
DMF-Activated Nrf2 Ameliorates Palmitic Acid Toxicity While Potentiates Ferroptosis Mediated Cell Death: Protective Role of the NO-Donor S-Nitroso-N-Acetylcysteine

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Line
2.3. Preparation of S-Nitrosothiols (RSNO)
2.4. Propidium Iodide Cell Viability Assay
2.5. MTT Cell Viability Assay
2.6. DNA Fragmentation Assay
2.7. Gene Expression
2.8. Nuclear Extracts Preparation
2.9. Western Blot Analysis
2.10. Lipid Peroxidation Analysis
2.11. Cellular Thiol Levels Analysis
2.12. Statistical Analysis
3. Results
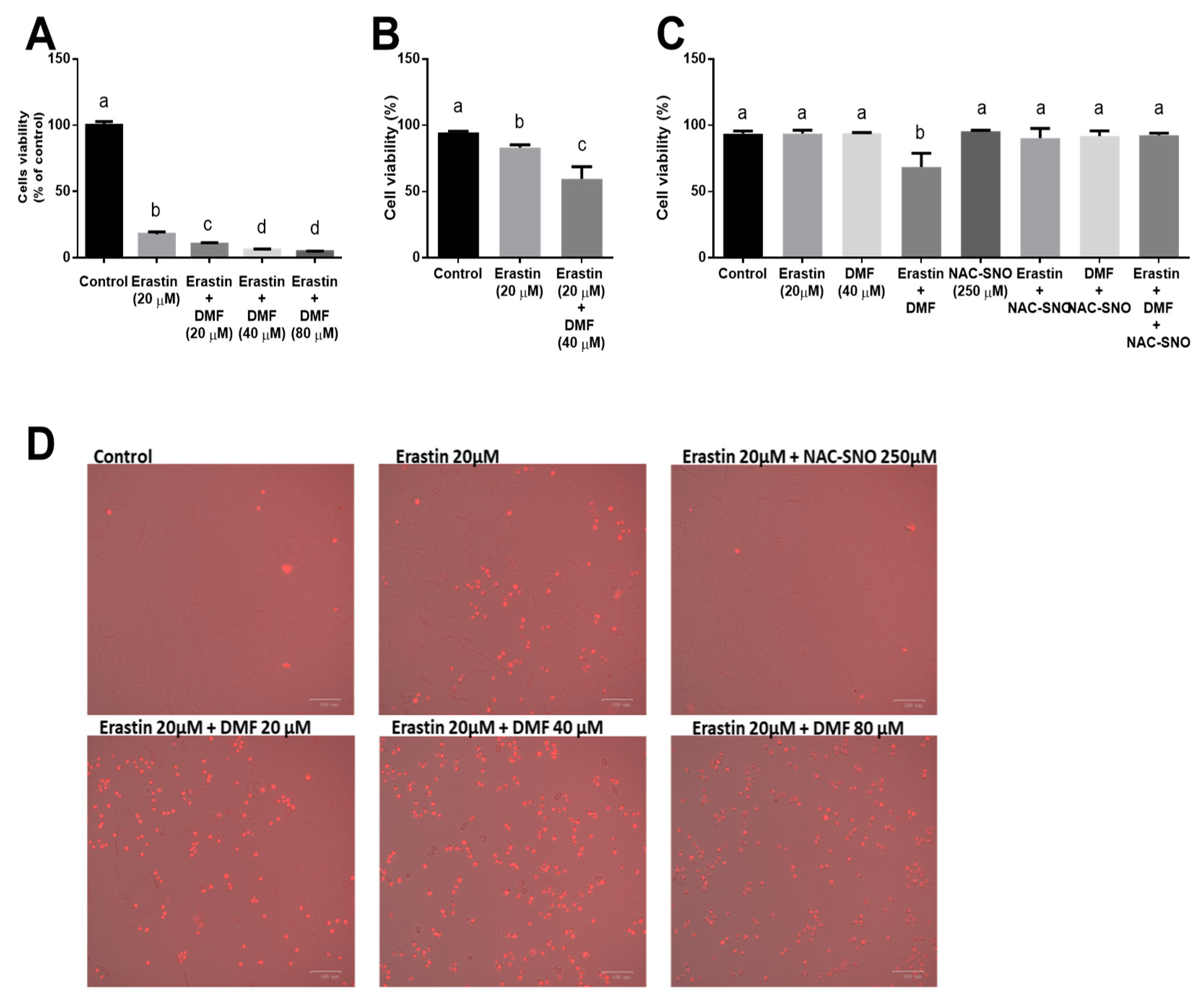
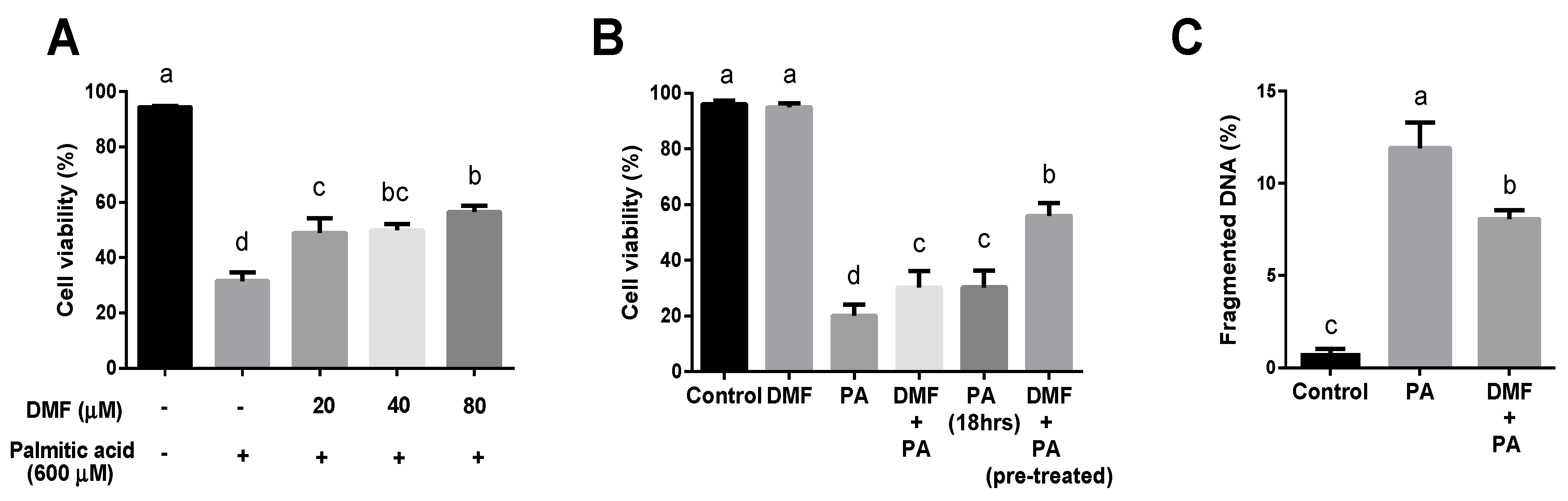
3.1. DMF Worsens Ferroptosis while Ameliorates Lipotoxicity in Hepatocytes
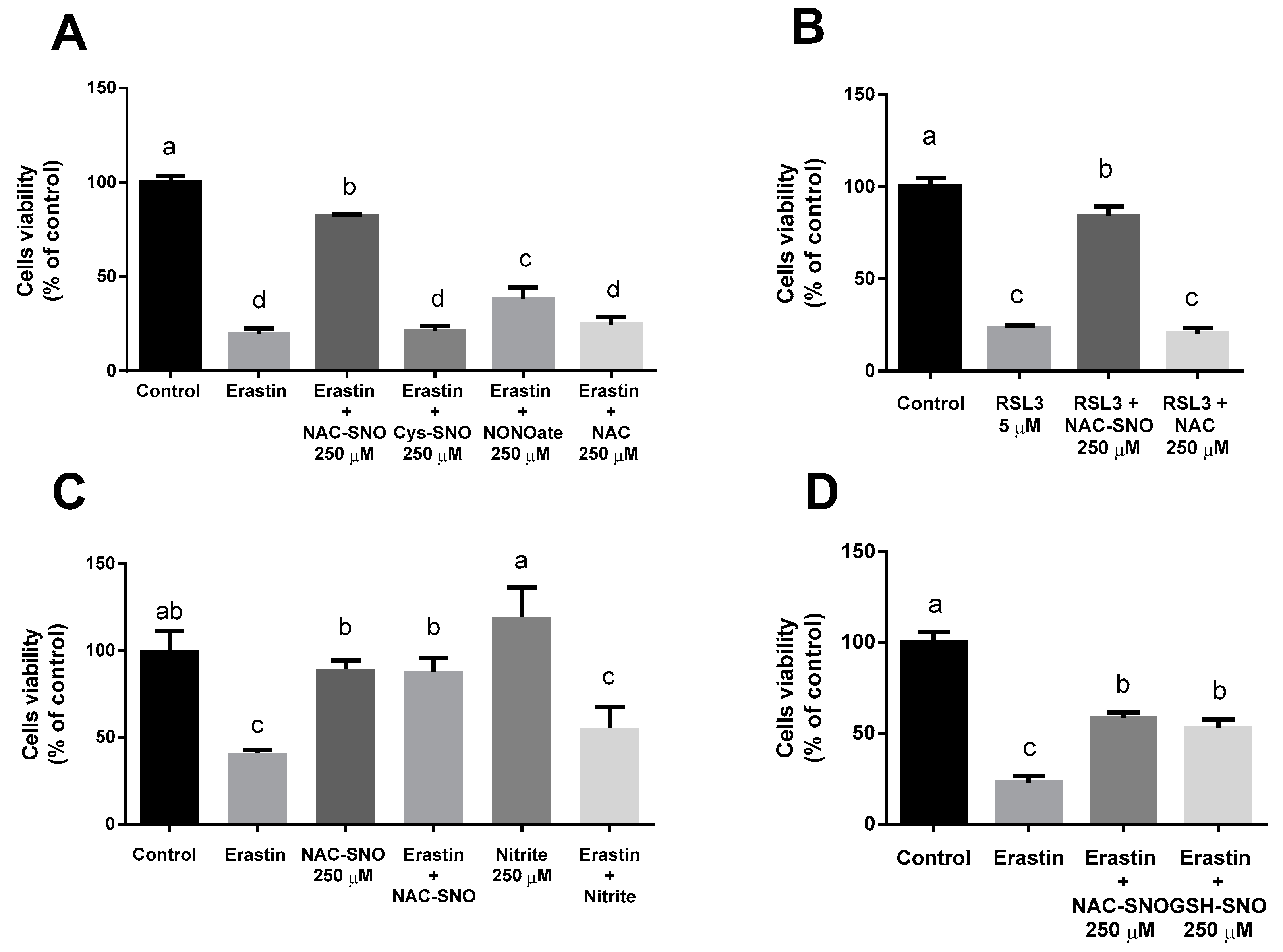
3.2. NAC-SNO Prevents Cell Death in Ferroptosis
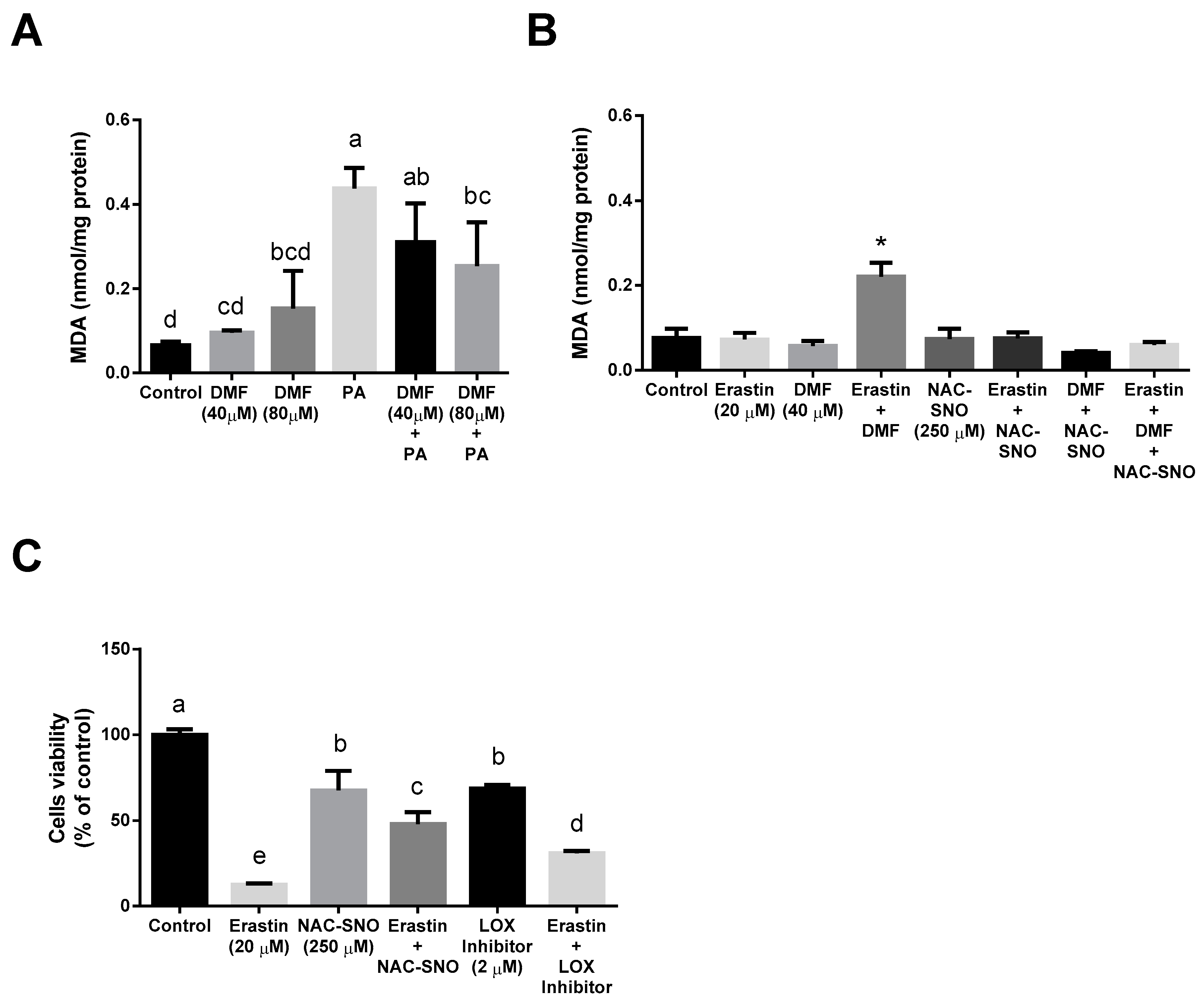
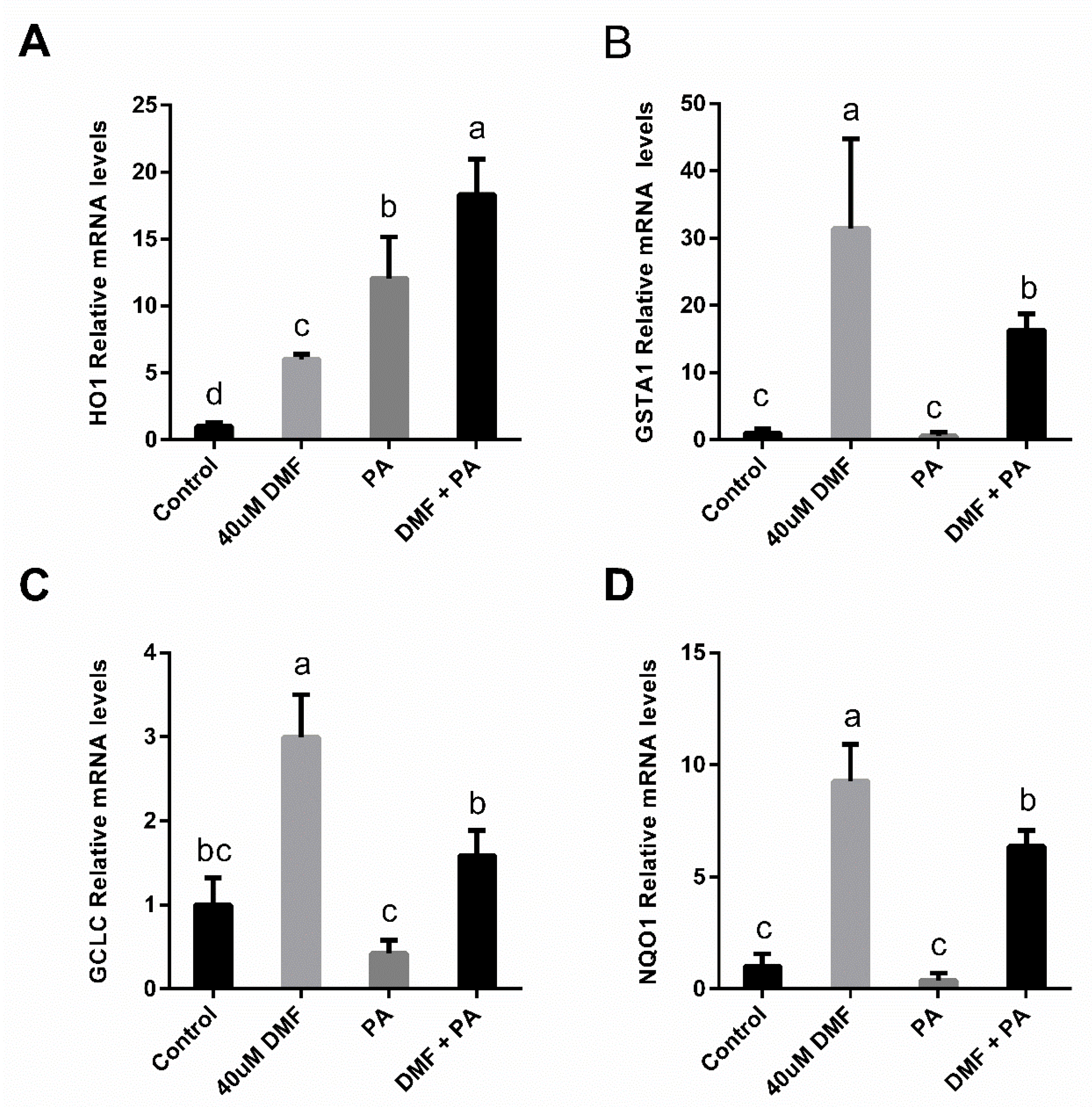
3.3. DMF Lowers the Extent of Oxidative Stress in the Lipotoxicity Model but Increases It in Ferroptosis
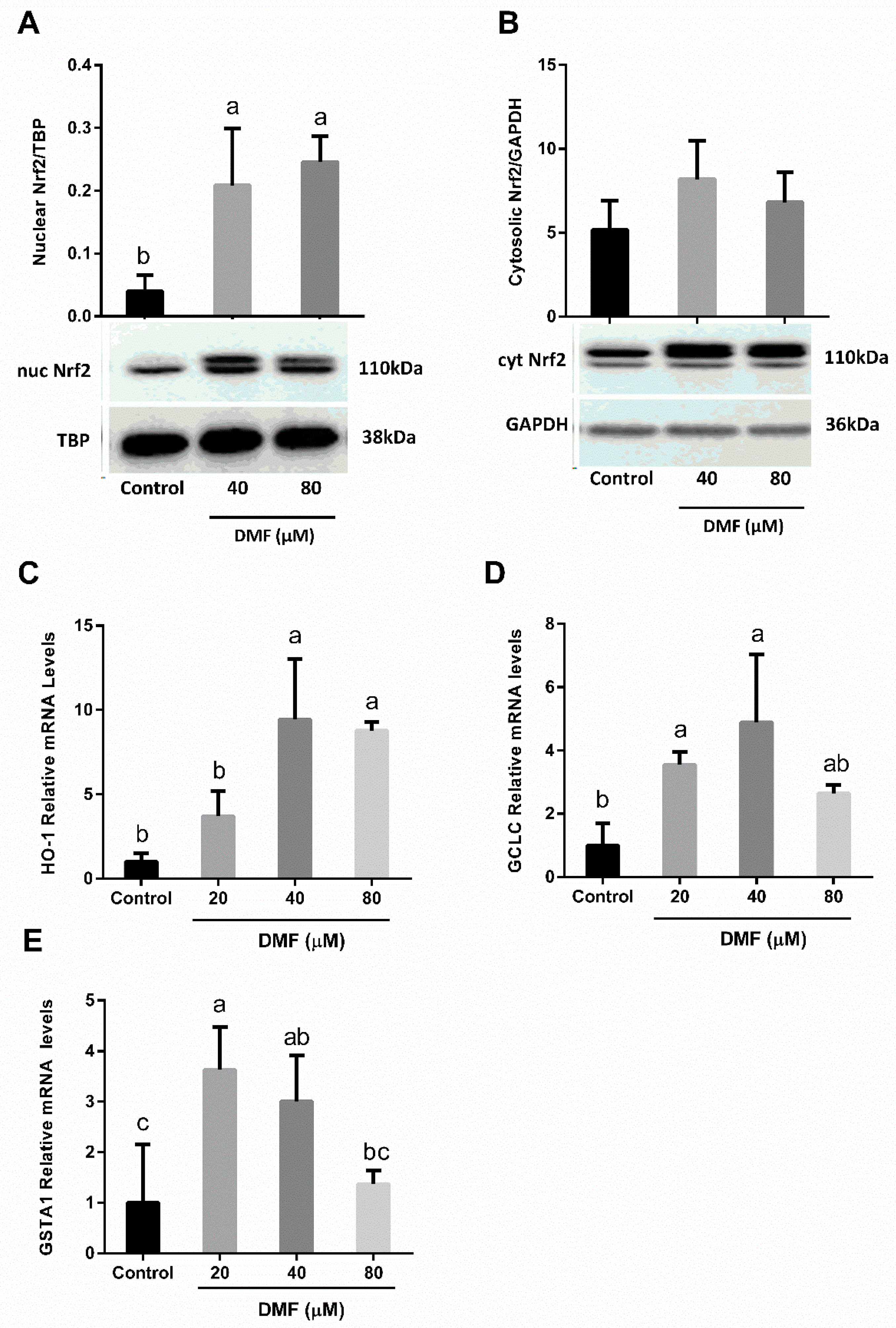
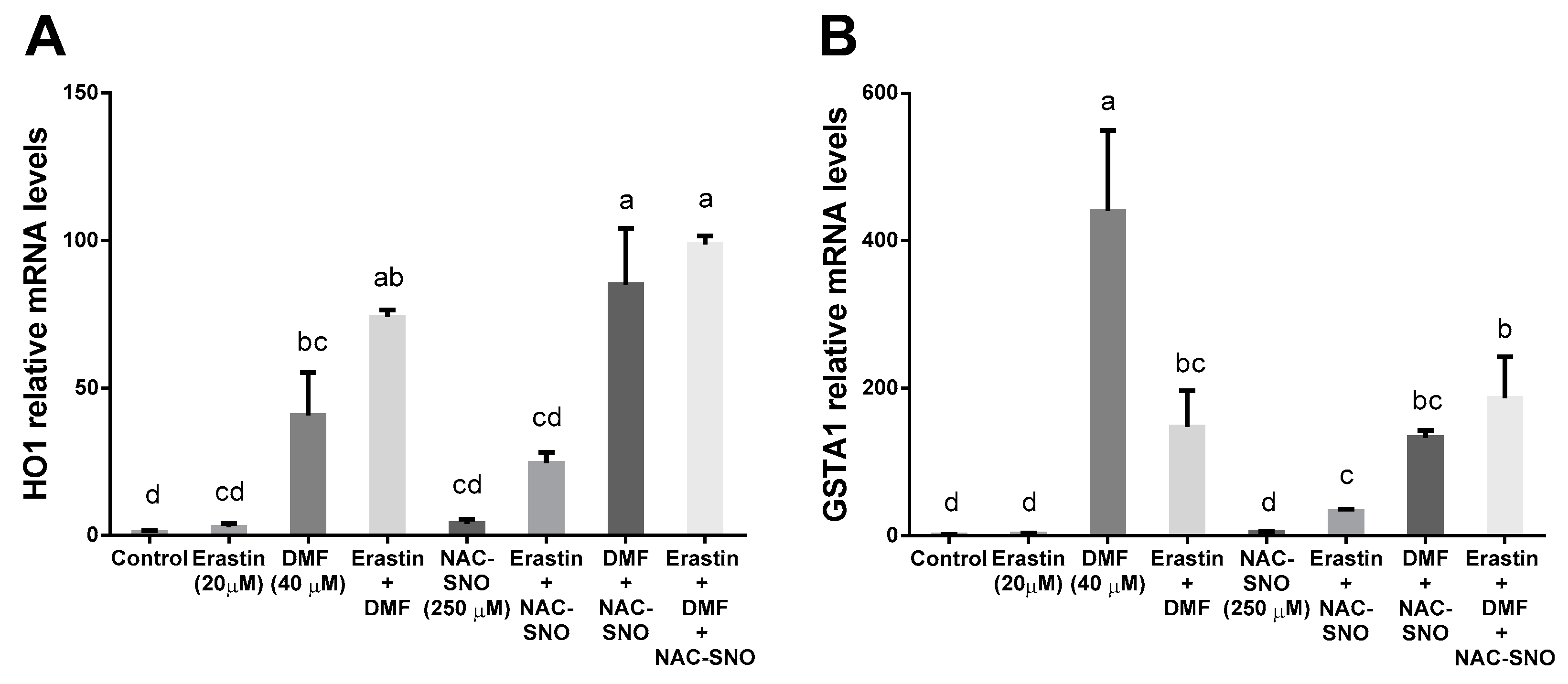
3.4. DMF Activates Nrf2-Related Genes in Both Lipotoxicity and Ferroptosis
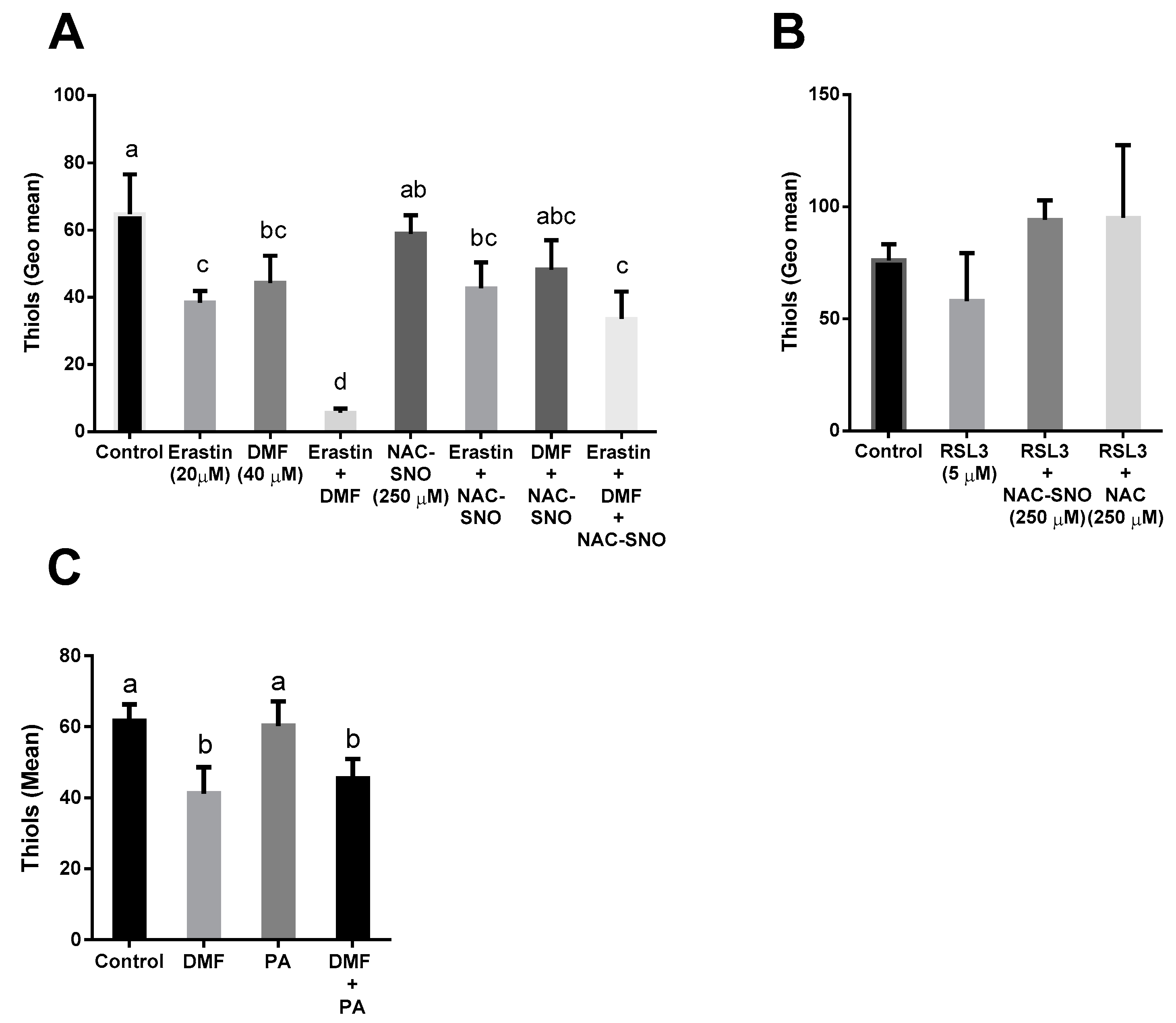
3.5. DMF Decreased Thiol Levels in Both Lipotoxicity and Ferroptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ajmera, V.; Loomba, R. Imaging Biomarkers of NAFLD, NASH, and Fibrosis. Mol. Metab. 2021, 50, 101167. