
Is Nuclear Factor Erythroid 2-Related Factor 2 a Target for the Intervention of Cytokine Storms?
 , , , and
, , , and 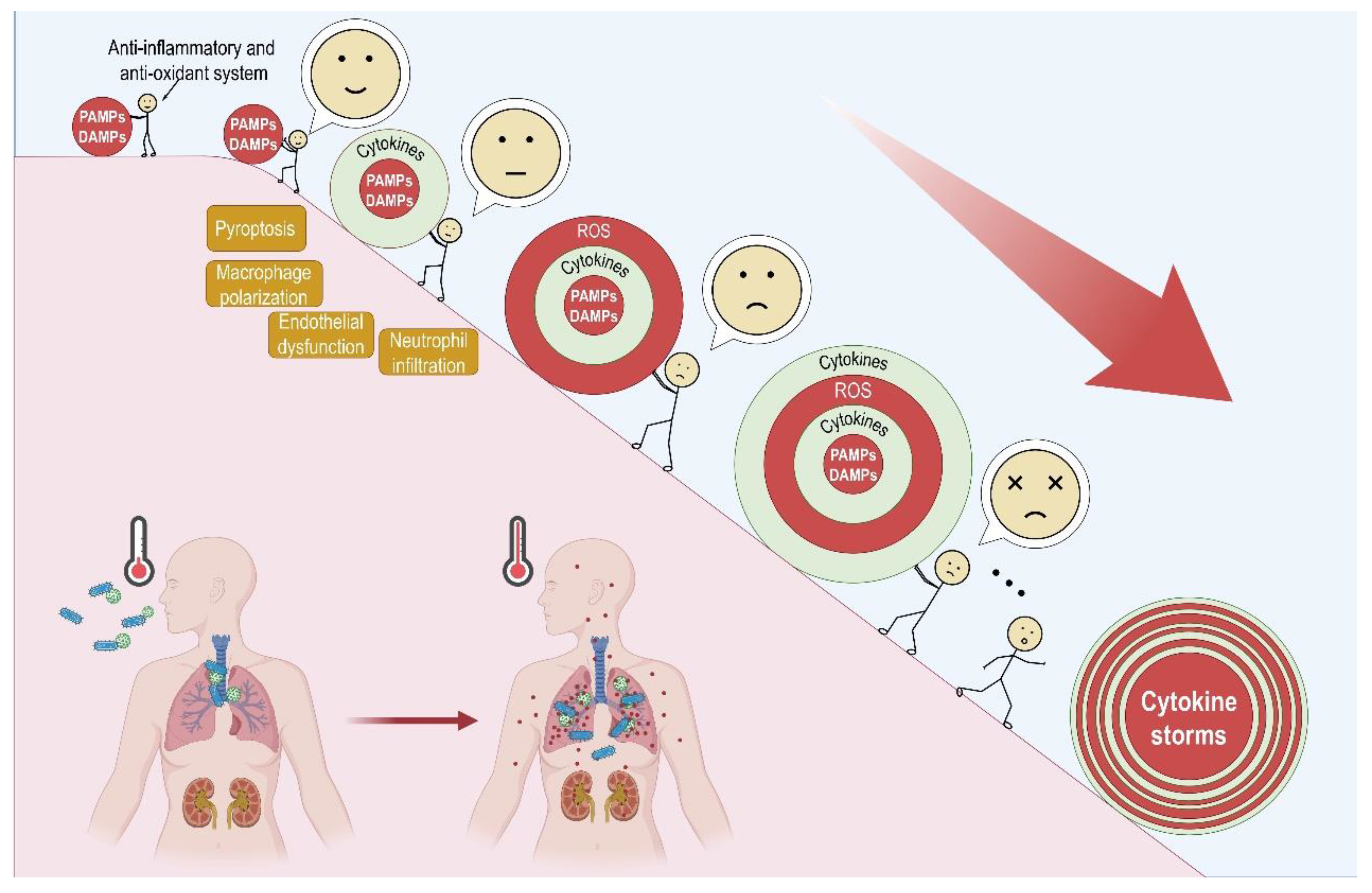{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cytokine Storms
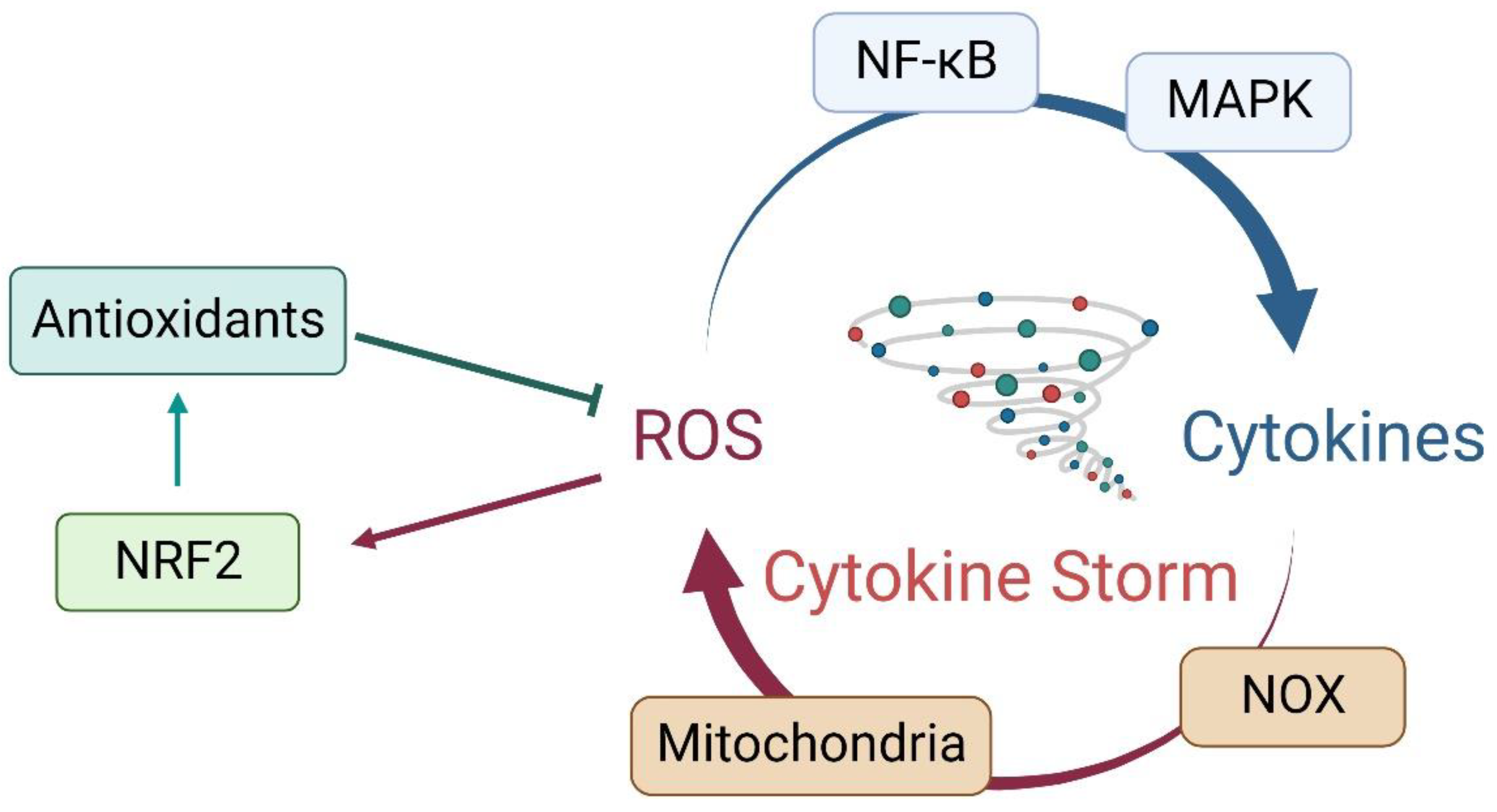
3. Crosstalk between Oxidative Stress and Inflammatory Response
4. NRF2 in Cytokine Storms
4.1. NRF2 Basics
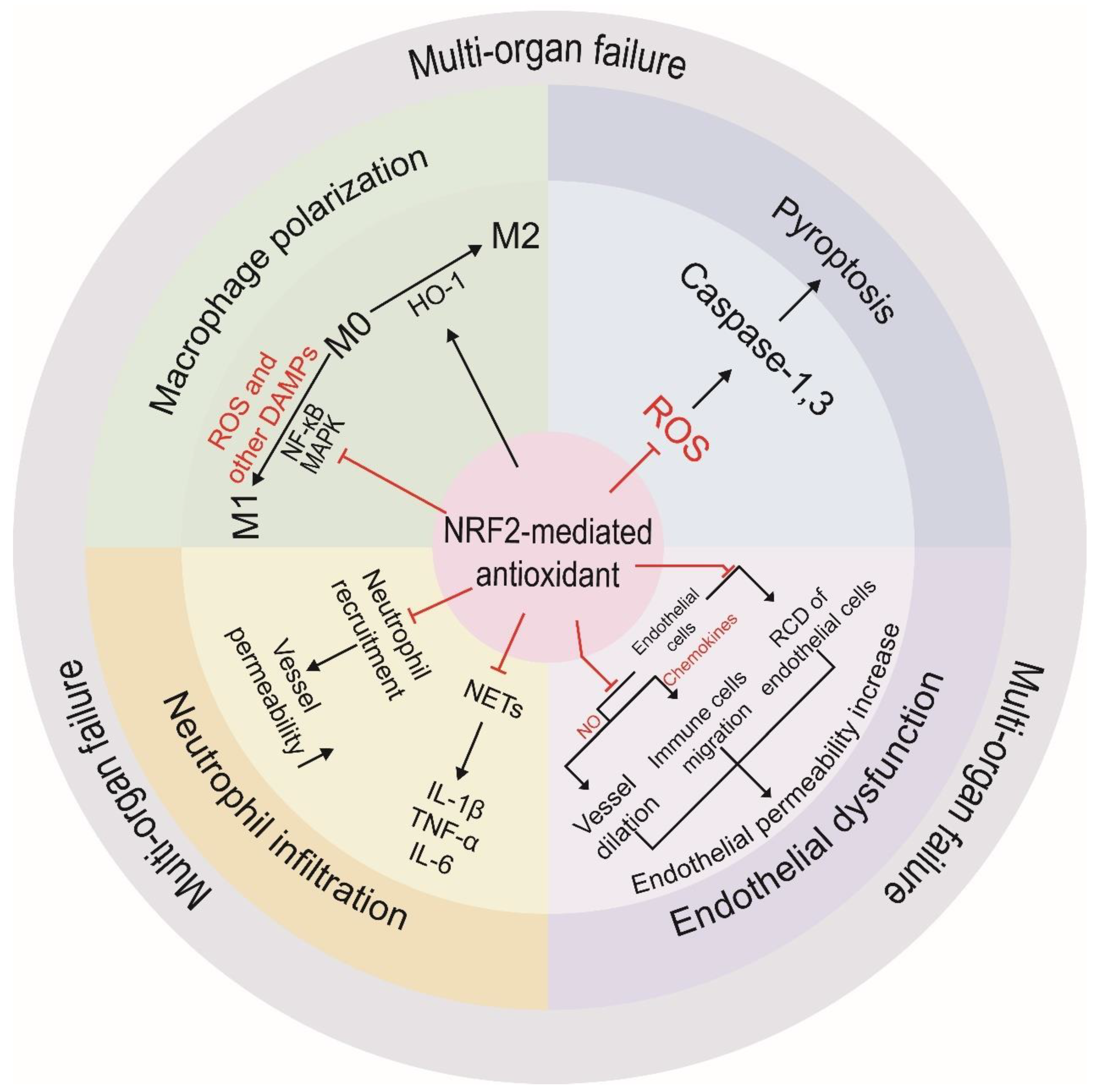
4.2. Key Cellular Events Regulated by NRF2 during Cytokine Storms
4.2.1. Macrophage Polarization
4.2.2. Pyroptosis
4.2.3. Endothelial Dysfunction
4.2.4. Neutrophil Infiltration
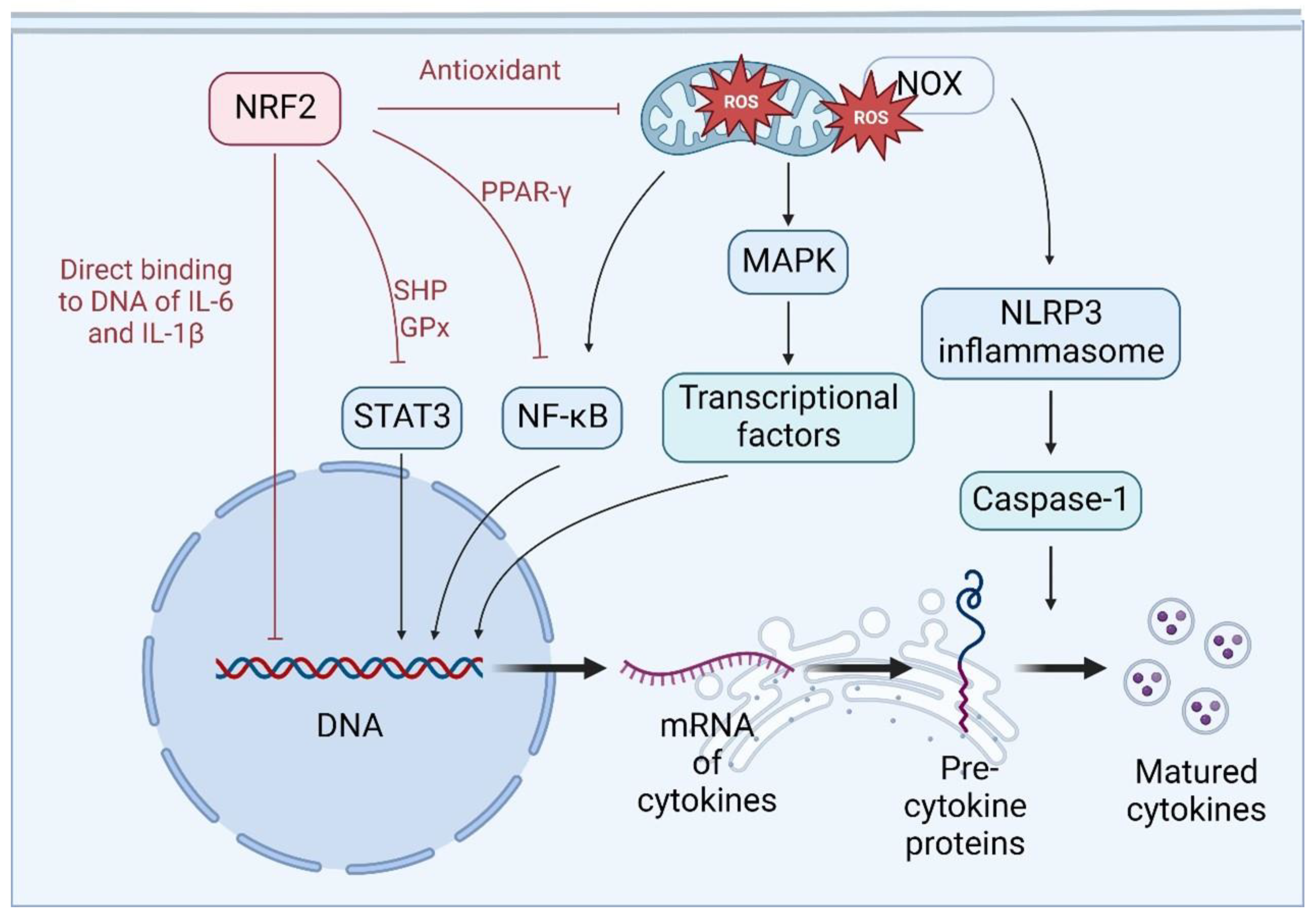
4.3. NRF2 Regulates the Transcription and Maturation of Cytokines
4.4. Strategies to Mitigate Cytokine Storms by Targeting NRF2
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ferrara, J.L.; Abhyankar, S.; Gilliland, D.G. Cytokine storm of graft-versus-host disease: A critical effector role for interleukin-1. Transplant. Proc. 1993, 25, 1216–1217. [Google Scholar]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, J.Y.; Yang, J.W.; Lee, K.H.; Effenberger, M.; Szpirt, W.; Kronbichler, A.; Shin, J.I. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics 2021, 11, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.D.; Ji, T.T.; Dong, J.R.; Feng, H.; Chen, F.Q.; Chen, X.; Zhao, H.Y.; Chen, D.K.; Ma, W.T. Pathogenesis and Treatment of Cytokine Storm Induced by Infectious Diseases. Int. J. Mol. Sci. 2021, 22, 13009. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Jia, Z.; Zhu, H. Regulation of Nrf2 Signaling. React. Oxyg. Species 2019, 8, 312–322. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Pi, J.; Zhang, Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol. 2022, 54, 102389. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Wang, H.; Zhu, L.; Mao, L.; Qiao, L.; Su, X. Depletion of Nrf2 enhances inflammation induced by oxyhemoglobin in cultured mice astrocytes. Neurochem. Res. 2011, 36, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Early, J.O.; Menon, D.; Wyse, C.A.; Cervantes-Silva, M.P.; Zaslona, Z.; Carroll, R.G.; Palsson-McDermott, E.M.; Angiari, S.; Ryan, D.G.; Corcoran, S.E.; et al. Circadian clock protein BMAL1 regulates IL-1beta in macrophages via NRF2. Proc. Natl. Acad. Sci. USA 2018, 115, E8460–E8468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhang, J.J.; Cheng, Y.T.; Ho, C.Y.; Yen, G.C. Monosodium urate crystals trigger Nrf2- and heme oxygenase-1-dependent inflammation in THP-1 cells. Cell. Mol. Immunol. 2015, 12, 424–434. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.M.; Haseeb, A.; Ansari, M.Y.; Devarapalli, P.; Haynie, S.; Haqqi, T.M. Wogonin, a plant derived small molecule, exerts potent anti-inflammatory and chondroprotective effects through the activation of ROS/ERK/Nrf2 signaling pathways in human Osteoarthritis chondrocytes. Free Radic. Biol. Med. 2017, 106, 288–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Link, H. The cytokine storm in multiple sclerosis. Mult. Scler. 1998, 4, 12–15. [Google Scholar] [CrossRef]
- Oldstone, M.B.; Rosen, H. Cytokine storm plays a direct role in the morbidity and mortality from influenza virus infection and is chemically treatable with a single sphingosine-1-phosphate agonist molecule. Curr. Top. Microbiol. Immunol. 2014, 378, 129–147. [Google Scholar] [CrossRef]
- Hantoushzadeh, S.; Norooznezhad, A.H. Possible Cause of Inflammatory Storm and Septic Shock in Patients Diagnosed with (COVID-19). Arch. Med. Res. 2020, 51, 347–348. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Godfred-Cato, S.; Bryant, B.; Leung, J.; Oster, M.E.; Conklin, L.; Abrams, J.; Roguski, K.; Wallace, B.; Prezzato, E.; Koumans, E.H.; et al. COVID-19-Associated Multisystem Inflammatory Syndrome in Children—United States, March-July 2020. MMWR Morb. Mortal. Wkl. Rep. 2020, 69, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.B.; Schwartz, N.G.; Patel, P.; Abbo, L.; Beauchamps, L.; Balan, S.; Lee, E.H.; Paneth-Pollak, R.; Geevarughese, A.; Lash, M.K.; et al. Case Series of Multisystem Inflammatory Syndrome in Adults Associated with SARS-CoV-2 Infection—United Kingdom and United States, March-August 2020. MMWR Morb. Mortal. Wkl. Rep. 2020, 69, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Wu, H.P.; Chen, C.K.; Chung, K.; Tseng, J.C.; Hua, C.C.; Liu, Y.C.; Chuang, D.Y.; Yang, C.H. Serial cytokine levels in patients with severe sepsis. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2009, 58, 385–393. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Stella, A.; Lamkanfi, M.; Portincasa, P. Familial Mediterranean Fever and COVID-19: Friends or Foes? Front. Immunol. 2020, 11, 574593. [Google Scholar] [CrossRef]
