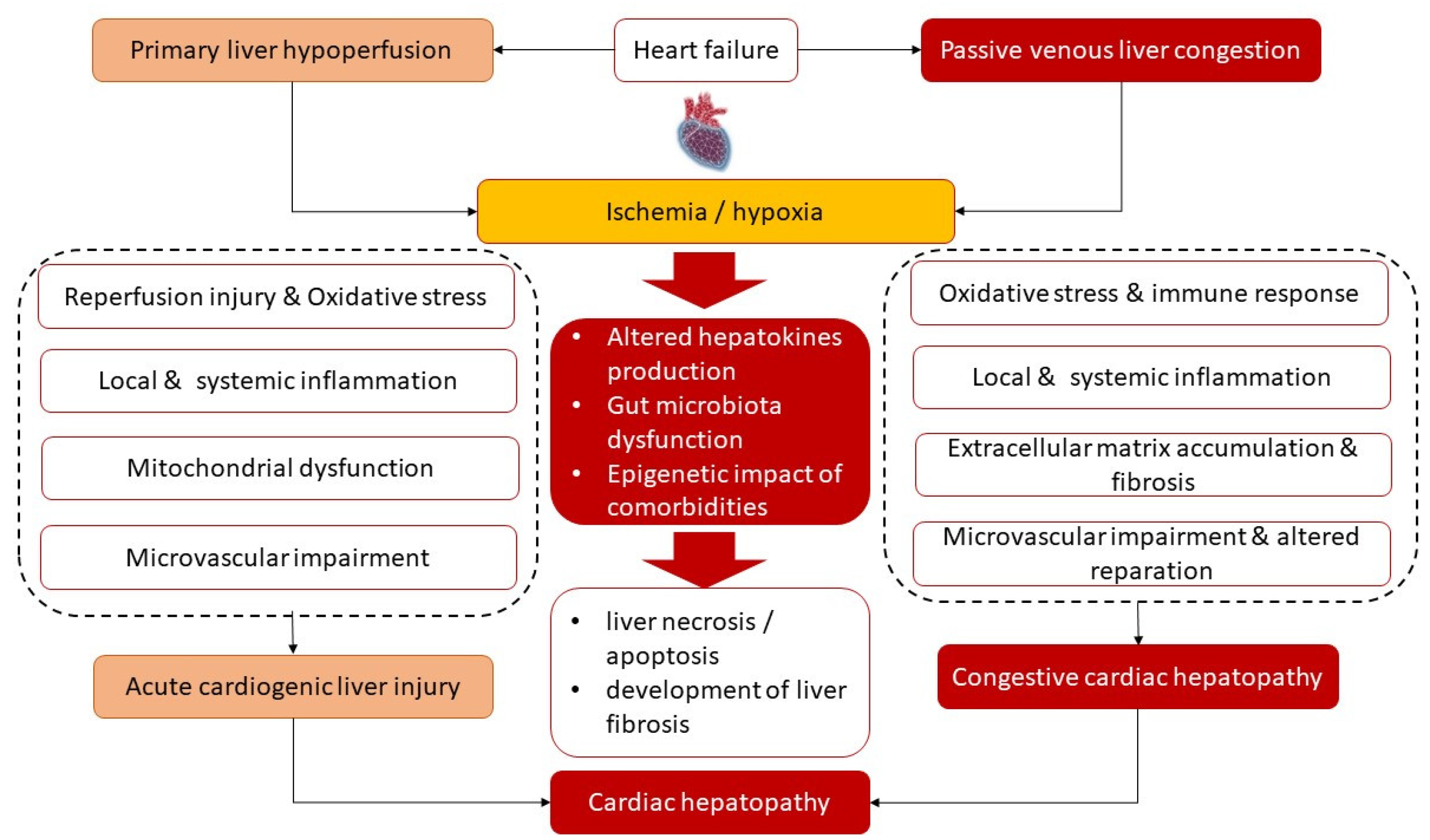
2. Definition, Morphological Criteria, and Biochemical Profiling of Liver Damage in HF
3. Common Underlying Molecular Mechanisms of Cardiac Hepatopathy
3.1. Ischemia/Reperfusion and Inflammation/Fibrosis Cascade
3.2. Intestinal Microbiota
3.3. Adipose Tissue Dysfunction
3.4. Impaired Skeletal Muscles Metabolism
3.5. Epigenetic Impact of Metabolic Comorbidities on Liver Tissue Modification
4. Plausible Role of Hepatokines in Cardiac Hepatopathy
4.1. Adropin
4.2. Fetuin-A
4.3. Alpha1-Microglobulin
4.4. Fibroblast Growth Factor-21
4.5. Selenoprotein P
4.6. Hepatokines and Liver Drug Metabolism
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Emmons-Bell, S.; Johnson, C.; Roth, G. Prevalence, incidence and survival of heart failure: A systematic review. Heart 2022, 108, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Van Riet, E.E.; Hoes, A.W.; Wagenaar, K.P.; Limburg, A.; Landman, M.A.; Rutten, F.H. Epidemiology of heart failure: The prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur. J. Heart Fail. 2016, 18, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Cvijic, M.; Rib, Y.; Danojevic, S.; Radulescu, C.I.; Nazghaidze, N.; Vardas, P. Heart failure with mildly reduced ejection fraction: From diagnosis to treatment. Gaps and dilemmas in current clinical practice. Heart Fail. Rev. 2022. [Google Scholar] [CrossRef] [PubMed]
- Echouffo-Tcheugui, J.B.; Erqou, S.; Butler, J.; Yancy, C.W.; Fonarow, G.C. Assessing the Risk of Progression from Asymptomatic Left Ventricular Dysfunction to Overt Heart Failure: A Systematic Overview and Meta-Analysis. JACC Heart Fail. 2016, 4, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Gori, M.; Redfield, M.M.; Calabrese, A.; Canova, P.; Cioffi, G.; De Maria, R.; Grosu, A.; Fontana, A.; Iacovoni, A.; Ferrari, P.; et al. Is mild asymptomatic left ventricular systolic dysfunction always predictive of adverse events in high-risk populations? Insights from the DAVID-Berg study. Eur. J. Heart Fail. 2018, 20, 1540–1548. [Google Scholar] [CrossRef] [Green Version]
- Pandhi, J.; Gottdiener, J.S.; Bartz, T.M.; Kop, W.J.; Mehra, M.R. Comparison of characteristics and outcomes of asymptomatic versus symptomatic left ventricular dysfunction in subjects 65 years old or older (from the Cardiovascular Health Study). Am. J. Cardiol. 2011, 107, 1667–1674. [Google Scholar] [CrossRef] [Green Version]
- Vyskocilova, K.; Spinarova, L.; Spinar, J.; Mikusova, T.; Vitovec, J.; Malek, J.; Malek, F.; Linhart, A.; Fedorco, M.; Widimsky, P.; et al. Prevalence and clinical significance of liver function abnormalities in patients with acute heart failure. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech Repub. 2015, 159, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Biegus, J.; Hillege, H.L.; Postmus, D.; Valente, M.A.; Bloomfield, D.M.; Cleland, J.G.; Cotter, G.; Davison, B.A.; Dittrich, H.C.; Fiuzat, M.; et al. Abnormal liver function tests in acute heart failure: Relationship with clinical characteristics and outcome in the PROTECT study. Eur. J. Heart Fail. 2016, 18, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Samsky, M.D.; Dunning, A.; DeVore, A.D.; Schulte, P.J.; Starling, R.C.; Tang, W.H.; Armstrong, P.W.; Ezekowitz, J.A.; Butler, J.; McMurray, J.J.; et al. Liver function tests in patients with acute heart failure and associated outcomes: Insights from ASCEND-HF. Eur. J. Heart Fail. 2016, 18, 424–432. [Google Scholar] [CrossRef]
- Ambrosy, A.P.; Vaduganathan, M.; Huffman, M.D.; Khan, S.; Kwasny, M.J.; Fought, A.J.; Maggioni, A.P.; Swedberg, K.; Konstam, M.A.; Zannad, F.; et al. Clinical course and predictive value of liver function tests in patients hospitalized for worsening heart failure with reduced ejection fraction: An analysis of the EVEREST trial. Eur. J. Heart Fail. 2012, 14, 302–311. [Google Scholar] [CrossRef]
- Wang, H.Y.; Huang, Y.; Chen, X.Z.; Zhang, Z.L.; Gui, C. Prognostic potential of liver injury in patients with dilated cardiomyopathy: A retrospective study. Eur. J. Med. Res. 2022, 27, 237. [Google Scholar] [CrossRef]
- Ambrosy, A.P.; Vaduganathan, M.; Mentz, R.J.; Greene, S.J.; Subačius, H.; Konstam, M.A.; Maggioni, A.P.; Swedberg, K.; Gheorghiade, M. Clinical profile and prognostic value of low systolic blood pressure in patients hospitalized for heart failure with reduced ejection fraction: Insights from the Efficacy of Vasopressin Antagonism in Heart Failure: Outcome Study with Tolvaptan (EVEREST) trial. Am. Heart J. 2013, 165, 216–225. [Google Scholar] [CrossRef]
- Meng, W.; Wang, L.; Fan, H.; Mao, S.; Song, X.; Zhang, Z. Total Bilirubin Level is Associated with the Risk of Left Atrial Appendage Thrombosis in Patients with Non-Valvular Atrial Fibrillation. Glob. Heart 2022, 17, 90. [Google Scholar] [CrossRef]
- Xanthopoulos, A.; Starling, R.C.; Kitai, T.; Triposkiadis, F. Heart Failure and Liver Disease: Cardiohepatic Interactions. JACC Heart Fail. 2019, 7, 87–97. [Google Scholar] [CrossRef]
- Laribi, S.; Mebazaa, A. Cardiohepatic syndrome: Liver injury in decompensated heart failure. Curr. Heart Fail. Rep. 2014, 11, 236–240. [Google Scholar] [CrossRef]
- Branchereau, M.; Burcelin, R.; Heymes, C. The gut microbiome and heart failure: A better gut for a better heart. Rev. Endocr. Metab. Disord. 2019, 20, 407–414. [Google Scholar] [CrossRef]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Relationship between Heart Disease and Liver Disease: A Two-Way Street. Cells 2020, 9, 567. [Google Scholar] [CrossRef] [Green Version]
- Gheorghiade, M.; Follath, F.; Ponikowski, P.; Barsuk, J.H.; Blair, J.E.; Cleland, J.G.; Dickstein, K.; Drazner, M.H.; Fonarow, G.C.; Jaarsma, T.; et al. Assessing and grading congestion in acute heart failure: A scientific statement from the acute heart failure committee of the heart failure association of the European Society of Cardiology and endorsed by the European Society of Intensive Care Medicine. Eur. J. Heart Fail. 2010, 12, 423–433. [Google Scholar] [CrossRef]
- Çağlı, K.; Başar, F.N.; Tok, D.; Turak, O.; Başar, Ö. How to interpret liver function tests in heart failure patients? Turk. J. Gastroenterol. 2015, 26, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Fortea, J.I.; Puente, Á.; Cuadrado, A.; Huelin, P.; Pellón, R.; González Sánchez, F.J.; Mayorga, M.; Cagigal, M.L.; García Carrera, I.; Cobreros, M.; et al. Congestive Hepatopathy. Int. J. Mol. Sci. 2020, 21, 49420. [Google Scholar] [CrossRef]
