
Role of Mitophagy in Regulating Intestinal Oxidative Damage


Abstract
:
1. Introduction
2. Oxidative Stress and Intestinal Oxidative Damage
Intestinal Oxidative Damage
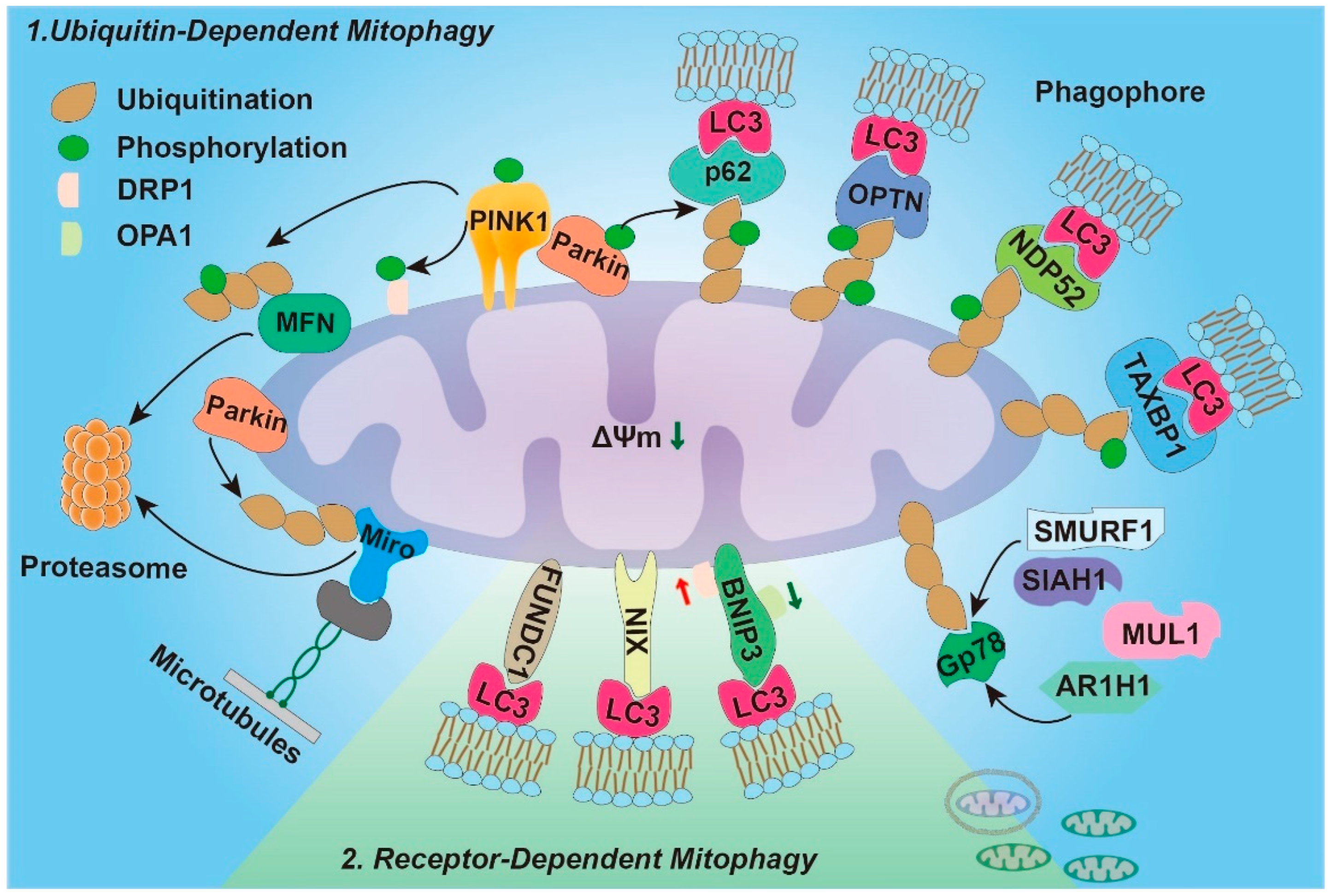
3. Mitophagy Pathway
3.1. Ubiquitin-Dependent Mitophagy
3.2. Ubiquitin-Independent Mitophagy (Receptor-Dependent)
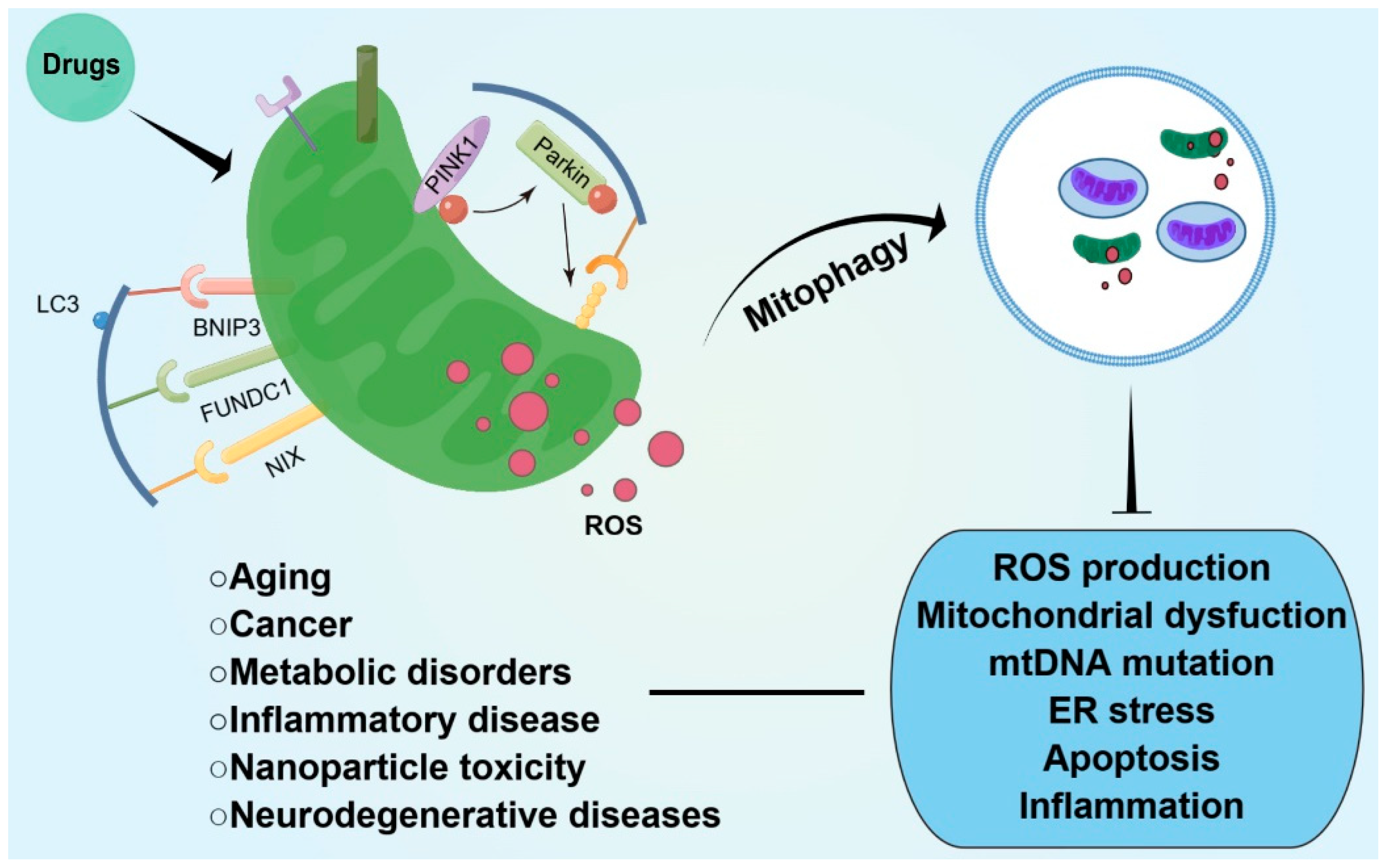
4. Mitophagy and Oxidative Stress in Intestinal Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Guo, Y.; Liu, Y.; Zhao, S.; Xu, W.; Li, Y.; Zhao, P.; Wang, D.; Cheng, H.; Ke, Y.; Zhang, X. Oxidative stress-induced FABP5 S-glutathionylation protects against acute lung injury by suppressing inflammation in macrophages. Nat. Commun. 2021, 12, 7094. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, J.Y.; Liu, X.S.; Chen, H.Z.; Ai, Y.L.; Cheng, K.; Sun, R.Y.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar] [CrossRef]
- Li, M.; Li, L.; Su, K.; Liu, X.; Zhang, T.; Liang, Y.; Jing, D.; Yang, X.; Zheng, D.; Cui, Z.; et al. Highly effective and noninvasive near-infrared eradication of a staphylococcus aureus biofilm on implants by a photoresponsive coating within 20 min. Adv. Sci 2019, 6, 1900599. [Google Scholar] [CrossRef]
- Faye, A.S.; Allin, K.H.; Iversen, A.T.; Agrawal, M.; Faith, J.; Colombel, J.-F.; Jess, T. Antibiotic use as a risk factor for inflammatory bowel disease across the ages: A population-based cohort study. Gut 2023, 1–8. [Google Scholar] [CrossRef]
- Kaplan, G.G.; Windsor, J.W. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 56–66. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of Oxidative stress in inflammatory bowel disease and potential antioxidant therapies. Oxid. Med. Cell Longev. 2017, 2017, 4535194. [Google Scholar] [CrossRef]
- Vo, M.T.; Smith, B.J.; Nicholas, J.; Choi, Y.B. Activation of NIX-mediated mitophagy by an interferon regulatory factor homologue of human herpesvirus. Nat. Commun. 2019, 10, 3203. [Google Scholar] [CrossRef]
- Krestinin, R.; Baburina, Y.; Odinokova, I.; Kruglov, A.; Fadeeva, I.; Zvyagina, A.; Sotnikova, L.; Krestinina, O. Isoproterenol-induced permeability transition pore-related dysfunction of heart mitochondria is attenuated by astaxanthin. Biomedicines 2020, 8, 437. [Google Scholar] [CrossRef]
- Lan, Q.; Lim, U.; Liu, C.S.; Weinstein, S.J.; Chanock, S.; Bonner, M.R.; Virtamo, J.; Albanes, D.; Rothman, N. A prospective study of mitochondrial DNA copy number and risk of non-Hodgkin lymphoma. Blood 2008, 112, 4247–4249. [Google Scholar] [CrossRef]
- Waltz, F.; Salinas-Giegé, T.; Englmeier, R.; Meichel, H.; Soufari, H.; Kuhn, L.; Pfeffer, S.; Förster, F.; Engel, B.D.; Giegé, P.; et al. How to build a ribosome from RNA fragments in Chlamydomonas mitochondria. Nat. Commun. 2021, 12, 7176. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2015, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Nie, P.; Zhou, C.; Hu, Y.; Duan, S.; Gu, M.; Jiang, D.; Wang, Y.; Deng, Z.; Chen, J.; et al. Oxidative stress-induced mitophagy is suppressed by the miR-106b-93-25 cluster in a protective manner. Cell Death Dis. 2021, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Deng, X.; Lim, G.G.Y.; Xie, S.; Zhou, Z.D.; Lim, K.L.; Tan, E.K. Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis. 2017, 8, e3097. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Zhang, Q.; Kuang, H.; Chen, C.; Yan, J.; Do-Umehara, H.C.; Liu, X.Y.; Dada, L.; Ridge, K.M.; Chandel, N.S.; Liu, J. The kinase Jnk2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor ARF for degradation. Nat. Immunol. 2015, 16, 458–466. [Google Scholar] [CrossRef]
- Zhu, C.L.; Yao, R.Q.; Li, L.X.; Li, P.; Xie, J.; Wang, J.F.; Deng, X.M. Mechanism of mitophagy and its role in sepsis induced organ dysfunction: A review. Front. Cell Dev. Biol. 2021, 9, 664896. [Google Scholar] [CrossRef]