- Savic, S.; Dickie, L.J.; Wittmann, M.; McDermott, M.F. Autoinflammatory syndromes and cellular responses to stress: Pathophysiology, diagnosis and new treatment perspectives. Best Pract. Res. Clin. Rheumatol. 2012, 26, 505–533. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.J.; Tang, Y.M.; Song, H.; Yang, S.L.; Xu, W.Q.; Zhao, N.; Shi, S.W.; Shen, H.P.; Mao, J.Q.; Zhang, L.Y.; et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J. Pediatr. 2012, 160, 984–990.e1. [Google Scholar] [CrossRef] [PubMed]
- Wrobleski, D.M.; Barth, M.M.; Oyen, L.J. Necrotizing pancreatitis: Pathophysiology, diagnosis, and acute care management. AACN Clin. Issues 1999, 10, 464–477. [Google Scholar] [CrossRef]
- Hegyi, P.; Szakacs, Z.; Sahin-Toth, M. Lipotoxicity and Cytokine Storm in Severe Acute Pancreatitis and COVID-19. Gastroenterology 2020, 159, 824–827. [Google Scholar] [CrossRef]
- Savarin, C.; Bergmann, C.C. Fine Tuning the Cytokine Storm by IFN and IL-10 Following Neurotropic Coronavirus Encephalomyelitis. Front. Immunol. 2018, 9, 3022. [Google Scholar] [CrossRef] [Green Version]
- de Candia, P.; Prattichizzo, F.; Garavelli, S.; Matarese, G. T Cells: Warriors of SARS-CoV-2 Infection. Trends Immunol. 2021, 42, 18–30. [Google Scholar] [CrossRef]
- Cagliero, J.; Villanueva, S.; Matsui, M. Leptospirosis Pathophysiology: Into the Storm of Cytokines. Front. Cell. Infect. Microbiol. 2018, 8, 204. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Channappanavar, R.; Kanneganti, T.D. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol. 2020, 41, 1083–1099. [Google Scholar] [CrossRef]
- Spaulding, A.R.; Salgado-Pabon, W.; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Kanneganti, T.D. The ‘cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef]
- Yang, Y.; Tang, H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell. Mol. Immunol. 2016, 13, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Krakauer, T. Staphylococcal Superantigens: Pyrogenic Toxins Induce Toxic Shock. Toxins 2019, 11, 178. [Google Scholar] [CrossRef] [Green Version]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Fara, A.; Mitrev, Z.; Rosalia, R.A.; Assas, B.M. Cytokine storm and COVID-19: A chronicle of pro-inflammatory cytokines. Open Biol. 2020, 10, 200160. [Google Scholar] [CrossRef]
- Wang, H.; Ma, S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am. J. Emerg. Med. 2008, 26, 711–715. [Google Scholar] [CrossRef]
- Kurahashi, K.; Kajikawa, O.; Sawa, T.; Ohara, M.; Gropper, M.A.; Frank, D.W.; Martin, T.R.; Wiener-Kronish, J.P. Pathogenesis of septic shock in Pseudomonas aeruginosa pneumonia. J. Clin. Investig. 1999, 104, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Pelaia, C.; Tinello, C.; Vatrella, A.; De Sarro, G.; Pelaia, G. Lung under attack by COVID-19-induced cytokine storm: Pathogenic mechanisms and therapeutic implications. Ther. Adv. Respir. Dis. 2020, 14, 1753466620933508. [Google Scholar] [CrossRef]
- Abbasifard, M.; Khorramdelazad, H. The bio-mission of interleukin-6 in the pathogenesis of COVID-19: A brief look at potential therapeutic tactics. Life Sci. 2020, 257, 118097. [Google Scholar] [CrossRef]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensiv. Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Jin, J.; Jeon, S.; Moon, S.H.; Park, M.Y.; Yum, D.Y.; Kim, J.H.; Kang, J.E.; Park, M.H.; Kim, E.J.; et al. SOD1 suppresses pro-inflammatory immune responses by protecting against oxidative stress in colitis. Redox Biol. 2020, 37, 101760. [Google Scholar] [CrossRef]
- Lee, W.; Ahn, J.H.; Park, H.H.; Kim, H.N.; Kim, H.; Yoo, Y.; Shin, H.; Hong, K.S.; Jang, J.G.; Park, C.G.; et al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal Transduct. Target. Ther. 2020, 5, 186. [Google Scholar] [CrossRef]