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Villani, R.; Facciorusso, A.; Vendemiale, G.; Serviddio, G. Lipid Oxidation Products in the Pathogenesis of Non-Alcoholic Steatohepatitis. Free Radic. Biol. Med. 2017, 111, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Polimeni, L.; Del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative Stress: New Insights on the Association of Non-Alcoholic Fatty Liver Disease and Atherosclerosis. World J. Hepatol. 2015, 7, 1325. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Gambino, R. Non-Alcoholic Steatohepatitis: Emerging Molecular Targets and Therapeutic Strategies. Nat. Rev. Drug Discov. 2016, 15, 249. [Google Scholar] [CrossRef]
- Engin, A.B. What Is Lipotoxicity? Obes. Lipotoxicity 2017, 960, 197–220. [Google Scholar]
- Hauck, A.K.; Bernlohr, D.A. Oxidative Stress and Lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- Giralt, M.; Díaz-Delfín, J.M.; Gallego-Escuredo, J.; Villarroya, J.; Domingo, P.; Villarroya, F. Lipotoxicity on the Basis of Metabolic Syndrome and Lipodystrophy in HIV-1-Infected Patients under Antiretroviral Treatment. Curr. Pharm. Des. 2010, 16, 3372–3378. [Google Scholar] [CrossRef]
- Li, B.; Leung, J.C.K.; Chan, L.Y.Y.; Yiu, W.H.; Tang, S.C.W. A Global Perspective on the Crosstalk between Saturated Fatty Acids and Toll-like Receptor 4 in the Etiology of Inflammation and Insulin Resistance. Prog. Lipid Res. 2020, 77, 101020. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Friedmann Angeli, J.P. Glutathione Peroxidase 4 (Gpx4) and Ferroptosis: What’s so Special about It? Mol. Cell Oncol. 