- De Gonzalez, A.K.K.; Lefkowitch, J.H. Heart Disease and the Liver: Pathologic Evaluation. Gastroenterol. Clin. North Am. 2017, 46, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.M.; Yehia, R. Hepato-cardiac disorders. World J. Hepatol. 2014, 6, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Hakuno, D.; Kimura, M.; Ito, S.; Satoh, J.; Nakashima, Y.; Horie, T.; Kuwabara, Y.; Nishiga, M.; Ide, Y.; Baba, O.; et al. Hepatokine α1-Microglobulin Signaling Exacerbates Inflammation and Disturbs Fibrotic Repair in Mouse Myocardial Infarction. Sci. Rep. 2018, 8, 16749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavoliuniene, A.; Vaitiekiene, A.; Cesnaite, G. Congestive hepatopathy and hypoxic hepatitis in heart failure: A cardiologist’s point of view. Int. J. Cardiol. 2013, 166, 554–558. [Google Scholar] [CrossRef]
- Birrer, R.; Takuda, Y.; Takara, T. Hypoxic hepatopathy: Pathophysiology and prognosis. Intern. Med. 2007, 46, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Mauriello, J.N.; Straughan, M.M. Right-Sided Heart Failure and the Liver. Crit. Care Nurs. Clin. North Am. 2022, 34, 341–350. [Google Scholar] [CrossRef]
- Correale, M.; Tarantino, N.; Petrucci, R.; Tricarico, L.; Laonigro, I.; Di Biase, M.; Brunetti, N.D. Liver disease and heart failure: Back and forth. Eur. J. Intern. Med. 2018, 48, 25–34. [Google Scholar] [CrossRef]
- Megalla, S.; Holtzman, D.; Aronow, W.S.; Nazari, R.; Korenfeld, S.; Schwarcz, A.; Goldberg, Y.; Spevack, D.M. Predictors of cardiac hepatopathy in patients with right heart failure. Med. Sci. Monit. 2011, 17, CR537–CR541. [Google Scholar] [CrossRef] [Green Version]
- Allen, L.A.; Felker, G.M.; Pocock, S.; McMurray, J.J.; Pfeffer, M.A.; Swedberg, K.; CHARM Investigators. Liver function abnormalities and outcome in patients with chronic heart failure: Data from the Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity (CHARM) program. Eur. J. Heart Fail. 2009, 11, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Benincasa, G.; Cuomo, O.; Vasco, M.; Vennarecci, G.; Canonico, R.; Della Mura, N.; Alfano, R.; Napoli, C. Epigenetic-sensitive challenges of cardiohepatic interactions: Clinical and therapeutic implications in heart failure patients. Eur. J. Gastroenterol. Hepatol. 2021, 33, 1247–1253. [Google Scholar] [CrossRef]
- Seeto, R.K.; Fenn, B.; Rockey, D.C. Ischemic hepatitis: Clinical presentation and pathogenesis. Am. J. Med. 2000, 109, 109–113. [Google Scholar] [CrossRef]
- Lightsey, J.M.; Rockey, D.C. Current concepts in ischemic hepatitis. Curr. Opin. Gastroenterol. 2017, 33, 158–163. [Google Scholar] [CrossRef]
- Wisse, E.; Jacobs, F.; Topal, B.; Frederik, P.; De Geest, B. The size of endothelial fenestrae in human liver sinusoids: Implications for hepatocyte-directed gene transfer. Gene. Ther. 2008, 15, 1193–1199. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, V.; Fang, J.C. Gastrointestinal and Liver Issues in Heart Failure. Circulation 2016, 133, 1696–1703. [Google Scholar] [CrossRef]
- Goncalvesova, E.; Kovacova, M. Heart failure affects liver morphology and function. What are the clinical implications? Bratisl. Lek. Listy. 2018, 119, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Silva, S.; Urbano, J.; Moura, M.C.; Ferreira-Coimbra, J.; Bettencourt, P.; Pimenta, J. Liver cytolysis in acute heart failure: What does it mean? Clinical profile and outcomes of a prospective hospital cohort. Int. J. Cardiol. 2016, 221, 422–427. [Google Scholar] [CrossRef]
- Møller, S.; Dümcke, C.W.; Krag, A. The heart and the liver. Expert. Rev. Gastroenterol. Hepatol. 2009, 3, 51–64. [Google Scholar] [CrossRef]
- Samsky, M.D.; Patel, C.B.; DeWald, T.A.; Smith, A.D.; Felker, G.M.; Rogers, J.G.; Hernandez, A.F. Cardiohepatic interactions in heart failure: An overview and clinical implications. J. Am. Coll. Cardiol. 2013, 61, 2397–2405. [Google Scholar] [CrossRef] [Green Version]
- Scalzo, N.; Canastar, M.; Lebovics, E. Part 2: Disease of the Heart and Liver: A Relationship That Cuts Both Ways. Cardiol. Rev. 2022, 30, 161–166. [Google Scholar] [CrossRef]
- Jaeschke, H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G15–G26. [Google Scholar] [CrossRef]
- Lee, W.Y.; Lee, J.S.; Lee, S.M. Protective effects of combined ischemic preconditioning and ascorbic acid on mitochondrial injury in hepatic ischemia/reperfusion. J. Surg. Res. 2007, 142, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Malle, E.; Farhood, A.; Jaeschke, H. Generation of hypochlorite-modified proteins by neutrophils during ischemia-reperfusion injury in rat liver: Attenuation by ischemic preconditioning. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G760–G767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.M.; Liu, Y.X.; Zhou, L.; Xie, H.Y.; Feng, X.W.; Li, H.; Zheng, S.S. Ischemic preconditioning attenuates morphological and biochemical changes in hepatic ischemia/reperfusion in rats. Pathobiology 2010, 77, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Braz, M.; Elias-Miró, M.; Jiménez-Castro, M.B.; Casillas-Ramírez, A.; Ramalho, F.S.; Peralta, C. The current state of knowledge of hepatic ischemia-reperfusion injury based on its study in experimental models. J. Biomed. Biotechnol. 2012, 2012, 298657. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.K.; Zhang, X.Y.; Zimmermann, A.; Davis, G.; Wheatley, A.M. Effect of ischemia-reperfusion injury on the microcirculation of the steatotic liver of the Zucker rat. Transplantation 2001, 72, 1625–1631. [Google Scholar] [CrossRef]
- Nastos, C.; Kalimeris, K.; Papoutsidakis, N.; Tasoulis, M.K.; Lykoudis, P.M.; Theodoraki, K.; Nastou, D.; Smyrniotis, V.; Arkadopoulos, N. Global consequences of liver ischemia/reperfusion injury. Oxid. Med. Cell. Longev. 2014, 2014, 906965. [Google Scholar] [CrossRef] [Green Version]
- Uhlmann, D.; Glasser, S.; Gaebel, G.; Armann, B.; Ludwig, S.; Tannapfel, A.; Hauss, J.; Witzigmann, H. Improvement of postischemic hepatic microcirculation after endothelin A receptor blockade—Endothelin antagonism influences platelet-endothelium interactions. J. Gastrointest. Surg. 2005, 9, 187–197. [Google Scholar] [CrossRef]
- Kuwano, A.; Kurokawa, M.; Kohjima, M.; Imoto, K.; Tashiro, S.; Suzuki, H.; Tanaka, M.; Okada, S.; Kato, M.; Ogawa, Y.; et al. Microcirculatory disturbance in acute liver injury. Exp. Ther. Med. 2021, 21, 596. [Google Scholar] [CrossRef]
- Tanaka, M.; Tanaka, K.; Masaki, Y.; Miyazaki, M.; Kato, M.; Kotoh, K.; Enjoji, M.; Nakamuta, M.; Takayanagi, R. Intrahepatic microcirculatory disorder, parenchymal hypoxia and NOX4 upregulation result in zonal differences in hepatocyte apoptosis following lipopolysaccharide- and D-galactosamine-induced acute liver failure in rats. Int. J. Mol. Med. 2014, 33, 254–262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jiang, W.; Zhou, A.L.; Zhao, M.; Jiang, D.R. Inhibitory effect of oxymatrine on hepatocyte apoptosis via TLR4/PI3K/Akt/GSK-3β signaling pathway. World J. Gastroenterol. 2017, 23, 3839–3849. [Google Scholar] [CrossRef]