- Hur, S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.M.; Go, Y.M.; Jones, D.P. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 215–234. [Google Scholar] [CrossRef]
- Liu, P.; Wang, X.; Sun, Y.; Zhao, H.; Cheng, F.; Wang, J.; Yang, F.; Hu, J.; Zhang, H.; Wang, C.C.; et al. SARS-CoV-2 ORF8 reshapes the ER through forming mixed disulfides with ER oxidoreductases. Redox Biol. 2022, 54, 102388. [Google Scholar] [CrossRef]
- Taylor, J.P.; Tse, H.M. The role of NADPH oxidases in infectious and inflammatory diseases. Redox Biol. 2021, 48, 102159. [Google Scholar] [CrossRef]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Cobley, J.N. Mechanisms of Mitochondrial ROS Production in Assisted Reproduction: The Known, the Unknown, and the Intriguing. Antioxidants 2020, 9, 933. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Fajgenbaum, D.C. Overview of Castleman disease. Blood 2020, 135, 1353–1364. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int. Immunopharmacol. 2020, 89, 107087. [Google Scholar] [CrossRef]
- Shirey, K.A.; Blanco, J.C.G.; Vogel, S.N. Targeting TLR4 Signaling to Blunt Viral-Mediated Acute Lung Injury. Front. Immunol. 2021, 12, 705080. [Google Scholar] [CrossRef]
- Liu, S.; Pi, J.; Zhang, Q. Mathematical modeling reveals quantitative properties of KEAP1-NRF2 signaling. Redox Biol. 2021, 47, 102139. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Benhar, M. Oxidants, Antioxidants and Thiol Redox Switches in the Control of Regulated Cell Death Pathways. Antioxidants 2020, 9, 309. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Trudler, D.; Oh, C.K.; Lipton, S.A. Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer’s Disease, Parkinson’s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome. Antioxidants 2022, 11, 124. [Google Scholar] [CrossRef]
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. CB 2016, 26, R568–R572. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Wu, D.; Ma, Y.; Cao, Y.; Liu, Q.; Tang, M.; Pu, Y.; Zhang, T. Reactive oxygen species trigger NF-kappaB-mediated NLRP3 inflammasome activation involvement in low-dose CdTe QDs exposure-induced hepatotoxicity. Redox Biol. 2021, 47, 102157. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Fakhouri, E.W.; Peterson, S.J.; Kothari, J.; Alex, R.; Shapiro, J.I.; Abraham, N.G. Genetic Polymorphisms Complicate COVID-19 Therapy: Pivotal Role of HO-1 in Cytokine Storm. Antioxidants 2020, 9, 636. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1alpha/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Sengupta, P.; Dutta, S.; Roychoudhury, S.; D’Souza, U.J.A.; Govindasamy, K.; Kolesarova, A. COVID-19, Oxidative Stress and Male Reproduction: Possible Role of Antioxidants. Antioxidants 2022, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Vadhan-Raj, S.; Nathan, C.F.; Sherwin, S.A.; Oettgen, H.F.; Krown, S.E. Phase I trial of recombinant interferon gamma by 1-hour i.v. infusion. Cancer Treat. Rep. 1986, 70, 609–614. [Google Scholar]
- Paniri, A.; Akhavan-Niaki, H. Emerging role of IL-6 and NLRP3 inflammasome as potential therapeutic targets to combat COVID-19: Role of lncRNAs in cytokine storm modulation. Life Sci. 2020, 257, 118114. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2013, 64, 409–421. [Google Scholar]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Wang, J.; Li, L.; Wu, Z.; Chen, X.; Yuan, J. ROS induced by spring viraemia of carp virus activate the inflammatory response via the MAPK/AP-1 and PI3K signaling pathways. Fish Shellfish Immunol. 2020, 101, 216–224. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.; Qiu, Z.; Huang, L.; Huang, L.; Liang, Y.; Liu, X.; Wang, M.; Zhou, B. Pyrogallol enhances therapeutic effect of human umbilical cord mesenchymal stem cells against LPS-mediated inflammation and lung injury via activation of Nrf2/HO-1 signaling. Free Radic. Biol. Med. 2022, 191, 66–81. [Google Scholar] [CrossRef]
- Yu, J.; Sun, X.; Goie, J.Y.G.; Zhang, Y. Regulation of Host Immune Responses against Influenza A Virus Infection by Mitogen-Activated Protein Kinases (MAPKs). Microorganisms 2020, 8, 1067. [Google Scholar] [CrossRef] [PubMed]