2015, 2, e995047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Hirschhorn, T.; Stockwell, B.R. The Development of the Concept of Ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Khanna, S.; Roy, S.; Ryu, H.; Bahadduri, P.; Swaan, P.W.; Ratan, R.R.; Sen, C.K. Molecular Basis of Vitamin E Action: Tocotrienol Modulates 12-Lipoxygenase, a Key Mediator of Glutamate-Induced Neurodegeneration. J. Biol. Chem. 2003, 278, 43508–43515. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, O.; Sen, C.K.; Roy, S.; Packer, L. Cellular and Mitochondrial Changes in Glutamate-Induced HT4 Neuronal Cell Death. Neuroscience 2000, 97, 531–541. [Google Scholar] [CrossRef]
- Tirosh, O.; Sen, C.K.; Roy, S.; Kobayashi, M.S.; Packer, L. Neuroprotective Effects of α-Lipoic Acid and Its Positively Charged Amide Analogue. Free Radic. Biol. Med. 1999, 26, 1418–1426. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Conrad, M.; Konno, H.; Yokoyama, C.; Fujii, J. Nitric Oxide Protects against Ferroptosis by Aborting the Lipid Peroxidation Chain Reaction. Nitric Oxide 2021, 115, 34–43. [Google Scholar] [CrossRef]
- Kanner, J.; Shpaizer, A.; Nelgas, L.; Tirosh, O. S-Nitroso-N-Acetylcysteine (NAC-SNO) as an Antioxidant in Cured Meat and Stomach Medium. J. Agric. Food Chem. 2019, 67, 10930–10936. [Google Scholar] [CrossRef]
- Kanner, J.; Ben-Gera, I.; Berman, S. Nitric-Oxide Myoglobin as an Inhibitor of Lipid Oxidation. Lipids 1980, 15, 944. [Google Scholar] [CrossRef]
- Kanner, J.; Juven, B.J. S-Nitrosocysteine as An Antioxidant, Color-Developing, and Anticlostridial Agent in Comminuted Turkey Meat. J. Food Sci. 1980, 45, 1105–1112. [Google Scholar] [CrossRef]
- Kanner, J.; Harel, S.; Shagalovich, J.; Berman, S. Antioxidative Effect of Nitrite in Cured Meat Products: Nitric Oxide-Iron Complexes of Low Molecular Weight. J. Agric. Food Chem. 1984, 32, 512–515. [Google Scholar] [CrossRef]