- Quesnelle, K.M.; Bystrom, P.V.; Toledo-Pereyra, L.H. Molecular responses to ischemia and reperfusion in the liver. Arch. Toxicol. 2015, 89, 651–657. [Google Scholar] [CrossRef]
- Wang, X.T.; Tang, Y.B.; Lin, Q.Q.; Wang, X.Y.; Song, Z.Y.; Hao, M.L.; Qian, W.; Wang, W.T. Role of autophagy in liver injury induced by lung ischemia/reperfusion in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2022, 38, 102–107. (In Chinese) [Google Scholar] [CrossRef]
- Zhao, J.; Chen, X.D.; Yan, Z.Z.; Huang, W.F.; Liu, K.X.; Li, C. Gut-Derived Exosomes Induce Liver Injury After Intestinal Ischemia/Reperfusion by Promoting Hepatic Macrophage Polarization. Inflammation 2022, 45, 2325–2338. [Google Scholar] [CrossRef]
- Ding, W.; Duan, Y.; Qu, Z.; Feng, J.; Zhang, R.; Li, X.; Sun, D.; Zhang, X.; Lu, Y. Acidic Microenvironment Aggravates the Severity of Hepatic Ischemia/Reperfusion Injury by Modulating M1-Polarization Through Regulating PPAR-γ Signal. Front. Immunol. 2021, 12, 697362. [Google Scholar] [CrossRef]
- Chen, L.Y.; Yang, B.; Zhou, L.; Ren, F.; Duan, Z.P.; Ma, Y.J. Promotion of mitochondrial energy metabolism during hepatocyte apoptosis in a rat model of acute liver failure. Mol. Med. Rep. 2015, 12, 5035–5041. [Google Scholar] [CrossRef] [Green Version]
- Teodoro, J.S.; Da Silva, R.T.; Machado, I.F.; Panisello-Roselló, A.; Roselló-Catafau, J.; Rolo, A.P.; Palmeira, C.M. Shaping of Hepatic Ischemia/Reperfusion Events: The Crucial Role of Mitochondria. Cells 2022, 11, 688. [Google Scholar] [CrossRef]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072. [Google Scholar] [CrossRef]
- Sun, Y.Y.; Li, X.F.; Meng, X.M.; Huang, C.; Zhang, L.; Li, J. Macrophage Phenotype in Liver Injury and Repair. Scand. J. Immunol. 2017, 85, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Möckel, D.; Baeck, C.; Baeck, C.; Hittatiya, K.; Eulberg, D.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971. [Google Scholar] [CrossRef] [Green Version]
- Bartneck, M.; Schrammen, P.L.; Möckel, D.; Govaere, O.; Liepelt, A.; Krenkel, O.; Ergen, C.; McCain, M.V.; Eulberg, D.; Luedde, T.; et al. The CCR2+ Macrophage Subset Promotes Pathogenic Angiogenesis for Tumor Vascularization in Fibrotic Livers. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 371–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, S.; Russell, J.O.; Molina, L.M.; Monga, S.P. Liver Progenitors and Adult Cell Plasticity in Hepatic Injury and Repair: Knowns and Unknowns. Annu. Rev. Pathol. 2020, 15, 23–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Z.T.; Liu, W.; Xue, C.R.; Wu, S.X.; Chen, W.N.; Lin, X.J.; Lin, X. AKT activator SC79 protects hepatocytes from TNF-α-mediated apoptosis and alleviates d-Gal/LPS-induced liver injury. Am. J. Physiol. Gastrointest. Liver. Physiol. 2019, 316, G387–G396. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Jing, Z.T.; Xue, C.R.; Wu, S.X.; Chen, W.N.; Lin, X.J.; Lin, X. PI3K/AKT inhibitors aggravate death receptor-mediated hepatocyte apoptosis and liver injury. Toxicol. Appl. Pharmacol. 2019, 381, 114729. [Google Scholar] [CrossRef]
- Matsuzaki, K. Smad phosphoisoform signals in acute and chronic liver injury: Similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. 2012, 347, 225–243. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Matsuzaki, K. Differential Regulation of TGF-β/Smad Signaling in Hepatic Stellate Cells between Acute and Chronic Liver Injuries. Front. Physiol. 2012, 3, 53. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.D.; Wu, R.H.; Huang, Y.Z.; Chen, M.X.; Zeng, A.M.; Zhuo, G.F.; Xu, F.S.; Liao, R.; Lin, Q.C. The role of ferroptosis in chronic intermittent hypoxia-induced liver injury in rats. Sleep Breath. 2020, 24, 1767–1773. [Google Scholar] [CrossRef]
- Liu, Z.L.; Huang, Y.P.; Wang, X.; He, Y.X.; Li, J.; Li, B. The role of ferroptosis in chronic intermittent hypoxia-induced cognitive impairment. Sleep Breath. 2023. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, B. The Gut-Liver Axis in Health and Disease: The Role of Gut Microbiota-Derived Signals in Liver Injury and Regeneration. Front. Immunol. 2021, 12, 775526. [Google Scholar] [CrossRef]
- Trebicka, J.; Macnaughtan, J.; Schnabl, B.; Shawcross, D.L.; Bajaj, J.S. The microbiota in cirrhosis and its role in hepatic decompensation. J. Hepatol. 2021, 75 (Suppl. 1), S67–S81. [Google Scholar] [CrossRef]
- Liu, H.X.; Keane, R.; Sheng, L.; Wan, Y.J. Implications of microbiota and bile acid in liver injury and regeneration. J. Hepatol. 2015, 63, 1502–1510. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.S.; Baker, R.D. Gut Microbiota and Liver Injury (II): Chronic Liver Injury. Adv. Exp. Med. Biol. 2020, 1238, 39–54. [Google Scholar] [CrossRef]
- Giuffrè, M.; Campigotto, M.; Campisciano, G.; Comar, M.; Crocè, L.S. A story of liver and gut microbes: How does the intestinal flora affect liver disease? A review of the literature. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G889–G906. [Google Scholar] [CrossRef]
- Behary, J.; Amorim, N.; Jiang, X.T.; Raposo, A.; Gong, L.; McGovern, E.; Ibrahim, R.; Chu, F.; Stephens, C.; Jebeili, H.; et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021, 12, 187. [Google Scholar] [CrossRef]
- Fukui, H.; Brauner, B.; Bode, J.C.; Bode, C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: Reevaluation with an improved chromogenic assay. J. Hepatol. 1991, 12, 162–169. [Google Scholar] [CrossRef]
- Bellot, P.; García-Pagán, J.C.; Francés, R.; Abraldes, J.G.; Navasa, M.; Pérez-Mateo, M.; Such, J.; Bosch, J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology 2010, 52, 2044–2052. [Google Scholar] [CrossRef]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- La Villa, G.; Gentilini, P. Hemodynamic alterations in liver cirrhosis. Mol. Aspects Med. 2008, 29, 112–118. [Google Scholar] [CrossRef]
- Chu, C.J.; Lee, F.Y.; Wang, S.S.; Chang, F.Y.; Lin, H.C.; Lu, R.H.; Chan, C.C.; Lee, S.D. Splanchnic endotoxin levels in cirrhotic rats induced by carbon tetrachloride. Zhonghua Yi Xue Za Zhi 2000, 63, 196–204. [Google Scholar]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B.; et al. Trimethylamine N-Oxide Binds and Activates PERK to Promote Metabolic Dysfunction. Cell Metab. 2019, 30, 1141–1151.e5. [Google Scholar] [CrossRef] [PubMed]