- Abroun, S.; Saki, N.; Ahmadvand, M.; Asghari, F.; Salari, F.; Rahim, F. STATs: An Old Story, Yet Mesmerizing. Cell J. 2015, 17, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Martin, L.; Mukawera, E.; Chartier, S.; De Deken, X.; Brochiero, E.; Miot, F.; Grandvaux, N. IFNbeta/TNFalpha synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH oxidase-mediated airway antiviral response. Cell Res. 2013, 23, 673–690. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crilly, M.J.; Tryon, L.D.; Erlich, A.T.; Hood, D.A. The role of Nrf2 in skeletal muscle contractile and mitochondrial function. J. Appl. Physiol. 2016, 121, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [Green Version]
- Lallemand, T.; Rouahi, M.; Swiader, A.; Grazide, M.H.; Geoffre, N.; Alayrac, P.; Recazens, E.; Coste, A.; Salvayre, R.; Negre-Salvayre, A.; et al. nSMase2 (Type 2-Neutral Sphingomyelinase) Deficiency or Inhibition by GW4869 Reduces Inflammation and Atherosclerosis in Apoe(-/-) Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1479–1492. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Du, R.; Huang, F.; Yin, S.; Yang, J.; Qin, S.; Cao, W. Sinomenine activation of Nrf2 signaling prevents hyperactive inflammation and kidney injury in a mouse model of obstructive nephropathy. Free Radic. Biol. Med. 2016, 92, 90–99. [Google Scholar] [CrossRef]
- Griess, B.; Mir, S.; Datta, K.; Teoh-Fitzgerald, M. Scavenging reactive oxygen species selectively inhibits M2 macrophage polarization and their pro-tumorigenic function in part, via Stat3 suppression. Free Radic. Biol. Med. 2020, 147, 48–60. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Jia, Y.; Wang, X.; Zhang, J.; Liu, K.; Wang, J.; Cai, W.; Li, J.; Li, S.; Zhao, M.; et al. Exosomes from adipose-derived stem cells alleviate the inflammation and oxidative stress via regulating Nrf2/HO-1 axis in macrophages. Free Radic. Biol. Med. 2021, 165, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Jingyuan, X.; Fangte, L.; Huijun, D.; Liu, Y.; Ren, J.; Jinyuan, L.; Linghui, P. Preconditioning with rHMGB1 ameliorates lung ischemia-reperfusion injury by inhibiting alveolar macrophage pyroptosis via the Keap1/Nrf2/HO-1 signaling pathway. J. Transl. Med. 2020, 18, 301. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear Factor E2-Related Factor-2 Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Chen, J.; Yang, H.; Chen, S.; Wang, Z. Overexpression of miR-200a-3p promoted inflammation in sepsis-induced brain injury through ROS-induced NLRP3. Int. J. Mol. Med. 2019, 44, 1811–1823. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Y.; Zhang, P.; Sun, X.; Chen, X.; Yu, J.; Shi, L.; Yin, Y.; Tao, K.; Li, R. Immune-responsive gene 1 protects against liver injury caused by concanavalin A via the activation Nrf2/HO-1 pathway and inhibition of ROS activation pathways. Free Radic. Biol. Med. 2022, 182, 108–118. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, Y.; Chen, X.; Wang, Z.; Liang, X.; Zhang, T.; Liu, M.; Zhou, N.; Lv, J.; Tang, K.; et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 2020, 5, eaax7969. [Google Scholar] [CrossRef]
- Chung, S.D.; Lai, T.Y.; Chien, C.T.; Yu, H.J. Activating Nrf-2 signaling depresses unilateral ureteral obstruction-evoked mitochondrial stress-related autophagy, apoptosis and pyroptosis in kidney. PLoS ONE 2012, 7, e47299. [Google Scholar] [CrossRef]
- Hong, M.K.; Hu, L.L.; Zhang, Y.X.; Xu, Y.L.; Liu, X.Y.; He, P.K.; Jia, Y.H. 6-Gingerol ameliorates sepsis-induced liver injury through the Nrf2 pathway. Int. Immunopharmacol. 2020, 80, 106196. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Saredy, J.; Xu, K.; Sun, Y.; Saaoud, F.; Drummer, C.T.; Lu, Y.; Luo, J.J.; Lopez-Pastrana, J.; Choi, E.T.; et al. Endothelial Immunity Trained by Coronavirus Infections, DAMP Stimulations and Regulated by Anti-Oxidant NRF2 May Contribute to Inflammations, Myelopoiesis, COVID-19 Cytokine Storms and Thromboembolism. Front. Immunol. 2021, 12, 653110. [Google Scholar] [CrossRef]
- Chen, H.; Xie, K.; Han, H.; Li, Y.; Liu, L.; Yang, T.; Yu, Y. Molecular hydrogen protects mice against polymicrobial sepsis by ameliorating endothelial dysfunction via an Nrf2/HO-1 signaling pathway. Int. Immunopharmacol. 2015, 28, 643–654. [Google Scholar] [CrossRef]