- Xing, W.; Yang, L.; Peng, Y.; Wang, Q.; Gao, M.; Yang, M.; Xiao, X. Ginsenoside Rg3 attenuates sepsis-induced injury and mitochondrial dysfunction in liver via AMPK-mediated autophagy flux. Biosci. Rep. 2017, 37, BSR20170934. [Google Scholar] [CrossRef] [PubMed]
- Munson, M.J.; Mathai, B.J.; Ng, M.Y.W.; Trachsel-Moncho, L.; de la Ballina, L.R.; Schultz, S.W.; Aman, Y.; Lystad, A.H.; Singh, S.; Singh, S.; et al. GAK and PRKCD are positive regulators of PRKN-independent mitophagy. Nat. Commun. 2021, 12, 6101. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Allen, R.G. Oxidative stress as a causal factor in differentiation and aging: A unifying hypothesis. Exp. Gerontol. 1990, 25, 499–522. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.X.; Liu, R.; Song, K.; Chen, L. Structures of human dual oxidase 1 complex in low-calcium and high-calcium states. Nat. Commun. 2021, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.H.; Huang, H.L.; Lin, Y.Y.; Tsui, K.H.; Chen, P.C.; Cheng, S.Y.; Chong, I.W.; Sung, P.J.; Tai, M.H.; Wen, Z.H.; et al. BA6 induces apoptosis via stimulation of reactive oxygen species and inhibition of oxidative phosphorylation in human lung cancer cells. Oxid. Med. Cell Longev. 2019, 2019, 6342104. [Google Scholar] [CrossRef]
- Thanas, C.; Ziros, P.G.; Chartoumpekis, D.V.; Renaud, C.O.; Sykiotis, G.P. The Keap1/Nrf2 signaling pathway in the Thyroid-2020 update. Antioxidants 2020, 9, 1082. [Google Scholar] [CrossRef]
- Sugiharto, S.; Yudiarti, T.; Isroli, I. Assay of antioxidant potential of two filamentous fungi isolated from the indonesian fermented dried cassava. Antioxidants 2016, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, S.; Calcaterra, V.; Pelizzo, G.; Bramanti, P.; Mazzon, E. Prenatal hypoxia and placental oxidative stress: Insights from animal models to clinical evidences. Antioxidants 2020, 9, 414. [Google Scholar] [CrossRef]
- Li, M.; Sun, T.; Wu, X.; An, P.; Wu, X.; Dang, H. Autophagy in the HTR-8/SVneo cell oxidative stress model is associated with the NLRP1 inflammasome. Oxid. Med. Cell Longev. 2021, 2021, 2353504. [Google Scholar] [CrossRef] [PubMed]
- Deters, E.L.; Stokes, R.S.; Genther-Schroeder, O.N.; Hansen, S.L. Effects of a Saccharomyces cerevisiae fermentation product in receiving diets of newly weaned beef steers. II. Digestibility and response to a vaccination challenge. J. Anim. Sci. 2018, 96, 3906–3915. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, F.S.F.; Clermont, L.; Tung, Q.N.; Antelmann, H.; Seibold, G.M. The industrial organism corynebacterium glutamicum requires mycothiol as antioxidant to resist against oxidative stress in bioreactor cultivations. Antioxidants 2020, 9, 969. [Google Scholar] [CrossRef]
- Chen, X.; Yi, H.; Liu, S.; Zhang, Y.; Su, Y.; Liu, X.; Bi, S.; Lai, H.; Zeng, Z.; Li, G. Probiotics improve eating disorders in mandarin fish (siniperca chuatsi) induced by a pellet feed diet via stimulating immunity and regulating gut microbiota. Microorganisms 2021, 9, 1288. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Sarkar, A.; Ryan, K.A.; Fox, S.; Berger, A.H.; Juncadella, I.J.; Bimczok, D.; Smythies, L.E.; Harris, P.R.; Ravichandran, K.S.; et al. Brain angiogenesis inhibitor 1 is expressed by gastric phagocytes during infection with Helicobacter pylori and mediates the recognition and engulfment of human apoptotic gastric epithelial cells. FASEB J. 2014, 28, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Laforgia, N.; Di Mauro, A.; Favia Guarnieri, G.; Varvara, D.; De Cosmo, L.; Panza, R.; Capozza, M.; Baldassarre, M.E.; Resta, N. The role of oxidative stress in the pathomechanism of congenital malformations. Oxid. Med. Cell Longev. 2018, 2018, 7404082. [Google Scholar] [CrossRef] [PubMed]