- Bessman, N.J.; Mathieu, J.R.R.; Renassia, C.; Zhou, L.; Fung, T.C.; Fernandez, K.C.; Austin, C.; Moeller, J.B.; Zumerle, S.; Louis, S.; et al. Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science 2020, 368, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.A.; Theriot, C.M. Diversification of host bile acids by members of the gut microbiota. Gut. Microbes 2020, 11, 158–171. [Google Scholar] [CrossRef]
- Weiss, T.S.; Lupke, M.; Ibrahim, S.; Buechler, C.; Lorenz, J.; Ruemmele, P.; Hofmann, U.; Melter, M.; Dayoub, R. Attenuated lipotoxicity and apoptosis is linked to exogenous and endogenous augmenter of liver regeneration by different pathways. PLoS ONE 2017, 12, e0184282. [Google Scholar] [CrossRef] [Green Version]
- Teshima, T.; Matsumoto, H.; Michishita, M.; Matsuoka, A.; Shiba, M.; Nagashima, T.; Koyama, H. Allogenic Adipose Tissue-Derived Mesenchymal Stem Cells Ameliorate Acute Hepatic Injury in Dogs. Stem Cells Int. 2017, 2017, 3892514. [Google Scholar] [CrossRef]
- Kim, M.D.; Kim, S.S.; Cha, H.Y.; Jang, S.H.; Chang, D.Y.; Kim, W.; Suh-Kim, H.; Lee, J.H. Therapeutic effect of hepatocyte growth factor-secreting mesenchymal stem cells in a rat model of liver fibrosis. Exp. Mol. Med. 2014, 46, e110. [Google Scholar] [CrossRef] [Green Version]
- Berezin, A.E.; Berezin, A.A. Impaired function of fibroblast growth factor 23/Klotho protein axis in prediabetes and diabetes mellitus: Promising predictor of cardiovascular risk. Diabetes Metab. Syndr. 2019, 13, 2549–2556. [Google Scholar] [CrossRef]
- Lakhani, H.V.; Sharma, D.; Dodrill, M.W.; Nawab, A.; Sharma, N.; Cottrill, C.L.; Shapiro, J.I.; Sodhi, K. Phenotypic Alteration of Hepatocytes in Non-Alcoholic Fatty Liver Disease. Int. J. Med. Sci. 2018, 15, 1591–1599. [Google Scholar] [CrossRef] [Green Version]
- Li, T.H.; Yang, Y.Y.; Huang, C.C.; Liu, C.W.; Tsai, H.C.; Lin, M.W.; Tsai, C.Y.; Huang, S.F.; Wang, Y.W.; Lee, T.Y.; et al. Elafibranor interrupts adipose dysfunction-mediated gut and liver injury in mice with alcoholic steatohepatitis. Clin. Sci. 2019, 133, 531–544. [Google Scholar] [CrossRef]
- Lin, Z.; Wu, F.; Lin, S.; Pan, X.; Jin, L.; Lu, T.; Shi, L.; Wang, Y.; Xu, A.; Li, X. Adiponectin protects against acetaminophen-induced mitochondrial dysfunction and acute liver injury by promoting autophagy in mice. J. Hepatol. 2014, 61, 825–831. [Google Scholar] [CrossRef]
- Sakane, S.; Hikita, H.; Shirai, K.; Myojin, Y.; Sasaki, Y.; Kudo, S.; Fukumoto, K.; Mizutani, N.; Tahata, Y.; Makino, Y.; et al. White Adipose Tissue Autophagy and Adipose-Liver Crosstalk Exacerbate Nonalcoholic Fatty Liver Disease in Mice. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1683–1699. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Zhang, Z.; Huang, B.; Cheng, X.; Wang, D.; la Gahu, Z.; Xue, Z.; Da, Y.; Li, D.; et al. Adiponectin-derived active peptide ADP355 exerts anti-inflammatory and anti-fibrotic activities in thioacetamide-induced liver injury. Sci. Rep. 2016, 6, 19445. [Google Scholar] [CrossRef]
- Clemens, M.M.; Kennon-McGill, S.; Vazquez, J.H.; Stephens, O.W.; Peterson, E.A.; Johann, D.J.; Allard, F.D.; Yee, E.U.; McCullough, S.S.; James, L.P.; et al. Exogenous phosphatidic acid reduces acetaminophen-induced liver injury in mice by activating hepatic interleukin-6 signaling through inter-organ crosstalk. Acta Pharm. Sin. B 2021, 11, 3836–3846. [Google Scholar] [CrossRef]
- Tilg, H.; Kaser, A.; Moschen, A.R. How to modulate inflammatory cytokines in liver diseases. Liver Int. 2006, 26, 1029–1039. [Google Scholar] [CrossRef]
- Wolf, A.M.; Wolf, D.; Avila, M.A.; Moschen, A.R.; Berasain, C.; Enrich, B.; Rumpold, H.; Tilg, H. Up-regulation of the anti-inflammatory adipokine adiponectin in acute liver failure in mice. J. Hepatol. 2006, 44, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Serbetçi, K.; Uysal, O.; Erkasap, N.; Köken, T.; Baydemir, C.; Erkasap, S. Anti-apoptotic and antioxidant effect of leptin on CCl₄-induced acute liver injury in rats. Mol. Biol. Rep. 2012, 39, 1173–1180. [Google Scholar] [CrossRef]
- Li, F.; Chen, J.; Liu, Y.; Gu, Z.; Jiang, M.; Zhang, L.; Chen, S.Y.; Deng, Z.; McClain, C.J.; Feng, W. Deficiency of Cathelicidin Attenuates High-Fat Diet Plus Alcohol-Induced Liver Injury through FGF21/Adiponectin Regulation. Cells 2021, 10, 3333. [Google Scholar] [CrossRef]
- Yasuzaki, H.; Yoshida, S.; Hashimoto, T.; Shibata, W.; Inamori, M.; Toya, Y.; Tamura, K.; Maeda, S.; Umemura, S. Involvement of the apelin receptor APJ in Fas-induced liver injury. Liver Int. 2013, 33, 118–126. [Google Scholar] [CrossRef]
- Garcia Whitlock, A.E.; Sostre-Colón, J.; Gavin, M.; Martin, N.D.; Baur, J.A.; Sims, C.A.; Titchenell, P.M. Loss of FOXO transcription factors in the liver mitigates stress-induced hyperglycemia. Mol. Metab. 2021, 51, 101246. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.H., Jr.; Yan, G.T.; Wang, L.H.; Zhang, J.Y.; Xue, H.; Zhang, K. Leptin relieves intestinal ischemia/reperfusion injury by promoting ERK1/2 phosphorylation and the NO signaling pathway. J. Trauma Acute Care Surg. 2012, 72, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Ikejima, K.; Honda, H.; Yoshikawa, M.; Hirose, M.; Kitamura, T.; Takei, Y.; Sato, N. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology 2001, 34, 288–297. [Google Scholar] [CrossRef]
- Berezin, A.E.; Berezin, A.A.; Lichtenauer, M. Myokines and Heart Failure: Challenging Role in Adverse Cardiac Remodeling, Myopathy, and Clinical Outcomes. Dis. Markers 2021, 2021, 6644631. [Google Scholar] [CrossRef]
- Pang, B.P.S.; Chan, W.S.; Chan, C.B. Mitochondria Homeostasis and Oxidant/Antioxidant Balance in Skeletal Muscle-Do Myokines Play a Role? Antioxidants 2021, 10, 179. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Bohovych, I.; Khalimonchuk, O. Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 2016, 4, 109. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Shao, Y.; Wu, F.; Wang, Y.; Xiong, R.; Zheng, J.; Tian, H.; Wang, B.; Wang, Y.; Zhang, Y.; et al. FGF21 Prevents Angiotensin II-Induced Hypertension and Vascular Dysfunction by Activation of ACE2/Angiotensin-(1-7) Axis in Mice. Cell Metab. 2018, 27, 1323–1337.e5. [Google Scholar] [CrossRef] [Green Version]
- Nagatomo, I.; Nakanishi, K.; Yamamoto, R.; Ide, S.; Ishibashi, C.; Moriyama, T.; Yamauchi-Takihara, K. Soluble angiotensin-converting enzyme 2 association with lipid metabolism. Front. Med. 2022, 9, 955928. [Google Scholar] [CrossRef]
- Landecho, M.F.; Tuero, C.; Valentí, V.; Bilbao, I.; de la Higuera, M.; Frühbeck, G. Relevance of Leptin and Other Adipokines in Obesity-Associated Cardiovascular Risk. Nutrients 2019, 11, 2664. [Google Scholar] [CrossRef] [Green Version]