- Kanner, J.; Harel, S.; Granit, R. Nitric Oxide as an Antioxidant. Arch. Biochem. Biophys. 1991, 289, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Kanner, J.; Harel, S.; Granit, R. Nitric Oxide, an Inhibitor of Lipid Oxidation by Lipoxygenase, Cyclooxygenase and Hemoglobin. Lipids 1992, 27, 46. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.G.; El-Emam, S.Z.; Mohamed, E.A.; Abd Ellah, M.F. Dimethyl Fumarate and Curcumin Attenuate Hepatic Ischemia/Reperfusion Injury via Nrf2/HO-1 Activation and Anti-Inflammatory Properties. Int. Immunopharmacol. 2020, 80, 106131. [Google Scholar] [CrossRef] [PubMed]
- Wilms, H.; Sievers, J.; Rickert, U.; Rostami-Yazdi, M.; Mrowietz, U.; Lucius, R. Dimethylfumarate Inhibits Microglial and Astrocytic Inflammation by Suppressing the Synthesis of Nitric Oxide, IL-1β, TNF-α and IL-6 in an in-Vitro Model of Brain Inflammation. J. Neuroinflam. 2010, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhao, S.; Fu, Y.; Yan, L.; Feng, Y.; Chen, Y.; Wu, Y.; Deng, Y.; Zhang, G.; Chen, Z. Computational Repositioning of Dimethyl Fumarate for Treating Alcoholic Liver Disease. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egnatchik, R.A.; Leamy, A.K.; Noguchi, Y.; Shiota, M.; Young, J.D. Palmitate-Induced Activation of Mitochondrial Metabolism Promotes Oxidative Stress and Apoptosis in H4IIEC3 Rat Hepatocytes. Metabolism 2014, 63, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Shpaizer, A.; Nussinovich, A.; Kanner, J.; Tirosh, O. S-Nitroso-N-Acetylcysteine Generates Less Carcinogenic N-Nitrosamines in Meat Products than Nitrite. J. Agric. Food Chem. 2018, 66, 11459–11467. [Google Scholar] [CrossRef]