- Girotti, A.W. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 1998, 39, 1529–1542. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.O.; Gerez, J.R.; Hohmann, M.S.N.; Verri, W.A.; Bracarense, A.P.F.R.L. Phytic acid decreases oxidative stress and intestinal lesions induced by fumonisin b₁ and deoxynivalenol in intestinal explants of pigs. Toxins 2019, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Zhang, F.; Chen, L.; Yang, Q.; Su, H.; Yang, X.; He, H.; Ling, M.; Zheng, J.; Duan, C.; et al. Dietary chenodeoxycholic acid improves growth performance and intestinal health by altering serum metabolic profiles and gut bacteria in weaned piglets. Anim. Nutr. 2021, 7, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Gago, S.; Overton, N.L.D.; Ben-Ghazzi, N.; Novak-Frazer, L.; Read, N.D.; Denning, D.W.; Bowyer, P. Lung colonization by Aspergillus fumigatus is controlled by ZNF77. Nat. Commun. 2018, 9, 3835. [Google Scholar] [CrossRef]
- Guttman, J.A.; Finlay, B.B. Tight junctions as targets of infectious agents. Biochim. Biophys. Acta 2009, 1788, 832–841. [Google Scholar] [CrossRef]
- Wilson, K.M.; Rodrigues, D.R.; Briggs, W.N.; Duff, A.F.; Chasser, K.M.; Bottje, W.G.; Bielke, L.R. Impact of in ovo administered pioneer colonizers on intestinal proteome on day of hatch. Poult. Sci. 2020, 99, 1254–1266. [Google Scholar] [CrossRef]
- Jiao, N.; Wu, Z.; Ji, Y.; Wang, B.; Dai, Z.; Wu, G. L-Glutamate enhances barrier and antioxidative functions in intestinal porcine epithelial cells. J. Nutr. 2015, 145, 2258–2264. [Google Scholar] [CrossRef]
- Elias, B.C.; Suzuki, T.; Seth, A.; Giorgianni, F.; Kale, G.; Shen, L.; Turner, J.R.; Naren, A.; Desiderio, D.M.; Rao, R. Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. J. Biol. Chem. 2009, 284, 1559–1569. [Google Scholar] [CrossRef] [Green Version]
- Kawauchiya, T.; Takumi, R.; Kudo, Y.; Takamori, A.; Sasagawa, T.; Takahashi, K.; Kikuchi, H. Correlation between the destruction of tight junction by patulin treatment and increase of phosphorylation of ZO-1 in Caco-2 human colon cancer cells. Toxicol. Lett. 2011, 205, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Dee, C.M.; Shen, J. Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front. Biosci. (Schol. Ed.) 2011, 3, 1216–1231. [Google Scholar] [CrossRef]
- Saha, A.; Sarkar, C.; Singh, S.P.; Zhang, Z.; Munasinghe, J.; Peng, S.; Chandra, G.; Kong, E.; Mukherjee, A.B. The blood-brain barrier is disrupted in a mouse model of infantile neuronal ceroid lipofuscinosis: Amelioration by resveratrol. Hum. Mol. Genet. 2012, 21, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- González-Mariscal, L.; Tapia, R.; Chamorro, D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 2008, 1778, 729–756. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, C.; Zhou, L.; Zhong, Z.; Zhu, W.; Huang, Y.; Zhang, Z.; Gorgels, T.G.M.F.; Berendschot, T.T.J.M. Effects of dietary supplementation with epidermal growth factor-expressing Saccharomyces cerevisiae on duodenal development in weaned piglets. Br. J. Nutr. 2016, 115, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Reese, A.T.; Cho, E.H.; Klitzman, B.; Nichols, S.P.; Wisniewski, N.A.; Villa, M.M.; Durand, H.K.; Jiang, S.; Midani, F.S.; Nimmagadda, S.N.; et al. Antibiotic-induced changes in the microbiota disrupt redox dynamics in the gut. Elife 2018, 7, e35987. [Google Scholar] [CrossRef]
- Ershler, W.B. A gripping reality: Oxidative stress, inflammation, and the pathway to frailty. J. Appl. Physiol. (1985) 2007, 103, 3–5. [Google Scholar] [CrossRef]
