
Macrophage Polarization and Reprogramming in Acute Inflammation: A Redox Perspective


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
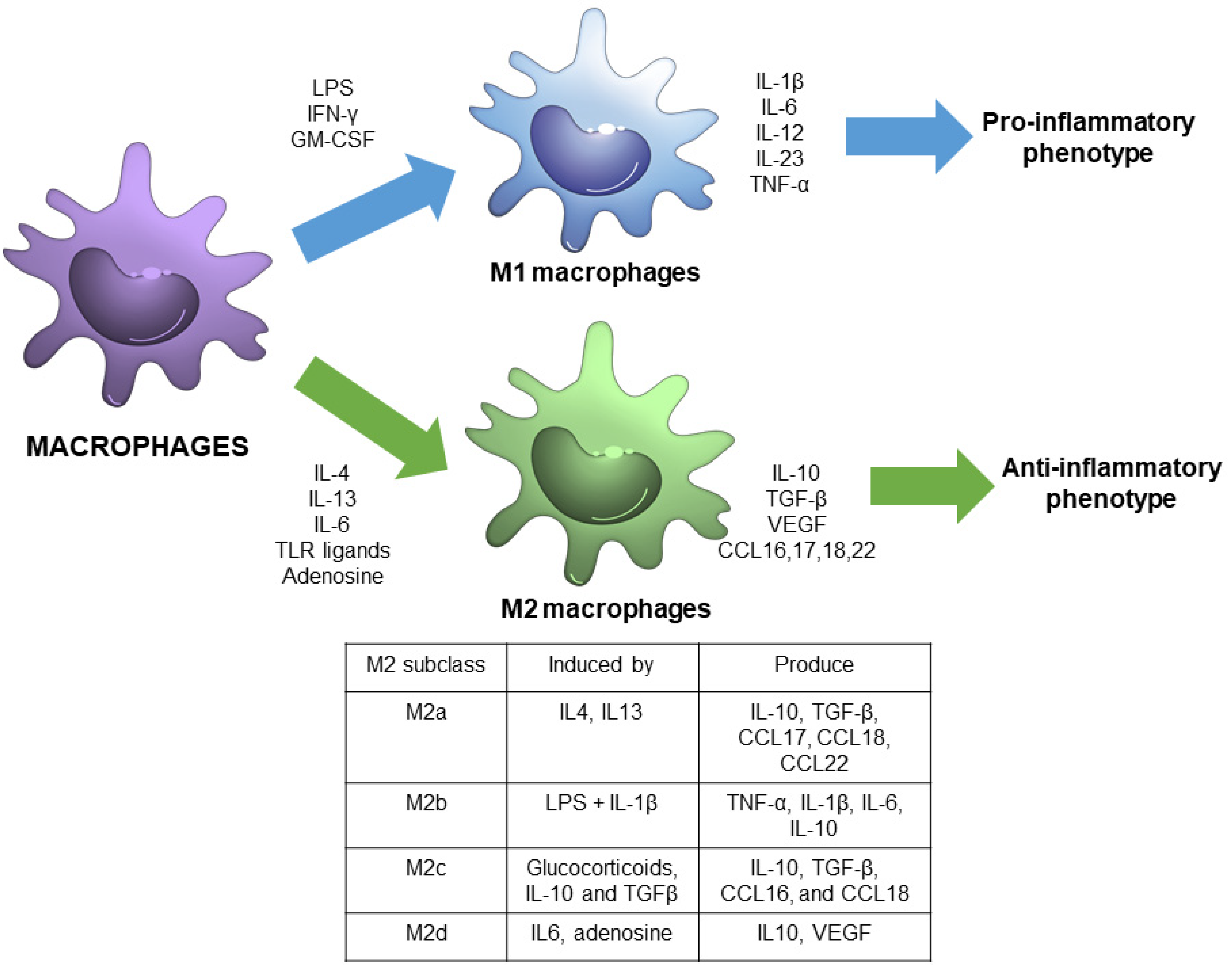
1.1. Macrophage Polarization
1.2. Role of Macrophage Polarization in Acute Inflammatory Processes
1.2.1. Sepsis
1.2.2. Viral Infections
1.2.3. Acute Kidney Injury
1.2.4. Acute Liver Injury
1.2.5. Acute Pancreatitis
1.2.6. Acute Respiratory Distress Syndrome
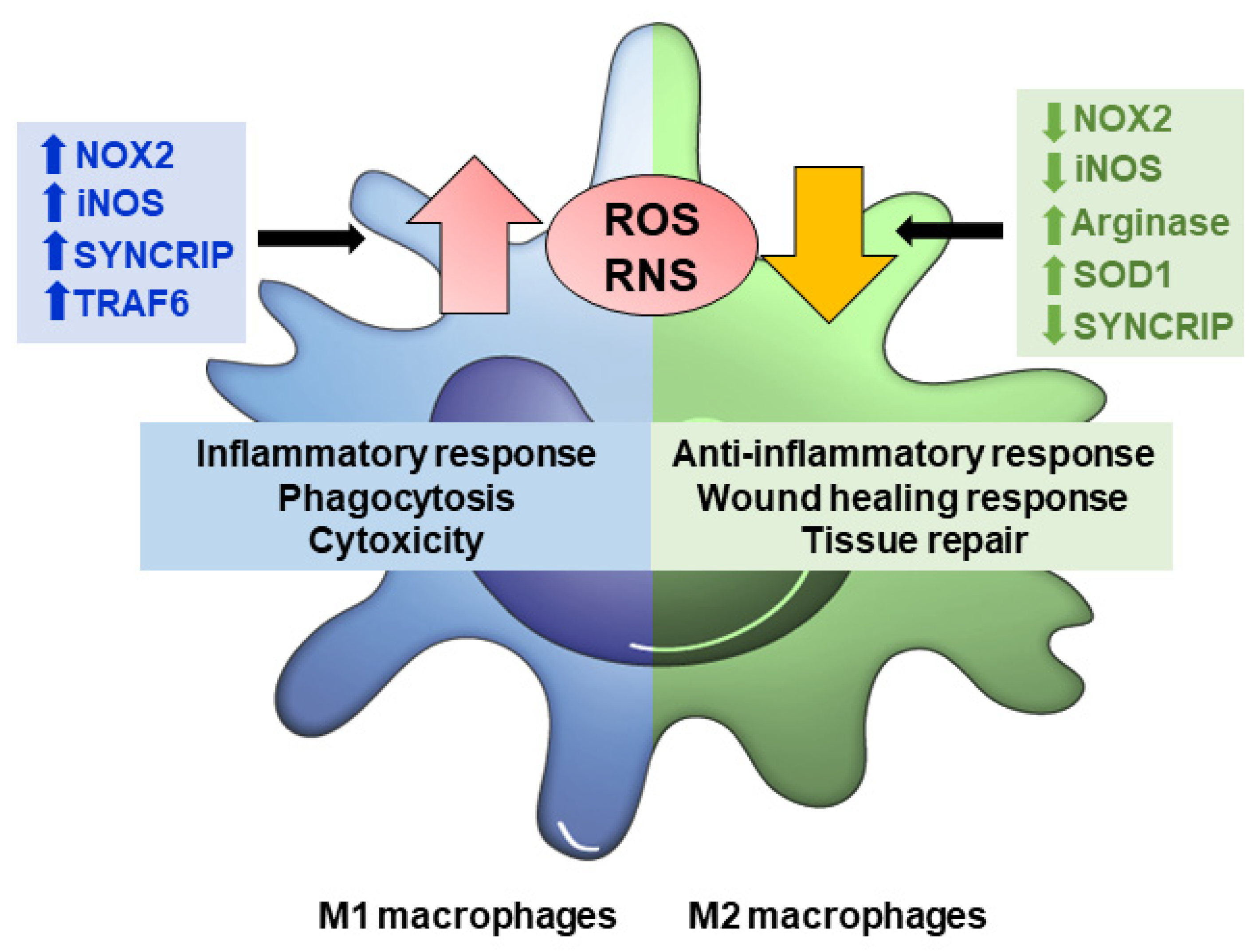
2. Redox Signaling in Macrophage Polarization
2.1. Redox Regulation of M1 Macrophages
2.2. Redox Regulation of M2 Macrophages
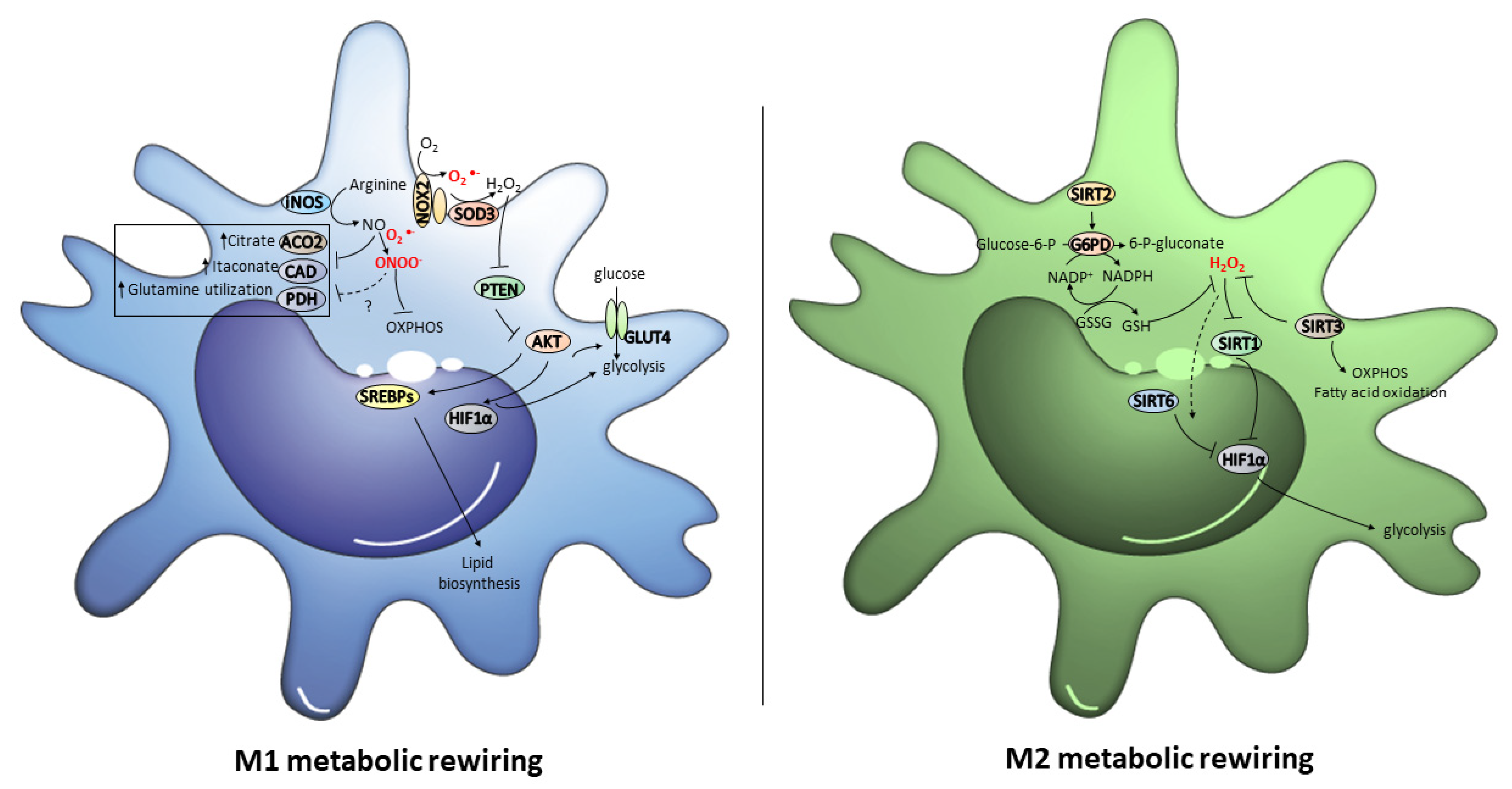
3. Role of Metabolic Reprogramming and Its Redox Control in Macrophage Polarization