- Ren, X.; Li, Y.; Zhou, Y.; Hu, W.; Yang, C.; Jing, Q.; Zhou, C.; Wang, X.; Hu, J.; Wang, L.; et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021, 46, 102122. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Geng, P.; Zhu, J.; Li, J.; Zhang, L.; Chen, W.; Zhang, D.; Lu, Y.; Xu, X. KLF2 regulates eNOS uncoupling via Nrf2/HO-1 in endothelial cells under hypoxia and reoxygenation. Chem.-Biol. Interact. 2019, 305, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.F.; Ward, P.A. Role of oxidants in lung injury during sepsis. Antioxid. Redox Signal. 2007, 9, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Holloway, P.M.; Gillespie, S.; Becker, F.; Vital, S.A.; Nguyen, V.; Alexander, J.S.; Evans, P.C.; Gavins, F.N.E. Sulforaphane induces neurovascular protection against a systemic inflammatory challenge via both Nrf2-dependent and independent pathways. Vasc. Pharmacol. 2016, 85, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Choi, Y.K.; Lee, K.S.; Cho, D.H.; Baek, Y.Y.; Lee, D.K.; Ha, K.S.; Choe, J.; Won, M.H.; Jeoung, D.; et al. Functional dissection of Nrf2-dependent phase II genes in vascular inflammation and endotoxic injury using Keap1 siRNA. Free Radic. Biol. Med. 2012, 53, 629–640. [Google Scholar] [CrossRef]
- Savla, S.R.; Prabhavalkar, K.S.; Bhatt, L.K. Cytokine storm associated coagulation complications in COVID-19 patients: Pathogenesis and Management. Expert Rev. Anti-Infect. Ther. 2021, 19, 1397–1413. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Connors, J.M.; Levy, J.H. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2020, 69, 1181–1189. [Google Scholar] [CrossRef]
- Cao, T.H.; Jin, S.G.; Fei, D.S.; Kang, K.; Jiang, L.; Lian, Z.Y.; Pan, S.H.; Zhao, M.R.; Zhao, M.Y. Artesunate Protects Against Sepsis-Induced Lung Injury Via Heme Oxygenase-1 Modulation. Inflammation 2016, 39, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wu, Y.; Jin, S.; Sun, J.; Wan, B.B.; Zhang, J.; Wang, Y.; Gao, Z.Q.; Chen, D.; Li, S.; et al. Itaconate ameliorates methicillin-resistant Staphylococcus aureus-induced acute lung injury through the Nrf2/ARE pathway. Ann. Transl. Med. 2021, 9, 712. [Google Scholar] [CrossRef]
- Giustina, A.D.; Bonfante, S.; Zarbato, G.F.; Danielski, L.G.; Mathias, K.; de Oliveira, A.N., Jr.; Garbossa, L.; Cardoso, T.; Fileti, M.E.; De Carli, R.J.; et al. Dimethyl Fumarate Modulates Oxidative Stress and Inflammation in Organs after Sepsis in Rats. Inflammation 2018, 41, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimmulappa, R.K.; Fuchs, R.J.; Malhotra, D.; Scollick, C.; Traore, K.; Bream, J.H.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Kensler, T.W.; et al. Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxid. Redox Signal. 2007, 9, 1963–1970. [Google Scholar] [CrossRef]
- Noel, S.; Zheng, L.; Navas-Acien, A.; Fuchs, R.J. The effect of ex vivo CDDO-Me activation on nuclear factor erythroid 2-related factor 2 pathway in white blood cells from patients with septic shock. Shock 2014, 42, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.Y.; Sheng, Z.X.; Yao, H. Pachymic acid ameliorates sepsis-induced acute kidney injury by suppressing inflammation and activating the Nrf2/HO-1 pathway in rats. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1924–1931. [Google Scholar] [PubMed]
- Huang, X.T.; Liu, W.; Zhou, Y.; Hao, C.X.; Zhou, Y.; Zhang, C.Y.; Sun, C.C.; Luo, Z.Q.; Tang, S.Y. Dihydroartemisinin attenuates lipopolysaccharide-induced acute lung injury in mice by suppressing NF-kappaB signaling in an Nrf2-dependent manner. Int. J. Mol. Med. 2019, 44, 2213–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, T.; Fu, S.; He, D.; Hu, G.; Gao, X.; Zhang, Y.; Huang, B.; Du, J.; Zhou, A.; Su, Y.; et al. Evodiamine Inhibits Lipopolysaccharide (LPS)-Induced Inflammation in BV-2 Cells via Regulating AKT/Nrf2-HO-1/NF-kappaB Signaling Axis. Cell. Mol. Neurobiol. 2021, 41, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Li, S.T.; Dai, Q.; Zhang, S.X.; Liu, Y.J.; Yu, Q.Q.; Tan, F.; Lu, S.H.; Wang, Q.; Chen, J.W.; Huang, H.Q.; et al. Ulinastatin attenuates LPS-induced inflammation in mouse macrophage RAW264.7 cells by inhibiting the JNK/NF-kappaB signaling pathway and activating the PI3K/Akt/Nrf2 pathway. Acta Pharmacol. Sin. 2018, 39, 1294–1304. [Google Scholar] [CrossRef]