- Winter, S.E.; Lopez, C.A.; Bäumler, A.J. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. 2013, 14, 319–327. [Google Scholar] [CrossRef]
- Ehrlich, A.M.; Pacheco, A.R.; Henrick, B.M.; Taft, D.; Xu, G.; Huda, M.N.; Mishchuk, D.; Goodson, M.L.; Slupsky, C.; Barile, D.; et al. Indole-3-lactic acid associated with Bifidobacterium-dominated microbiota significantly decreases inflammation in intestinal epithelial cells. BMC Microbiol. 2020, 20, 357. [Google Scholar] [CrossRef]
- Gao, D.; Gao, Z.; Zhu, G. Antioxidant effects of Lactobacillus plantarum via activation of transcription factor Nrf2. Food Funct. 2013, 4, 982–989. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Wang, Y.; Xu, H.; Mei, X.; Yu, D.; Wang, Y.; Li, W. Antioxidant properties of probiotic bacteria. Nutrients 2017, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Prado, C.; Michels, M.; Ávila, P.; Burger, H.; Milioli, M.V.M.; Dal-Pizzol, F. The protective effects of fecal microbiota transplantation in an experimental model of necrotizing enterocolitis. J. Pediatr. Surg. 2019, 54, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Stojanovski, S.; Maechler, P. Mitochondrial hormesis in pancreatic β cells: Does uncoupling protein 2 play a role? Oxid. Med. Cell Longev. 2012, 2012, 740849. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Li, Y.J.; Lei, Y.H.; Hu, N.; Chen, W.M.; Yao, Z.; Yu, M.; Liu, J.S.; Ye, W.C.; Zhang, D.M. A piperazidine derivative of 23-hydroxy betulinic acid induces a mitochondria-derived ROS burst to trigger apoptotic cell death in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2016, 35, 192. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.K.; Melendez, J.A. Mitochondrial redox control of matrix metalloproteinases. Free Radic. Biol. Med. 2004, 37, 768–784. [Google Scholar] [CrossRef]
- Chien, C.-C.; Wu, M.-S.; Shen, S.-C.; Ko, C.-H.; Chen, C.-H.; Yang, L.-L.; Chen, Y.-C. Activation of JNK contributes to evodiamine-induced apoptosis and G2/M arrest in human colorectal carcinoma cells: A structure-activity study of evodiamine. PLoS ONE 2014, 9, e99729. [Google Scholar] [CrossRef]
- Saita, S.; Shirane, M.; Nakayama, K.I. Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun. 2013, 4, 1410. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen. Res. 2019, 14, 749–756. [Google Scholar] [CrossRef]
- Vernucci, E.; Tomino, C.; Molinari, F.; Limongi, D.; Aventaggiato, M.; Sansone, L.; Tafani, M.; Russo, M.A. Mitophagy and oxidative stress in cancer and aging: Focus on sirtuins and nanomaterials. Oxid. Med. Cell Longev. 2019, 2019, 6387357. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, A.; Gibellini, L.; Zanini, G.; Nasi, M.; Cossarizza, A.; Pinti, M. Mitophagy and oxidative stress: The role of aging. Antioxidants 2021, 10, 794. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Park, M.H.; Hwang, C.J.; Kim, Y.; Hwang, D.Y.; Han, S.B.; Hong, J.T. Parkin deficiency prevents chronic ethanol-induced hepatic lipid accumulation through β-catenin accumulation. Cell Commun. Signal. 2019, 17, 104. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Lorenz, H.; Hehn, B.; Lemberg, M.K. Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 2015, 11, 1484–1498. [Google Scholar] [CrossRef]
- Zhuang, N.; Li, L.; Chen, S.; Wang, T. PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 2016, 7, e2501. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Aguirre, J.D.; Dunkerley, K.M.; Mercier, P.; Shaw, G.S. Structure of phosphorylated UBL domain and insights into PINK1-orchestrated parkin activation. Proc. Natl. Acad. Sci. USA 2017, 114, 298–303. [Google Scholar] [CrossRef]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Luciani, A.; Schumann, A.; Berquez, M.; Chen, Z.; Nieri, D.; Failli, M.; Debaix, H.; Festa, B.P.; Tokonami, N.; Raimondi, A.; et al. Impaired mitophagy links mitochondrial disease to epithelial stress in methylmalonyl-CoA mutase deficiency. Nat. Commun. 2020, 11, 970. [Google Scholar] [CrossRef]