- Gupta, D.; Harvey, S.A.K.; Kenchegowda, D.; Swamynathan, S.; Swamynathan, S.K. Regulation of Mouse Lens Maturation and Gene Expression by Krüppel-like Factor 4. Exp. Eye Res. 2013, 116, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Takashi, Y.; Ouchi, Y.; Kuwahara, Y.; Igarashi, K.; Nagasawa, T.; Nabika, H.; Kurimasa, A.; Fukumoto, M.; Nishitani, Y. Lipid Peroxidation Increases Hydrogen Peroxide Permeability Leading to Cell Death in Cancer Cell Lines That Lack MtDNA. Cancer Sci. 2019, 110, 2856–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze-Topphoff, U.; Varrin-Doyer, M.; Pekarek, K.; Spencer, C.M.; Shetty, A.; Sagan, S.A.; Cree, B.A.C.; Sobel, R.A.; Wipke, B.T.; Steinman, L. Dimethyl Fumarate Treatment Induces Adaptive and Innate Immune Modulation Independent of Nrf2. Proc. Natl. Acad. Sci. USA 2016, 113, 4777–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasu, C.; Vaziri, N.D.; Li, S.; Robles, L.; Vo, K.; Takasu, M.; Pham, C.; Farzaneh, S.H.; Shimada, M.; Stamos, M.J. Treatment with Dimethyl Fumarate Ameliorates Liver Ischemia/Reperfusion Injury. World J. Gastroenterol. 2017, 23, 4508. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wu, S.; Ye, S.; Huang, H.; Zhou, Y.; Zhou, H.; Wu, S.; Mao, Y.; Shangguan, F.; Lan, L. Dimethyl Fumarate Induces Metabolic Crisie to Suppress Pancreatic Carcinoma. Front. Pharmacol. 2021, 12, 617714. [Google Scholar] [CrossRef]