- Feng, Y.; Cui, R.; Li, Z.; Zhang, X.; Jia, Y.; Zhang, X.; Shi, J.; Qu, K.; Liu, C.; Zhang, J. Methane Alleviates Acetaminophen-Induced Liver Injury by Inhibiting Inflammation, Oxidative Stress, Endoplasmic Reticulum Stress, and Apoptosis through the Nrf2/HO-1/NQO1 Signaling Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 7067619. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Nikam, A.; Manin, S.; Ollivier, A.; Wilson, J.L.; Djouadi, S.; Muchova, L.; Martens, T.; Rivard, M.; Foresti, R. HYCO-3, a dual CO-releaser/Nrf2 activator, reduces tissue inflammation in mice challenged with lipopolysaccharide. Redox Biol. 2019, 20, 334–348. [Google Scholar] [CrossRef]
- Wruck, C.J.; Streetz, K.; Pavic, G.; Gotz, M.E.; Tohidnezhad, M.; Brandenburg, L.O.; Varoga, D.; Eickelberg, O.; Herdegen, T.; Trautwein, C.; et al. Nrf2 induces interleukin-6 (IL-6) expression via an antioxidant response element within the IL-6 promoter. J. Biol. Chem. 2011, 286, 4493–4499. [Google Scholar] [CrossRef] [Green Version]
- Lv, P.; Xue, P.; Dong, J.; Peng, H.; Clewell, R.; Wang, A.; Wang, Y.; Peng, S.; Qu, W.; Zhang, Q.; et al. Keap1 silencing boosts lipopolysaccharide-induced transcription of interleukin 6 via activation of nuclear factor kappaB in macrophages. Toxicol. Appl. Pharmacol. 2013, 272, 697–702. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Du, F.; Su, X.; Sun, G.; Zhou, G.; Bian, X.; Liu, N. Epigallocatechin-3-Gallate Attenuates Oxidative Stress and Inflammation in Obstructive Nephropathy via NF-kappaB and Nrf2/HO-1 Signalling Pathway Regulation. Basic Clin. Pharmacol. Toxicol. 2015, 117, 164–172. [Google Scholar] [CrossRef]
- Pedruzzi, L.M.; Stockler-Pinto, M.B.; Leite, M., Jr.; Mafra, D. Nrf2-keap1 system versus NF-kappaB: The good and the evil in chronic kidney disease? Biochimie 2012, 94, 2461–2466. [Google Scholar] [CrossRef]
- Park, S.Y.; Jin, M.L.; Wang, Z.; Park, G.; Choi, Y.W. 2,3,4’,5-tetrahydroxystilbene-2-O-beta-d-glucoside exerts anti-inflammatory effects on lipopolysaccharide-stimulated microglia by inhibiting NF-kappaB and activating AMPK/Nrf2 pathways. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2016, 97, 159–167. [Google Scholar] [CrossRef]
- Tan, Y.; Wan, H.H.; Sun, M.M.; Zhang, W.J.; Dong, M.; Ge, W.; Ren, J.; Peng, H. Cardamonin protects against lipopolysaccharide-induced myocardial contractile dysfunction in mice through Nrf2-regulated mechanism. Acta Pharmacol. Sin. 2021, 42, 404–413. [Google Scholar] [CrossRef]
- Zhong, W.; Qian, K.; Xiong, J.; Ma, K.; Wang, A.; Zou, Y. Curcumin alleviates lipopolysaccharide induced sepsis and liver failure by suppression of oxidative stress-related inflammation via PI3K/AKT and NF-kappaB related signaling. Biomed. Pharmacother. Biomed. Pharmacother. 2016, 83, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.P.; Wang, Q.W.; Su, Y.; Gu, L.M.; Zhao, Y.; Chen, X.X.; Chen, C.; Li, W.Z.; Wang, G.F.; Li, K.S. Emodin Inhibition of Influenza A Virus Replication and Influenza Viral Pneumonia via the Nrf2, TLR4, p38/JNK and NF-kappaB Pathways. Molecules 2017, 22, 1754. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.; Zhang, X.J.; Chen, H.M. Bardoxolone treatment alleviates lipopolysaccharide (LPS)-induced acute lung injury through suppressing inflammation and oxidative stress regulated by Nrf2 signaling. Biochem. Biophys. Res. Commun. 2019, 516, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Dai, J.; Gu, L.; Su, Y.; Wang, Q.; Zhao, Y.; Chen, X.; Deng, H.; Li, W.; Wang, G.; Li, K. Inhibition of curcumin on influenza A virus infection and influenzal pneumonia via oxidative stress, TLR2/4, p38/JNK MAPK and NF-kappaB pathways. Int. Immunopharmacol. 2018, 54, 177–187. [Google Scholar] [CrossRef]
- Kim, T.H.; Yoon, S.J.; Lee, S.M. Genipin attenuates sepsis by inhibiting Toll-like receptor signaling. Mol. Med. 2012, 18, 455–465. [Google Scholar] [CrossRef]