- Scheffer, D.d.L.; Garcia, A.A.; Lee, L.; Mochly-Rosen, D.; Ferreira, J.C.B. Mitochondrial fusion, fission, and mitophagy in cardiac diseases: Challenges and therapeutic opportunities. Antioxid. Redox. Signal. 2022, 36, 844–863. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Bai, J.; Tian, X.; Zhao, X.; Liu, W.; Duan, X.; Shang, W.; Fan, H.-Y.; Tong, C. Mitoguardin regulates mitochondrial fusion through MitoPLD and is required for neuronal homeostasis. Mol. Cell 2016, 61, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Smith, H.L.; Chau, K.-Y.; Schapira, A.H.V. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Kagan, V.E.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Desbourdes, C.; Cottet-Rousselle, C.; Dar, H.H.; Verma, M.; Tyurin, V.A.; Kapralov, A.A.; et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016, 23, 1140–1151. [Google Scholar] [CrossRef]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444. [Google Scholar] [CrossRef] [PubMed]
- Montava-Garriga, L.; Ganley, I.G. Outstanding questions in mitophagy: What we do and do not know. J. Mol. Biol. 2020, 432, 206–230. [Google Scholar] [CrossRef]
- Praharaj, P.P.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Sethi, G.; Bhutia, S.K. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: Its implication in cancer therapeutics. Cell Mol. Life Sci. 2019, 76, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Behera, B.P.; Mishra, S.R.; Bhutia, S.K. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol. 2020, 66, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, Å.B.; Lamark, T.; Johansen, T. The LIR motif—Crucial for selective autophagy. J. Cell Sci 2013, 126, 3237–3247. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, Q.; Sun, H.; Liu, L.; Wang, C.; Li, Z.; Xu, Y.; Wang, L.; Zhang, L.; Zhang, H.; et al. BNIP3 (BCL2 interacting protein 3) regulates pluripotency by modulating mitochondrial homeostasis via mitophagy. Cell Death Dis. 2022, 13, 334. [Google Scholar] [CrossRef]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, P.; Li, R.; Ren, J.; Zhou, H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox. Biol. 2020, 30, 101415. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 2017, 168, 224–238.e10. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Hu, R.; Wang, Y.; Liu, L.; You, H.; Zhang, J.; Wu, X.; Pei, T.; Wang, F.; Lu, L.; et al. Atractylenolide III attenuates muscle wasting in chronic kidney disease via the oxidative stress-mediated PI3K/AKT/mTOR pathway. Oxid. Med. Cell Longev. 2019, 2019, 1875471. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hou, Y.; Yi, D.; Wang, L.; Ding, B.; Chen, X.; Long, M.; Liu, Y.; Wu, G. Protective effects of N-acetylcysteine on acetic acid-induced colitis in a porcine model. BMC Gastroenterol. 2013, 13, 133. [Google Scholar] [CrossRef]
- Arab, H.H.; Al-Shorbagy, M.Y.; Abdallah, D.M.; Nassar, N.N. Telmisartan attenuates colon inflammation, oxidative perturbations and apoptosis in a rat model of experimental inflammatory bowel disease. PLoS ONE 2014, 9, e97193. [Google Scholar] [CrossRef]
- Mangerich, A.; Dedon, P.C.; Fox, J.G.; Tannenbaum, S.R.; Wogan, G.N. Chemistry meets biology in colitis-associated carcinogenesis. Free Radic. Res. 2013, 47, 958–986. [Google Scholar] [CrossRef]
- Bouzid, D.; Gargouri, B.; Mansour, R.B.; Amouri, A.; Tahri, N.; Lassoued, S.; Masmoudi, H. Oxidative stress markers in intestinal mucosa of Tunisian inflammatory bowel disease patients. Saudi J. Gastroenterol. 2013, 19, 131–135. [Google Scholar] [CrossRef]