- Saidu, N.E.B.; Noé, G.; Cerles, O.; Cabel, L.; Kavian-Tessler, N.; Chouzenoux, S.; Bahuaud, M.; Chéreau, C.; Nicco, C.; Leroy, K. Dimethyl Fumarate Controls the NRF2/DJ-1 Axis in Cancer Cells: Therapeutic ApplicationsAnticancer Properties of High Concentration DMF. Mol. Cancer Ther. 2017, 16, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme Oxygenase-1 Accelerates Erastin-Induced Ferroptotic Cell Death. Oncotarget 2015, 6, 24393. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M.K. Mechanisms of Cell Death in Oxidative Stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef]
- Britton, L.J.; Subramaniam, V.N.; Crawford, D.H.G. Iron and Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2016, 22, 8112. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, J.; Shen, C.; Zhang, X.; Sun, S.; Cho, M.; Sun, C.; Song, Z. Tert-Butylhydroquinone (TBHQ) Protects Hepatocytes against Lipotoxicity via Inducing Autophagy Independently of Nrf2 Activation. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2014, 1841, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the Negative Regulator of Nrf2, Keap1: A Historical Overview. Antioxid. Redox Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Nemecz, M.; Constantin, A.; Dumitrescu, M.; Alexandru, N.; Filippi, A.; Tanko, G.; Georgescu, A. The Distinct Effects of Palmitic and Oleic Acid on Pancreatic Beta Cell Function: The Elucidation of Associated Mechanisms and Effector Molecules. Front. Pharmacol. 2019, 9, 1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, V.; González-Rodríguez, Á.; Muntané, J.; Kozma, S.C.; Valverde, Á.M. Role of Hepatocyte S6K1 in Palmitic Acid-Induced Endoplasmic Reticulum Stress, Lipotoxicity, Insulin Resistance and in Oleic Acid-Induced Protection. Food Chem. Toxicol. 