- Kotiadis, V.N.; Duchen, M.R.; Osellame, L.D. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta 2014, 1840, 1254–1265. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Gambino, R.; De Michieli, F.; Durazzo, M.; Pagano, G.; Cassader, M. Adiponectin gene polymorphisms modulate acute adiponectin response to dietary fat: Possible pathogenetic role in NASH. Hepatology 2008, 47, 1167–1177. [Google Scholar] [CrossRef]
- Gambino, R.; Cassader, M.; Pagano, G.; Durazzo, M.; Musso, G. Polymorphism in microsomal triglyceride transfer protein: A link between liver disease and atherogenic postprandial lipid profile in NASH? Hepatology 2007, 45, 1097–1107. [Google Scholar] [CrossRef]
- Stasinou, E.; Argyraki, M.; Sotiriadou, F.; Lambropoulos, A.; Fotoulaki, M. Association between rs738409 and rs2896019 single-nucleotide polymorphisms of phospholipase domain-containing protein 3 and susceptibility to nonalcoholic fatty liver disease in Greek children and adolescents. Ann. Gastroenterol. 2022, 35, 297–306. [Google Scholar] [CrossRef]
- Zhang, R.N.; Shen, F.; Pan, Q.; Cao, H.X.; Chen, G.Y.; Fan, J.G. PPARGC1A rs8192678 G>A polymorphism affects the severity of hepatic histological features and nonalcoholic steatohepatitis in patients with nonalcoholic fatty liver disease. World J. Gastroenterol. 2021, 27, 3863–3876. [Google Scholar] [CrossRef]
- Ali, I.I.; D’Souza, C.; Singh, J.; Adeghate, E. Adropin’s Role in Energy Homeostasis and Metabolic Disorders. Int. J. Mol. Sci. 2022, 23, 58318. [Google Scholar] [CrossRef]
- Kumar, K.G.; Trevaskis, J.L.; Lam, D.D.; Sutton, G.M.; Koza, R.A.; Chouljenko, V.N.; Kousoulas, K.G.; Rogers, P.M.; Kesterson, R.A.; Thearle, M.; et al. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 2008, 8, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Mushala, B.A.S.; Scott, I. Adropin: A hepatokine modulator of vascular function and cardiac fuel metabolism. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H238–H244. [Google Scholar] [CrossRef]
- Jasaszwili, M.; Billert, M.; Strowski, M.Z.; Nowak, K.W.; Skrzypski, M. Adropin as A Fat-Burning Hormone with Multiple Functions-Review of a Decade of Research. Molecules 2020, 25, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maudsley, S.; Walter, D.; Schrauwen, C.; Van Loon, N.; Harputluoğlu, İ.; Lenaerts, J.; McDonald, P. Intersection of the Orphan G Protein-Coupled Receptor, GPR19, with the Aging Process. Int. J. Mol. Sci. 2022, 23, 13598. [Google Scholar] [CrossRef] [PubMed]
- Kalaany, N.Y.; Mangelsdorf, D.J. LXRS and FXR: The yin and yang of cholesterol and fat metabolism. Annu. Rev. Physiol. 2006, 68, 159–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, L.; Wei, Y.; Fang, C.; Liu, S.; Zhou, F.; Li, Y.; Zhao, G.; Guo, Z.; Luo, Y.; et al. Exercise suppresses NLRP3 inflammasome activation in mice with diet-induced NASH: A plausible role of adropin. Lab. Investig. 2021, 101, 369–380. [Google Scholar] [CrossRef]
- Thapa, D.; Stoner, M.W.; Zhang, M.; Xie, B.; Manning, J.R.; Guimaraes, D.; Shiva, S.; Jurczak, M.J.; Scott, I. Adropin regulates pyruvate dehydrogenase in cardiac cells via a novel GPCR-MAPK-PDK4 signaling pathway. Redox Biol. 2018, 18, 25–32. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Al-Omran, M.; Teoh, H.; Verma, S. Adropin is a novel regulator of endothelial function. Circulation 2010, 122 (Suppl. 11), S185–S192. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Fang, J.; Yuan, X.; Xiong, C.; Chen, L. Adropin reduces hypoxia/reoxygenation-induced myocardial injury via the reperfusion injury salvage kinase pathway. Exp. Ther. Med. 2019, 18, 3307–3314. [Google Scholar] [CrossRef] [Green Version]
- Soltani, S.; Kolahdouz-Mohammadi, R.; Aydin, S.; Yosaee, S.; Clark, C.C.T.; Abdollahi, S. Circulating levels of adropin and overweight/obesity: A systematic review and meta-analysis of observational studies. Hormones 2022, 21, 15–22. [Google Scholar] [CrossRef]
- Yosaee, S.; Soltani, S.; Sekhavati, E.; Jazayeri, S. Adropin—A Novel Biomarker of Heart Disease: A Systematic Review Article. Iran. J. Public Health 2016, 45, 1568–1576. [Google Scholar]
- Li, L.; Xie, W.; Zheng, X.L.; Yin, W.D.; Tang, C.K. A novel peptide adropin in cardiovascular diseases. Clin. Chim. Acta 2016, 453, 107–113. [Google Scholar] [CrossRef]
- Maciorkowska, M.; Musiałowska, D.; Małyszko, J. Adropin and irisin in arterial hypertension, diabetes mellitus and chronic kidney disease. Adv. Clin. Exp. Med. 2019, 28, 1571–1575. [Google Scholar] [CrossRef]
- Aydin, S.; Eren, M.N.; Yilmaz, M.; Kalayci, M.; Yardim, M.; Alatas, O.D.; Kuloglu, T.; Balaban, H.; Cakmak, T.; Kobalt, M.A.; et al. Adropin as a potential marker of enzyme-positive acute coronary syndrome. Cardiovasc. J. Afr. 2017, 28, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Cui, B.; Zhao, X.; Wu, Y.; Qin, H.; Guo, Y.; Wang, H.; Lu, M.; Zhang, S.; Shen, J.; et al. Correlation of Serum Adropin Levels with Risk Factors of Cardiovascular Disease in Hemodialysis Patients. Metab. Syndr. Relat. Disord. 2021, 19, 401–408. [Google Scholar] [CrossRef]
- Berezin, A.A.; Obradovic, Z.; Novikov, E.V.; Boxhammer, E.; Lichtenauer, M.; Berezin, A.E. Interplay between Myokine Profile and Glycemic Control in Type 2 Diabetes Mellitus Patients with Heart Failure. Diagnostics 2022, 12, 2940. [Google Scholar] [CrossRef]
- Jurrissen, T.J.; Ramirez-Perez, F.I.; Cabral-Amador, F.J.; Soares, R.N.; Pettit-Mee, R.J.; Betancourt-Cortes, E.E.; McMillan, N.J.; Sharma, N.; Rocha HN, M.; Fujie, S.; et al. Role of adropin in arterial stiffening associated with obesity and type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H879–H891. [Google Scholar] [CrossRef]
- Zhao, L.P.; You, T.; Chan, S.P.; Chen, J.C.; Xu, W.T. Adropin is associated with hyperhomocysteine and coronary atherosclerosis. Exp. Ther. Med. 2016, 11, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xue, H.; Fang, W.; Chen, K.; Chen, S.; Yang, W.; Shen, T.; Chen, X.; Zhang, P.; Ling. Adropin protects against liver injury in nonalcoholic steatohepatitis via the Nrf2 mediated antioxidant capacity. Redox Biol. 2019, 21, 101068. [Google Scholar] [CrossRef]
- Chen, X.; Sun, X.; Shen, T.; Chen, Q.; Chen, S.; Pang, J.; Mi, J.; Tang, Y.; You, Y.; Xu, H.; et al. Lower adropin expression is associated with oxidative stress and severity of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 160, 191–198. [Google Scholar] [CrossRef]
- Eser Karlidag, G.; Arslan Solmaz, O. Are adropin, apelin, elabela, asprosin and betatrophin biomarkers for chronic hepatitis and staging of fibrosis? Biotech. Histochem. 2020, 95, 152–159. [Google Scholar] [CrossRef]