- Dudzińska, E.; Gryzinska, M.; Ognik, K.; Gil-Kulik, P.; Kocki, J. Oxidative stress and effect of treatment on the oxidation product decomposition processes in IBD. Oxid. Med. Cell Longev. 2018, 2018, 7918261. [Google Scholar] [CrossRef]
- Dashdorj, A.; Jyothi, K.R.; Lim, S.; Jo, A.; Nguyen, M.N.; Ha, J.; Yoon, K.S.; Kim, H.J.; Park, J.H.; Murphy, M.P.; et al. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013, 11, 178. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Yang, S.; Zhao, D.; Wang, M. A polysaccharide from cultured mycelium of Hericium erinaceus relieves ulcerative colitis by counteracting oxidative stress and improving mitochondrial function. Int. J. Biol. Macromol. 2019, 125, 572–579. [Google Scholar] [CrossRef]
- Mai, C.T.; Wu, M.M.; Wang, C.L.; Su, Z.R.; Cheng, Y.Y.; Zhang, X.J. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol. Immunol. 2019, 105, 76–85. [Google Scholar] [CrossRef]
- Lopez-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-Garcia, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- McGary, C.T.; Lowe, M.C. Educational case: Idiopathic inflammatory bowel disease. Acad. Pathol. 2020, 7, 2374289520937433. [Google Scholar] [CrossRef]
- Veres-Székely, A.; Bernáth, M.; Pap, D.; Rokonay, R.; Szebeni, B.; Takács, I.M.; Lippai, R.; Cseh, Á.; Szabó, A.J.; Vannay, Á. PARK7 diminishes oxidative stress-induced mucosal damage in celiac disease. Oxid. Med. Cell Longev. 2020, 2020, 4787202. [Google Scholar] [CrossRef]
- Szaflarska-Poplawska, A.; Siomek, A.; Czerwionka-Szaflarska, M.; Gackowski, D.; Rózalski, R.; Guz, J.; Szpila, A.; Zarakowska, E.; Olinski, R. Oxidatively damaged DNA/oxidative stress in children with celiac disease. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1960–1965. [Google Scholar] [CrossRef]
- Alzoghaibi, M.A. Concepts of oxidative stress and antioxidant defense in Crohn’s disease. World J. Gastroenterol. 2013, 19, 6540–6547. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, L.; Wang, Z.; Chen, S.; Feng, S.; He, Y.; Zhang, S. Network pharmacology-based strategy to identify the pharmacological mechanisms of pulsatilla decoction against Crohn’s disease. Front. Pharmacol. 2022, 13, 844685. [Google Scholar] [CrossRef]
- Khaloian, S.; Rath, E.; Hammoudi, N.; Gleisinger, E.; Blutke, A.; Giesbertz, P.; Berger, E.; Metwaly, A.; Waldschmitt, N.; Allez, M.; et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 2020, 69, 1939–1951. [Google Scholar] [CrossRef]
- Jackson, D.N.; Panopoulos, M.; Neumann, W.L.; Turner, K.; Cantarel, B.L.; Thompson-Snipes, L.; Dassopoulos, T.; Feagins, L.A.; Souza, R.F.; Mills, J.C.; et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 2020, 69, 1928–1938. [Google Scholar] [CrossRef]
- Pierre, N.; Salée, C.; Massot, C.; Blétard, N.; Mazzucchelli, G.; Smargiasso, N.; Morsa, D.; Baiwir, D.; De Pauw, E.; Reenaers, C.; et al. Proteomics Highlights Common and Distinct Pathophysiological Processes Associated with Ileal and Colonic Ulcers in Crohn’s Disease. J. Crohns. Colitis. 2020, 14, 205–215. [Google Scholar] [CrossRef]
- Kugathasan, S.; Denson, L.A.; Walters, T.D.; Kim, M.O.; Marigorta, U.M.; Schirmer, M.; Mondal, K.; Liu, C.; Griffiths, A.; Noe, J.D.; et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: A multicentre inception cohort study. Lancet 2017, 389, 1710–1718. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, J.; Yu, J.T.; Tan, Y.J.; Ng, A.; De Guzman, M.F.; Natividad, B.; Daroy, M.L.; Cano, J.; Yu, J.; Lian, M.M.; et al. Novel optineurin frameshift insertion in a family with frontotemporal dementia and parkinsonism without amyotrophic lateral sclerosis. Front. Neurol. 2021, 12, 645913. [Google Scholar] [CrossRef] [PubMed]