- Lu, H.; Wang, B.; Cui, N.; Zhang, Y. Artesunate suppresses oxidative and inflammatory processes by activating Nrf2 and ROS-dependent p38 MAPK and protects against cerebral ischemia-reperfusion injury. Mol. Med. Rep. 2018, 17, 6639–6646. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Xue, P.; Bai, Y.; Liu, D.; Woods, C.G.; Yarborough, K.; Fu, J.; Zhang, Q.; Sun, G.; Collins, S.; et al. Nuclear factor erythroid-derived factor 2-related factor 2 regulates transcription of CCAAT/enhancer-binding protein beta during adipogenesis. Free Radic. Biol. Med. 2012, 52, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Leung, L.; Xue, P.; Wang, W.; Hou, Y.; Liu, D.; Yehuda-Shnaidman, E.; Lee, C.; Lau, J.; Kurtz, T.W.; et al. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J. Biol. Chem. 2010, 285, 9292–9300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Fu, J.; Liu, D.; Sun, J.; Hou, Y.; Chen, C.; Shao, J.; Wang, L.; Wang, X.; Zhao, R.; et al. Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: Involvement of reduced PPARgamma expression. Redox Biol. 2020, 30, 101412. [Google Scholar] [CrossRef]
- Cho, H.Y.; Gladwell, W.; Wang, X.; Chorley, B.; Bell, D.; Reddy, S.P.; Kleeberger, S.R. Nrf2-regulated PPARgamma expression is critical to protection against acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2010, 182, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Hovsepian, E.; Penas, F.; Goren, N.B. 15-deoxy-Delta12,14 prostaglandin GJ2 but not rosiglitazone regulates metalloproteinase 9, NOS-2, and cyclooxygenase 2 expression and functions by peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms in cardiac cells. Shock 2010, 34, 60–67. [Google Scholar] [CrossRef]
- von Knethen, A.; Soller, M.; Brune, B. Peroxisome proliferator-activated receptor gamma (PPAR gamma) and sepsis. Arch. Immunol. Ther. Exp. 2007, 55, 19–25. [Google Scholar] [CrossRef]
- Ciavarella, C.; Motta, I.; Valente, S.; Pasquinelli, G. Pharmacological (or Synthetic) and Nutritional Agonists of PPAR-gamma as Candidates for Cytokine Storm Modulation in COVID-19 Disease. Molecules 2020, 25, 2076. [Google Scholar] [CrossRef] [PubMed]
- Mansour, H.H.; El Kiki, S.M.; Galal, S.M. Metformin and low dose radiation modulates cisplatin-induced oxidative injury in rat via PPAR-gamma and MAPK pathways. Arch. Biochem. Biophys. 2017, 616, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhang, B.; Di, J.; Jiang, G.; Chen, F.; Li, H.; Li, L.; Pei, D.; Zheng, J. Keap1: One stone kills three birds Nrf2, IKKbeta and Bcl-2/Bcl-xL. Int. J. Mol. Sci. 2012, 325, 26–34. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; You, D.J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell. Signal. 2010, 22, 1645–1654. [Google Scholar] [CrossRef]
- Zhao, R.; Hou, Y.; Zhang, Q.; Woods, C.G.; Xue, P.; Fu, J.; Yarborough, K.; Guan, D.; Andersen, M.E.; Pi, J. Cross-regulations among NRFs and KEAP1 and effects of their silencing on arsenic-induced antioxidant response and cytotoxicity in human keratinocytes. Environ. Health Perspect. 2012, 120, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Tai, H.; Huang, N.; Xiao, P.; Mo, C.; Wang, X.; Han, X.; Zhou, J.; Chen, H.; Tang, X.; et al. Nrf2-SHP Cascade-Mediated STAT3 Inactivation Contributes to AMPK-Driven Protection Against Endotoxic Inflammation. Front. Immunol. 2020, 11, 414. [Google Scholar] [CrossRef] [Green Version]
- Yuk, J.M.; Jin, H.S.; Jo, E.K. Small Heterodimer Partner and Innate Immune Regulation. Endocrinol. Metab. 2016, 31, 17–24. [Google Scholar] [CrossRef]
- Gawish, R.A.; Fahmy, H.A.; Abd El Fattah, A.I.; Nada, A.S. The potential effect of methylseleninic acid (MSA) against gamma-irradiation induced testicular damage in rats: Impact on JAK/STAT pathway. Arch. Biochem. Biophys. 2020, 679, 108205. [Google Scholar] [CrossRef]
- Netea, M.G.; van de Veerdonk, F.L.; van der Meer, J.W.; Dinarello, C.A.; Joosten, L.A. Inflammasome-independent regulation of IL-1-family cytokines. Annu. Rev. Immunol. 2015, 33, 49–77. [Google Scholar] [CrossRef]
- Rahim, I.; Sayed, R.K.; Fernandez-Ortiz, M.; Aranda-Martinez, P.; Guerra-Librero, A.; Fernandez-Martinez, J.; Rusanova, I.; Escames, G.; Djerdjouri, B.; Acuna-Castroviejo, D. Melatonin alleviates sepsis-induced heart injury through activating the Nrf2 pathway and inhibiting the NLRP3 inflammasome. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Zhang, Y.; Wang, Y.; Meng, X.; Wang, Y.; Yu, Y.; Chen, H. Hydrogen attenuates sepsis-associated encephalopathy by NRF2 mediated NLRP3 pathway inactivation. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2020, 69, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Neyaz, A.; Szabolcs, A.; Shih, A.R.; Chen, J.H.; Thapar, V.; Nieman, L.T.; Solovyov, A.; Mehta, A.; Lieb, D.J.; et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020, 11, 6319. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarbato, G.F.; de Souza Goldim, M.P.; Giustina, A.D.; Danielski, L.G.; Mathias, K.; Florentino, D.; de Oliveira Junior, A.N.; da Rosa, N.; Laurentino, A.O.; Trombetta, T.; et al. Dimethyl Fumarate Limits Neuroinflammation and Oxidative Stress and Improves Cognitive Impairment After Polymicrobial Sepsis. Neurotox. Res. 2018, 34, 418–430. [Google Scholar] [CrossRef]