2015, 80, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Mikulska-Ruminska, K.; Anthonymuthu, T.S.; Levkina, A.; Shrivastava, I.H.; Kapralov, A.A.; Bayır, H.; Kagan, V.E.; Bahar, I. NO● Represses the Oxygenation of Arachidonoyl PE by 15LOX/PEBP1: Mechanism and Role in Ferroptosis. Int. J. Mol. Sci. 2021, 22, 5253. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Rai, G.; Joshi, N.; Perry, S.; Yasgar, A.; Schultz, L.; Jung, J.E.; Liu, Y.; Terasaki, Y.; Diaz, G.; Kenyon, V. Discovery of ML351, a Potent and Selective Inhibitor of Human 15-Lipoxygenase-1. In Probe Reports from the NIH Molecular Libraries Program [Internet]; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2014. [Google Scholar]
- Martínez-Clemente, M.; Ferré, N.; Titos, E.; Horrillo, R.; González-Périz, A.; Morán-Salvador, E.; López-Vicario, C.; Miquel, R.; Arroyo, V.D.; Funk, C. Disruption of the 12/15-lipoxygenase Gene (Alox15) Protects Hyperlipidemic Mice from Nonalcoholic Fatty Liver Disease. Hepatology 2010, 52, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, R.G.; Brüne, B. Regulation and Functions of 15-Lipoxygenases in Human Macrophages. Front. Pharmacol. 2019, 10, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoetzel, A.; Vagts, D.A.; Loop, T.; Humar, M.; Bauer, M.; Pahl, H.L.; Geiger, K.K.; Pannen, B.H.J. Effect of Nitric Oxide on Shock-induced Hepatic Heme Oxygenase-1 Expression in the Rat. Hepatology 2001, 33, 925–937. [Google Scholar] [CrossRef]