- Weil, R.; Laplantine, E.; Curic, S.; Génin, P. Role of optineurin in the mitochondrial dysfunction: Potential implications in neurodegenerative diseases and cancer. Front. Immunol. 2018, 9, 1243. [Google Scholar] [CrossRef]
- Cao, S.; Xiao, H.; Li, X.; Zhu, J.; Gao, J.; Wang, L.; Hu, C. AMPK-PINK1/Parkin mediated mitophagy is necessary for alleviating oxidative stress-induced intestinal epithelial barrier damage and mitochondrial energy metabolism dysfunction in IPEC-J2. Antioxidants 2021, 10, 2010. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, J.; Peng, J.; Wei, H. Oregano essential oil induces SOD1 and GSH expression through Nrf2 activation and alleviates hydrogen peroxide-induced oxidative damage in IPEC-J2 Cells. Oxid. Med. Cell Longev. 2016, 2016, 5987183. [Google Scholar] [CrossRef]
- Yin, J.; Wu, M.; Li, Y.; Ren, W.; Xiao, H.; Chen, S.; Li, C.; Tan, B.; Ni, H.; Xiong, X.; et al. Toxicity assessment of hydrogen peroxide on Toll-like receptor system, apoptosis, and mitochondrial respiration in piglets and IPEC-J2 cells. Oncotarget 2017, 8, 3124–3131. [Google Scholar] [CrossRef]
- Wen, Z.S.; Ma, L.; Xiang, X.W.; Tang, Z.; Guan, R.F.; Qu, Y.L. Protective effect of low molecular-weight seleno-aminopolysaccharides against H2O2-induecd oxidative stress in intestinal epithelial cells. Int. J. Biol. Macromol. 2018, 112, 745–753. [Google Scholar] [CrossRef]
- Liang, D.; Zhuo, Y.; Guo, Z.; He, L.; Wang, X.; He, Y.; Li, L.; Dai, H. SIRT1/PGC-1 pathway activation triggers autophagy/mitophagy and attenuates oxidative damage in intestinal epithelial cells. Biochimie 2020, 170, 10–20. [Google Scholar] [CrossRef]
- Cao, S.; Wang, C.; Yan, J.; Li, X.; Wen, J.; Hu, C. Curcumin ameliorates oxidative stress-induced intestinal barrier injury and mitochondrial damage by promoting Parkin dependent mitophagy through AMPK-TFEB signal pathway. Free Radic. Biol. Med. 2020, 147, 8–22. [Google Scholar] [CrossRef]
- Li, X.; Wang, C.; Zhu, J.; Lin, Q.; Yu, M.; Wen, J.; Feng, J.; Hu, C. Sodium butyrate ameliorates oxidative stress-induced intestinal epithelium barrier injury and mitochondrial damage through AMPK-Mitophagy pathway. Oxid. Med. Cell Longev. 2022, 2022, 3745135. [Google Scholar] [CrossRef]
- Yan, S.; Qiao, L.; Dou, X.; Song, X.; Chen, Y.; Zhang, B.; Xu, C. Biogenic selenium nanoparticles by Lactobacillus casei ATCC 393 alleviate the intestinal permeability, mitochondrial dysfunction and mitophagy induced by oxidative stress. Food Funct. 2021, 12, 7068–7080. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Ji, S.; Jia, P.; Chen, Y.; Li, Y.; Wang, T. Resveratrol and its derivative pterostilbene attenuate oxidative stress-induced intestinal injury by improving mitochondrial redox homeostasis and function via SIRT1 signaling. Free Radic. Biol. Med. 2021, 177, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Shen, Z.; Wang, C.; Zhang, Q.; Hong, Q.; He, Y.; Hu, C. Resveratrol improves intestinal barrier function, alleviates mitochondrial dysfunction and induces mitophagy in diquat challenged piglets. Food Funct. 2019, 10, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Wu, H.; Wang, C.; Zhang, Q.; Jiao, L.; Lin, F.; Hu, C.H. Diquat-induced oxidative stress increases intestinal permeability, impairs mitochondrial function, and triggers mitophagy in piglets. J. Anim. Sci. 2018, 96, 1795–1805. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar] [CrossRef]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef]
- Fang, W.J.; Wang, C.J.; He, Y.; Zhou, Y.L.; Peng, X.D.; Liu, S.K. Resveratrol alleviates diabetic cardiomyopathy in rats by improving mitochondrial function through PGC-1α deacetylation. Acta Pharmacol. Sin. 2018, 39, 59–73. [Google Scholar] [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef]