- Skrzypski, M.; Kołodziejski, P.A.; Pruszyńska-Oszmałek, E.; Wojciechowicz, T.; Janicka, P.; Krążek, M.; Małek, E.; Strowski, M.Z.; Nowak, K.W. Daily Treatment of Mice with Type 2 Diabetes with Adropin for Four Weeks Improves Glucolipid Profile, Reduces Hepatic Lipid Content and Restores Elevated Hepatic Enzymes in Serum. Int. J. Mol. Sci. 2022, 23, 79807. [Google Scholar] [CrossRef]
- Li, Y.X.; Cheng, K.C.; Liu, I.M.; Niu, H.S. Myricetin Increases Circulating Adropin Level after Activation of Glucagon-like Peptide 1 (GLP-1) Receptor in Type-1 Diabetic Rats. Pharmaceuticals 2022, 15, 173. [Google Scholar] [CrossRef] [PubMed]
- Tičinović Kurir, T.; Miličević, T.; Novak, A.; Vilović, M.; Božić, J. Adropin—Potential link in cardiovascular protection for obese male type 2 diabetes mellitus patients treated with liraglutide. Acta Clin. Croat. 2020, 59, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Emoto, M.; Inaba, M. Fetuin-A: A multifunctional protein. Recent Pat. Endocr. Metab. Immune Drug Discov. 2011, 5, 124–146. [Google Scholar] [CrossRef] [PubMed]
- Khadir, A.; Kavalakatt, S.; Madhu, D.; Tiss, A. Fetuin-a expression profile in mouse and human adipose tissue. Lipids Health Dis. 2020, 19, 38. [Google Scholar] [CrossRef] [Green Version]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520. [Google Scholar] [CrossRef]
- Sardana, O.; Goyal, R.; Bedi, O. Molecular and pathobiological involvement of fetuin-A in the pathogenesis of NAFLD. Inflammopharmacology 2021, 29, 1061–1074. [Google Scholar] [CrossRef]
- Mukhuty, A.; Fouzder, C.; Kundu, R. Fetuin-A excess expression amplifies lipid induced apoptosis and β-cell damage. J. Cell. Physiol. 2022, 237, 532–550. [Google Scholar] [CrossRef]
- Shen, X.; Yang, L.; Yan, S.; Zheng, H.; Liang, L.; Cai, X.; Liao, M. Fetuin A promotes lipotoxicity in β cells through the TLR4 signaling pathway and the role of pioglitazone in anti-lipotoxicity. Mol. Cell Endocrinol. 2015, 412, 1–11. [Google Scholar] [CrossRef]
- Mukhuty, A.; Fouzder, C.; Kundu, R. Fetuin-A secretion from β-cells leads to accumulation of macrophages in islets, aggravates inflammation and impairs insulin secretion. J. Cell Sci. 2021, 134, jcs258507. [Google Scholar] [CrossRef]
- Rudloff, S.; Jahnen-Dechent, W.; Huynh-Do, U. Tissue chaperoning-the expanded functions of fetuin-A beyond inhibition of systemic calcification. Pflugers. Arch. 2022, 474, 949–962. [Google Scholar] [CrossRef]
- Guo, V.Y.; Cao, B.; Cai, C.; Cheng, K.K.; Cheung, B.M.Y. Fetuin-A levels and risk of type 2 diabetes mellitus: A systematic review and meta-analysis. Acta Diabetol. 2018, 55, 87–98. [Google Scholar] [CrossRef]
- Jirak, P.; Stechemesser, L.; Moré, E.; Franzen, M.; Topf, A.; Mirna, M.; Paar, V.; Pistulli, R.; Kretzschmar, D.; Wernly, B.; et al. Clinical implications of fetuin-A. Adv. Clin. Chem. 2019, 89, 79–130. [Google Scholar] [CrossRef]
- Sujana, C.; Huth, C.; Zierer, A.; Meesters, S.; Sudduth-Klinger, J.; Koenig, W.; Herder, C.; Peters, A.; Thorand, B. Association of fetuin-A with incident type 2 diabetes: Results from the MONICA/KORA Augsburg study and a systematic meta-analysis. Eur. J. Endocrinol. 2018, 178, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Yamasandhi, P.G.; Dharmalingam, M.; Balekuduru, A. Fetuin-A in newly detected type 2 diabetes mellitus as a marker of non-alcoholic fatty liver disease. Indian J. Gastroenterol. 2021, 40, 556–562. [Google Scholar] [CrossRef]
- Ossareh, S.; Rayatnia, M.; Vahedi, M.; Jafari, H.; Zebarjadi, M. Association of Serum Fetuin-A with Vascular Calcification in Hemodialysis Patients and Its’ Impact on 3-year Mortality. Iran. J. Kidney Dis. 2020, 14, 500–509. [Google Scholar] [PubMed]
- Lebensztejn, D.M.; Flisiak-Jackiewicz, M.; Białokoz-Kalinowska, I.; Bobrus-Chociej, A.; Kowalska, I. Hepatokines and non-alcoholic fatty liver disease. Acta Biochim. Pol. 2016, 63, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Kröger, J.; Meidtner, K.; Stefan, N.; Guevara, M.; Kerrison, N.D.; Ardanaz, E.; Aune, D.; Boeing, H.; Dorronsoro, M.; Dow, C.; et al. Circulating Fetuin-A and Risk of Type 2 Diabetes: A Mendelian Randomization Analysis. Diabetes 2018, 67, 1200–1205. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.K.; Jensen, R.A.; Mukamal, K.J.; Guo, X.; Yao, J.; Sun, Q.; Cornelis, M.; Liu, Y.; Chen, M.H.; Kizer, J.R.; et al. Detection of genetic loci associated with plasma fetuin-A: A meta-analysis of genome-wide association studies from the CHARGE Consortium. Hum. Mol. Genet. 2017, 26, 2156–2163. [Google Scholar] [CrossRef] [Green Version]
- Umapathy, D.; Subramanyam, P.V.; Krishnamoorthy, E.; Viswanathan, V.; Ramkumar, K.M. Association of Fetuin-A with Thr256Ser exon polymorphism of α2-Heremans Schmid Glycoprotein (AHSG) gene in type 2 diabetic patients with overt nephropathy. J. Diabetes Complicat. 2022, 36, 108074. [Google Scholar] [CrossRef]
- Von Loeffelholz, C.; Horn, P.; Birkenfeld, A.L.; Claus, R.A.; Metzing, B.U.; Döcke, S.; Jahreis, G.; Heller, R.; Hoppe, S.; Stockmann, M.; et al. Fetuin A is a Predictor of Liver Fat in Preoperative Patients with Nonalcoholic Fatty Liver Disease. J. Investig. Surg. 2016, 29, 266–274. [Google Scholar] [CrossRef]
- Sato, M.; Kamada, Y.; Takeda, Y.; Kida, S.; Ohara, Y.; Fujii, H.; Akita, M.; Mizutani, K.; Yoshida, Y.; Yamada, M.; et al. Fetuin-A negatively correlates with liver and vascular fibrosis in nonalcoholic fatty liver disease subjects. Liver Int. 2015, 35, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Celebi, G.; Genc, H.; Gurel, H.; Sertoglu, E.; Kara, M.; Tapan, S.; Acikel, C.; Karslioglu, Y.; Ercin, C.N.; Dogru, T. The relationship of circulating fetuin-a with liver histology and biomarkers of systemic inflammation in nondiabetic subjects with nonalcoholic fatty liver disease. Saudi J. Gastroenterol. 2015, 21, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Toprak, K.; Görpelioğlu, S.; Özsoy, A.; Özdemir, Ş.; Ayaz, A. Does fetuin-A mediate the association between pro-inflammatory diet and type-2 diabetes mellitus risk? Nutr. Hosp. 2022, 39, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sama, A.E. Anti-inflammatory role of fetuin-A in injury and infection. Curr. Mol. Med. 2012, 12, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Misaka, T.; Yoshihisa, A.; Ichijo, Y.; Ishibashi, S.; Matsuda, M.; Yamadera, Y.; Ohara, H.; Sugawara, Y.; Hotsuki, Y.; et al. Decreases in hepatokine Fetuin-A levels are associated with hepatic hypoperfusion and predict cardiac outcomes in patients with heart failure. Clin. Res. Cardiol. 2022, 111, 1104–1112. [Google Scholar] [CrossRef]
- Åkerström, B.; Gram, M. A1M, an extravascular tissue cleaning and housekeeping protein. Free Radic. Biol. Med. 2014, 74, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Bergwik, J.; Kristiansson, A.; Allhorn, M.; Gram, M.; Åkerström, B. Structure, Functions, and Physiological Roles of the Lipocalin α1-Microglobulin (A1M). Front. Physiol. 2021, 12, 645650. [Google Scholar] [CrossRef]