- Qi, T.; Xu, F.; Yan, X.; Li, S.; Li, H. Sulforaphane exerts anti-inflammatory effects against lipopolysaccharide-induced acute lung injury in mice through the Nrf2/ARE pathway. Int. J. Mol. Med. 2016, 37, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.C.; Kim, D.Y.; Bae, J.S. Sulforaphane Reduces HMGB1-Mediated Septic Responses and Improves Survival Rate in Septic Mice. Am. J. Chin. Med. 2017, 45, 1253–1271. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.K.; Han, M.S.; Bae, J.S. Sulforaphane inhibits endothelial protein C receptor shedding in vitro and in vivo. Vasc. Pharmacol. 2014, 63, 13–18. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, L. Curcumin alleviates macrophage activation and lung inflammation induced by influenza virus infection through inhibiting the NF-kappaB signaling pathway. Influ. Respir. Viruses 2017, 11, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Valizadeh, H.; Abdolmohammadi-Vahid, S.; Danshina, S.; Ziya Gencer, M.; Ammari, A.; Sadeghi, A.; Roshangar, L.; Aslani, S.; Esmaeilzadeh, A.; Ghaebi, M.; et al. Nano-curcumin therapy, a promising method in modulating inflammatory cytokines in COVID-19 patients. Int. Immunopharmacol. 2020, 89, 107088. [Google Scholar] [CrossRef]
- Ali, F.E.M.; Bakr, A.G.; Abo-Youssef, A.M.; Azouz, A.A.; Hemeida, R.A.M. Targeting Keap-1/Nrf-2 pathway and cytoglobin as a potential protective mechanism of diosmin and pentoxifylline against cholestatic liver cirrhosis. Life Sci. 2018, 207, 50–60. [Google Scholar] [CrossRef]
- Patangrao Renushe, A.; Kumar Banothu, A.; Kumar Bharani, K.; Mekala, L.; Mahesh Kumar, J.; Neeradi, D.; Durga Veera Hanuman, D.; Gadige, A.; Khurana, A. Vincamine, an active constituent of Vinca rosea ameliorates experimentally induced acute lung injury in Swiss albino mice through modulation of Nrf-2/NF-kappaB signaling cascade. Int. Immunopharmacol. 2022, 108, 108773. [Google Scholar] [CrossRef]
- Khurana, A.; Sikha, M.S.; Ramesh, K.; Venkatesh, P.; Godugu, C. Modulation of cerulein-induced pancreatic inflammation by hydroalcoholic extract of curry leaf (Murraya koenigii). Phytother. Res. PTR 2019, 33, 1510–1525. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.; Pi, J.; Yehuda-Shnaidman, E. Uncoupling and reactive oxygen species (ROS)—A double-edged sword for beta-cell function? “Moderation in all things”. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Zhang, Q.; Fu, J.; Woods, C.G.; Hou, Y.; Corkey, B.E.; Collins, S.; Andersen, M.E. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol. Appl. Pharmacol. 2010, 244, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The Role of Reactive Oxygen Species in Arsenic Toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Yan, Y.; Shi, Q.; Kong, Y.; Gao, L.; Bao, H.; Li, Y. Recuperating lung decoction attenuates inflammation and oxidation in cigarette smoke-induced COPD in rats via activation of ERK and Nrf2 pathways. Cell Biochem. Funct. 2017, 35, 278–286. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Deng, P.; Liu, S.; Bian, Y.; Xu, Y.; Zhang, Q.; Wang, H.; Pi, J. Is Nuclear Factor Erythroid 2-Related Factor 2 a Target for the Intervention of Cytokine Storms? Antioxidants 2023, 12, 172. https://doi.org/10.3390/antiox12010172
Liu Z, Deng P, Liu S, Bian Y, Xu Y, Zhang Q, Wang H, Pi J. Is Nuclear Factor Erythroid 2-Related Factor 2 a Target for the Intervention of Cytokine Storms? Antioxidants. 2023; 12(1):172. https://doi.org/10.3390/antiox12010172
Chicago/Turabian StyleLiu, Zihang, Panpan Deng, Shengnan Liu, Yiying Bian, Yuanyuan Xu, Qiang Zhang, Huihui Wang, and Jingbo Pi. 2023. "Is Nuclear Factor Erythroid 2-Related Factor 2 a Target for the Intervention of Cytokine Storms?" Antioxidants 12, no. 1: 172. https://doi.org/10.3390/antiox12010172