- Kim, S.J.; Jang, J.Y.; Kim, E.J.; Cho, E.K.; Ahn, D.G.; Kim, C.; Park, H.S.; Jeong, S.W.; Lee, S.H.; Kim, S.G.; et al. Ginsenoside Rg3 restores hepatitis C virus-induced aberrant mitochondrial dynamics and inhibits virus propagation. Hepatology 2017, 66, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Han, Y.; Wang, C.; Zhan, Z.; Lu, M.; Jin, J.; Liu, X. Mechanisms of panax notoginseng saponins pretreatment on reducing H/R injury in cardiomyocytes by activating HIF-1α/BNIP3 mitochondrial autophagy signaling pathways. China Mod. Dr. 2022, 60, 15–19. [Google Scholar]
- Cai, C.; Guo, Z.; Chang, X.; Li, Z.; Wu, F.; He, J.; Cao, T.; Wang, K.; Shi, N.; Zhou, H.; et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox. Biol. 2022, 52, 102288. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drugs | Disease | Model | Effect and Mechanism |
|---|---|---|---|
| Urolithin A | Alzheimer’s disease | human neuronal SH-SY5Y cells; AD mice | Urolithin A induces neuronal mitophagy by increasing key mitophagy-related proteins. It can also enhance the phagocytic efficiency of microglia, and mitigate NLRP3/caspase-1-dependent neuroinflammation [124] |
| Rapamycin | Parkinson’s disease | SH-SY5Y cells treated with rotenone | Rapamycin enhances mitophagy through inhibition of mTOR to clear the Cytochrome c, thereby inhibiting the occurrence of apoptosis caused by rotenone [125] |
| Pifithrin-α | Diabetes | streptozotocin-treated and db/db mice | Pifithrin-α induces mitophagy by promoting Parkin activity through p53 downregulation, then ameliorates mitochondrial dysfunction and glucose intolerance [126] |
| Resveratrol | Diabetic cardiomyopathy | rats by a high-fat diet combined with STZ injection | Resveratrol alleviates cardiac dysfunction in diabetes by improving mitochondrial function via SIRT1-mediated PGC-1α deacetylation [127] |
| Rapamycin | Leigh syndrome | Ndufs4−/− mice | Rapamycin delays the onset of neurological symptoms, reduces neuroinflammation, and prevents brain lesions [128] |
| Ginsenoside Rg3 | Hepatitis C Virus | HCV-infected Huh7 and Huh7.5.1 cells | Ginsenoside Rg3 restores the HCV-induced dynamin-related protein 1-mediated aberrant mitochondrial fission process, thereby resulting in the suppression of persistent HCV infection [129] |
| Panax notoginseng saponins | Hypoxia/reoxygenation | H9c2 cells with H/R injury | Panax notoginseng saponins reduce H/R injury in cardiomyocytes by activating HIF-1α/BNIP3 mitochondrial autophagy signaling pathways [130] |
| Empagliflozin | Cardiac microvascular ischemia/reperfusion | myocardial ischemia (45 min)/reperfusion (2 h) injury mice | Empagliflozin normalizes mitochondrial fission and fusion, neutralizes supraphysiologic ROS concentrations, and suppresses mitochondrial apoptosis by activating FUNDC1-dependent mitophagy through the AMPKα1/ULK1 pathway [131] |
| Spermidine | Aging | yeast, flies, nematodes, and human immune cells | Spermidine plays an important role in mitophagy-mediated cytoprotective and anti-aging effects through energy metabolism restoration [132] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, X.; Tang, L.; Zhong, R.; Liu, L.; Chen, L.; Zhang, H. Role of Mitophagy in Regulating Intestinal Oxidative Damage. Antioxidants 2023, 12, 480. https://doi.org/10.3390/antiox12020480
Wen X, Tang L, Zhong R, Liu L, Chen L, Zhang H. Role of Mitophagy in Regulating Intestinal Oxidative Damage. Antioxidants. 2023; 12(2):480. https://doi.org/10.3390/antiox12020480
Chicago/Turabian StyleWen, Xiaobin, Lixin Tang, Ruqing Zhong, Lei Liu, Liang Chen, and Hongfu Zhang. 2023. "Role of Mitophagy in Regulating Intestinal Oxidative Damage" Antioxidants 12, no. 2: 480. https://doi.org/10.3390/antiox12020480