- Shigetomi, H.; Onogi, A.; Kajiwara, H.; Yoshida, S.; Furukawa, N.; Haruta, S.; Tanase, Y.; Kanayama, S.; Noguchi, T.; Yamada, Y.; et al. Anti-inflammatory actions of serine protease inhibitors containing the Kunitz domain. Inflamm. Res. 2010, 59, 679–687. [Google Scholar] [CrossRef]
- Kristiansson, A.; Gram, M.; Flygare, J.; Hansson, S.R.; Åkerström, B.; Storry, J.R. The Role of α1-Microglobulin (A1M) in Erythropoiesis and Erythrocyte Homeostasis-Therapeutic Opportunities in Hemolytic Conditions. Int. J. Mol. Sci. 2020, 21, 97234. [Google Scholar] [CrossRef]
- Tyagi, S.; Salier, J.P.; Lal, S.K. The liver-specific human alpha(1)-microglobulin/bikunin precursor (AMBP) is capable of self-association. Arch. Biochem. Biophys. 2002, 399, 66–72. [Google Scholar] [CrossRef]
- Akerström, B.; Lögdberg, L.; Berggård, T.; Osmark, P.; Lindqvist, A. alpha(1)-Microglobulin: A yellow-brown lipocalin. Biochim. Biophys. Acta 2000, 1482, 172–184. [Google Scholar] [CrossRef]
- Rutardottir, S.; Nilsson, E.J.; Pallon, J.; Gram, M.; Åkerström, B. The cysteine 34 residue of A1M/α1-microglobulin is essential for protection of irradiated cell cultures and reduction of carbonyl groups. Free Radic. Res. 2013, 47, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Bergwik, J.; Kristiansson, A.; Welinder, C.; Göransson, O.; Hansson, S.R.; Gram, M.; Erlandsson, L.; Åkerström, B. Knockout of the radical scavenger α1-microglobulin in mice results in defective bikunin synthesis, endoplasmic reticulum stress and increased body weight. Free Radic. Biol. Med. 2021, 162, 160–170. [Google Scholar] [CrossRef]
- Kristiansson, A.; Ahlstedt, J.; Holmqvist, B.; Brinte, A.; Tran, T.A.; Forssell-Aronsson, E.; Strand, S.E.; Gram, M.; Åkerström, B. Protection of Kidney Function with Human Antioxidation Protein α1-Microglobulin in a Mouse 177Lu-DOTATATE Radiation Therapy Model. Antioxid. Redox Signal. 2019, 30, 1746–1759. [Google Scholar] [CrossRef] [Green Version]
- Kristiansson, A.; Örbom, A.; Timmermand, O.V.; Ahlstedt, J.; Strand, S.E.; Åkerström, B. Kidney Protection with the Radical Scavenger α1-Microglobulin (A1M) during Peptide Receptor Radionuclide and Radioligand Therapy. Antioxidants 2021, 10, 81271. [Google Scholar] [CrossRef]
- Cederlund, M.; Deronic, A.; Pallon, J.; Sørensen, O.E.; Åkerström, B. A1M/α1-microglobulin is proteolytically activated by myeloperoxidase, binds its heme group and inhibits low density lipoprotein oxidation. Front. Physiol. 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Fukao, Y.; Nagasawa, H.; Nihei, Y.; Hiki, M.; Naito, T.; Kihara, M.; Gohda, T.; Ueda, S.; Suzuki, Y. COVID-19-induced acute renal tubular injury associated with elevation of serum inflammatory cytokine. Clin. Exp. Nephrol. 2021, 25, 1240–1246. [Google Scholar] [CrossRef]
- Tang, J.; Shi, Y.; Deng, R.; Zhang, J.; An, Y.; Li, Y.; Wang, L. Cytokine Profile in Calcineurin Inhibitor-Induced Chronic Nephrotoxicity in Chinese Liver Transplant Recipients. Transplant. Proc. 2016, 48, 2756–2762. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Ramos, D.; Aguilar-Salinas, C.A. Modulation of energy balance by fibroblast growth factor 21. Horm. Mol. Biol. Clin. Investig. 2016, 30. [Google Scholar] [CrossRef]
- Giralt, M.; Gavaldà-Navarro, A.; Villarroya, F. Fibroblast growth factor-21, energy balance and obesity. Mol. Cell. Endocrinol. 2015, 418 Pt 1, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Lam, K.S.L.; Xu, A. The therapeutic potential of FGF21 in metabolic diseases: From bench to clinic. Nat. Rev. Endocrinol. 2020, 16, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Seok, S.; Kim, Y.C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarei, M.; Pizarro-Delgado, J.; Barroso, E.; Palomer, X.; Vázquez-Carrera, M. Targeting FGF21 for the Treatment of Nonalcoholic Steatohepatitis. Trends. Pharmacol. Sci. 2020, 41, 199–208. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, D.; Long, X.X.; Fang, Q.C.; Jia, W.P.; Li, H.T. The role of FGF21 in the pathogenesis of cardiovascular disease. Chin. Med. J. 2021, 134, 2931–2943. [Google Scholar] [CrossRef]
- Yang, X.; Jin, Z.; Lin, D.; Shen, T.; Zhang, J.; Li, D.; Wang, X.; Zhang, C.; Lin, Z.; Li, X.; et al. FGF21 alleviates acute liver injury by inducing the SIRT1-autophagy signalling pathway. J. Cell. Mol. Med. 2022, 26, 868–879. [Google Scholar] [CrossRef]
- Zhang, J.; Cheng, Y.; Gu, J.; Wang, S.; Zhou, S.; Wang, Y.; Tan, Y.; Feng, W.; Fu, Y.; Mellen, N.; et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin. Sci. 2016, 130, 625–641. [Google Scholar] [CrossRef]
- Li, Y.; Wong, K.; Giles, A.; Jiang, J.; Lee, J.W.; Adams, A.C.; Kharitonenkov, A.; Yang, Q.; Gao, B.; Guarente, L.; et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 2014, 146, 539–549.e7. [Google Scholar] [CrossRef]
- Wu, A.; Feng, B.; Yu, J.; Yan, L.; Che, L.; Zhuo, Y.; Luo, Y.; Yu, B.; Wu, D.; Chen, D. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021, 46, 102131. [Google Scholar] [CrossRef]
- Wu, L.; Mo, W.; Feng, J.; Li, J.; Yu, Q.; Li, S.; Zhang, J.; Chen, K.; Ji, J.; Dai, W.; et al. Astaxanthin attenuates hepatic damage and mitochondrial dysfunction in non-alcoholic fatty liver disease by up-regulating the FGF21/PGC-1α pathway. Br. J. Pharmacol. 2020, 177, 3760–3777. [Google Scholar] [CrossRef]
- Prida, E.; Álvarez-Delgado, S.; Pérez-Lois, R.; Soto-Tielas, M.; Estany-Gestal, A.; Fernø, J.; Seoane, L.M.; Quiñones, M.; Al-Massadi, O. Liver Brain Interactions: Focus on FGF21 a Systematic Review. Int. J. Mol. Sci. 2022, 23, 13318. [Google Scholar] [CrossRef]
- Abu-Odeh, M.; Zhang, Y.; Reilly, S.M.; Ebadat, N.; Keinan, O.; Valentine, J.M.; Hafezi-Bakhtiari, M.; Ashayer, H.; Mamoun, L.; Zhou, X.; et al. FGF21 promotes thermogenic gene expression as an autocrine factor in adipocytes. Cell Rep. 2021, 35, 109331. [Google Scholar] [CrossRef]
- Lin, Z.; Tian, H.; Lam, K.S.; Lin, S.; Hoo, R.C.; Konishi, M.; Itoh, N.; Wang, Y.; Bornstein, S.R.; Xu, A.; et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013, 17, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Takamura, T. Hepatokine Selenoprotein P-Mediated Reductive Stress Causes Resistance to Intracellular Signal Transduction. Antioxid. Redox Signal. 2020, 33, 517–524. [Google Scholar] [CrossRef]
- Arora, A.S.; Gores, G.J. The role of metals in ischemia/reperfusion injury of the liver. Semin. Liver Dis. 1996, 16, 31–38. [Google Scholar] [CrossRef]