- Choi, A.M.; Alam, J. Heme Oxygenase-1: Function, Regulation, and Implication of a Novel Stress-Inducible Protein in Oxidant-Induced Lung Injury. Am. J. Respir. Cell Mol. Biol. 1996, 15, 9–19. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Accession Number | Reverse | Forward |
|---|---|---|---|
| 18S | NR_003278.3 | 5′-CCTCAGTTCCGAAAACCAAC-3′ | 5′-ACCGCAGCTAGGAATAATGG-3′ |
| HO-1 | NM_010442.2 | 5′-CTTCCAGGGCCGTGTAGAT-3′ | 5′-CAGAAGGGTCAGGTGTCCA-3′ |
| GCLC | NM_010295.2 | 5′-TCGCCTCCATTCAGTAACAA-3′ | 5′-CGAGGTGGAGTACATGTTGG-3′ |
| GSTA1 | NM_008181.3 | 5′-TGCAGCTTCACTGAATCTTGAAAG-3′ | 5′-CCCCTTTCCCTCTGCTGAAG-3′ |
| NQO1 | NM_008706.5 | 5′-CCTTTCAGAATGGCTGGCA-3′ | 5′-GGAAGCTGCAGACCTGGTGA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu-Halaka, D.; Shpaizer, A.; Zeigerman, H.; Kanner, J.; Tirosh, O. DMF-Activated Nrf2 Ameliorates Palmitic Acid Toxicity While Potentiates Ferroptosis Mediated Cell Death: Protective Role of the NO-Donor S-Nitroso-N-Acetylcysteine. Antioxidants 2023, 12, 512. https://doi.org/10.3390/antiox12020512
Abu-Halaka D, Shpaizer A, Zeigerman H, Kanner J, Tirosh O. DMF-Activated Nrf2 Ameliorates Palmitic Acid Toxicity While Potentiates Ferroptosis Mediated Cell Death: Protective Role of the NO-Donor S-Nitroso-N-Acetylcysteine. Antioxidants. 2023; 12(2):512. https://doi.org/10.3390/antiox12020512
Chicago/Turabian StyleAbu-Halaka, Diana, Adi Shpaizer, Haim Zeigerman, Joseph Kanner, and Oren Tirosh. 2023. "DMF-Activated Nrf2 Ameliorates Palmitic Acid Toxicity While Potentiates Ferroptosis Mediated Cell Death: Protective Role of the NO-Donor S-Nitroso-N-Acetylcysteine" Antioxidants 12, no. 2: 512. https://doi.org/10.3390/antiox12020512