- Saito, Y. Selenoprotein P as an in vivo redox regulator: Disorders related to its deficiency and excess. J. Clin. Biochem. Nutr. 2020, 66, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y. Selenoprotein P as a significant regulator of pancreatic β cell function. J. Biochem. 2020, 167, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y. Selenium Transport Mechanism via Selenoprotein P-Its Physiological Role and Related Diseases. Front. Nutr. 2021, 8, 685517. [Google Scholar] [CrossRef]
- Hill, K.E.; Chittum, H.S.; Lyons, P.R.; Boeglin, M.E.; Burk, R.F. Effect of selenium on selenoprotein P expression in cultured liver cells. Biochim. Biophys. Acta 1996, 1313, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Chadani, H.; Usui, S.; Inoue, O.; Kusayama, T.; Takashima, S.I.; Kato, T.; Murai, H.; Furusho, H.; Nomura, A.; Misu, H.; et al. Endogenous Selenoprotein, P, a Liver-Derived Secretory Protein, Mediates Myocardial Ischemia/Reperfusion Injury in Mice. Int. J. Mol. Sci. 2018, 19, 30878. [Google Scholar] [CrossRef] [Green Version]
- Caviglia, G.P.; Rosso, C.; Armandi, A.; Gaggini, M.; Carli, F.; Abate, M.L.; Olivero, A.; Ribaldone, D.G.; Saracco, G.M.; Gastaldelli, A.; et al. Interplay between Oxidative Stress and Metabolic Derangements in Non-Alcoholic Fatty Liver Disease: The Role of Selenoprotein P. Int. J. Mol. Sci. 2020, 21, 28838. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mavrouli, M.; Katsinelos, P.; Doulberis, M.; Gavana, E.; Duntas, L. Selenoprotein P in Patients with Nonalcoholic Fatty Liver Disease. Exp. Clin. Endocrinol. Diabetes 2019, 127, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Takeishi, R.; Misaka, T.; Ichijo, Y.; Ishibashi, S.; Matsuda, M.; Yamadera, Y.; Ohara, H.; Sugawara, Y.; Hotsuki, Y.; Watanabe, K.; et al. Increases in Hepatokine Selenoprotein P Levels Are Associated with Hepatic Hypoperfusion and Predict Adverse Prognosis in Patients with Heart Failure. J. Am. Heart Assoc. 2022, 11, e024901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, Y.; Qiu, C.; Deng, G.; Guo, M. Protective Action of Se-Supplement Against Acute Alcoholism Is Regulated by Selenoprotein P (SelP) in the Liver. Biol. Trace Elem. Res. 2017, 175, 375–387. [Google Scholar] [CrossRef] [PubMed]
- di Giuseppe, R.; Koch, M.; Schlesinger, S.; Borggrefe, J.; Both, M.; Müller, H.P.; Kassubek, J.; Jacobs, G.; Nöthlings, U.; Lieb, W. Circulating selenoprotein P levels in relation to MRI-derived body fat volumes, liver fat content, and metabolic disorders. Obesity 2017, 25, 1128–1135. [Google Scholar] [CrossRef] [Green Version]
- di Giuseppe, R.; Plachta-Danielzik, S.; Koch, M.; Nöthlings, U.; Schlesinger, S.; Borggrefe, J.; Both, M.; Müller, H.P.; Kassubek, J.; Jacobs, G.; et al. Dietary pattern associated with selenoprotein P and MRI-derived body fat volumes, liver signal intensity, and metabolic disorders. Eur. J. Nutr. 2019, 58, 1067–1079. [Google Scholar] [CrossRef]
- Pendyal, A.; Gelow, J.M. Cardiohepatic Interactions: Implications for Management in Advanced Heart Failure. Heart Fail. Clin. 2016, 12, 349–361. [Google Scholar] [CrossRef]
- Verbeeck, R.K. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur. J. Clin. Pharmacol. 2008, 64, 1147–1161. [Google Scholar] [CrossRef]
- Mangoni, A.A.; Jarmuzewska, E.A. The influence of heart failure on the pharmacokinetics of cardiovascular and non-cardiovascular drugs: A critical appraisal of the evidence. Br. J. Clin. Pharmacol. 2019, 85, 20–36. [Google Scholar] [CrossRef]
- Berezin, A.A.; Obradovic, Z.; Fushtey, I.M.; Berezina, T.A.; Novikov, E.V.; Schmidbauer, L.; Lichtenauer, M.; Berezin, A.E. The Impact of SGLT2 Inhibitor Dapagliflozin on Adropin Serum Levels in Men and Women with Type 2 Diabetes Mellitus and Chronic Heart Failure. Biomedicines 2023, 11, 457. [Google Scholar] [CrossRef]


{kind=link}
| Hepatokines | Local Liver Effect | Systemic Effects | References |
|---|---|---|---|
| Adropin | ↓ activation of hepatic stellate cells, ↓NLRP3 inflammasome, ↓ inflammatory gene expression, ↓ oxidative stress, ↓ lipid peroxidation, ↓ lipid toxicity, ↓ liver injury and fibrosis, ↓ autophagy, ↑ glucose metabolism, ↑ insulin sensitivity, ↑ anti-apoptotic and antioxidant effects, ↑ pre-conditioning | ↓ adverse cardiac remodeling, ↓ systemic inflammatory reaction, ↑ vascular integrity/endothelial function, ↑ renal and splanchnic blood flow, ↑ angiogenesis, ↑ NO production | [118,122,125,126,138,139,140,141,142] |
| Fetuin-A | ↓ liver inflammation, injury and fibrosis, ↓ necrosis and apoptosis, ↓ fasting glucose levels, ↑ mitochondrial function, ↑ lipid metabolism, ↑ angiogenesis | ↑ antioxidant capacity, ↑ endothelial function, ↓ WAT dysfunction, ↓ inflammation, ↓ pancreatic beta-cell damage/apoptosis, ↓ adverse cardiac remodeling, ↓ vessel calcification, ↑ skeletal muscle energy metabolism, ↑ NO production | [144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165] |
| Alpha1-microglobulin | Anti-oxidative effects, ↓ apoptosis, ↓ mitochondrial damage, ↓ autophagy, ↓ hepatocyte damage, ↓ activation of hepatic stellate cells, | ↓ cardiac, lung and kidney injury, ↑ anti-ischemic protection | [170,171,172,173,174,175,176,177,178] |
| Fibroblast growth factor-21 | ↓ mitochondrial oxidative stress, ↓ autophagy, ↓ fasting glucose levels, ↑ gluconeogenesis, ↑ insulin sensitivity | ↓ WAT inflammation, ↓ lipolysis in WAT, ↓ fibrosis in myocardium, ↑ endothelial function, ↓ microvascular inflammation, ↑ NO production | [179,180,181,182,183,184,185,186,187,188,189,190,191,192,193] |
| Selenoprotein P | ↓ oxidative stress, ↓ lipid peroxidation, ↓ lipid toxicity, ↓ liver injury and fibrosis | ↓ pancreatic beta-cell apoptosis, ↑ antioxidant capacity, ↑ angiogenesis, ↑ insulin sensitivity | [194,195,196,197,198,199,200,201,202,203,204,205,206] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berezin, A.A.; Obradovic, Z.; Berezina, T.A.; Boxhammer, E.; Lichtenauer, M.; Berezin, A.E. Cardiac Hepatopathy: New Perspectives on Old Problems through a Prism of Endogenous Metabolic Regulations by Hepatokines. Antioxidants 2023, 12, 516. https://doi.org/10.3390/antiox12020516
Berezin AA, Obradovic Z, Berezina TA, Boxhammer E, Lichtenauer M, Berezin AE. Cardiac Hepatopathy: New Perspectives on Old Problems through a Prism of Endogenous Metabolic Regulations by Hepatokines. Antioxidants. 2023; 12(2):516. https://doi.org/10.3390/antiox12020516
Chicago/Turabian StyleBerezin, Alexander A., Zeljko Obradovic, Tetiana A. Berezina, Elke Boxhammer, Michael Lichtenauer, and Alexander E. Berezin. 2023. "Cardiac Hepatopathy: New Perspectives on Old Problems through a Prism of Endogenous Metabolic Regulations by Hepatokines" Antioxidants 12, no. 2: 516. https://doi.org/10.3390/antiox12020516