3.1. Redox Regulation of the Metabolic Switch to the M1 Phenotype
3.2. Redox Regulation of the Metabolic Switch to the M2 Phenotype
4. Modulating Macrophage Function by Redox Signals: A Therapeutic Point of View
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- He, C.; Carter, A.B. The Metabolic Prospective and Redox Regulation of Macrophage Polarization. J. Clin. Cell. Immunol. 2015, 6, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Han, H.I.; Skvarca, L.B.; Espiritu, E.B.; Davidson, A.J.; Hukriede, N.A. The role of macrophages during acute kidney injury: Destruction and repair. Pediatr. Nephrol. 2019, 34, 561–569. [Google Scholar] [CrossRef]
- Ruytinx, P.; Proost, P.; Van Damme, J.; Struyf, S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 2018, 9, 1930. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Sica, A.; Lewis, C.E. Plasticity of macrophage function during tumor progression: Regulation by distinct molecular mechanisms. J. Immunol. 2008, 180, 2011–2017. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Sinha, M.; Datta, S.; Abas, M.; Chaffee, S.; Sen, C.K.; Roy, S. Monocyte and macrophage plasticity in tissue repair and regeneration. Am. J. Pathol. 2015, 185, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Marion, T.N.; Cao, X.; Wang, W.; Cao, Y. Park 7: A Novel Therapeutic Target for Macrophages in Sepsis-Induced Immunosuppression. Front. Immunol. 2018, 9, 2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomão, R.; Martins, P.S.; Brunialti, M.K.; Fernandes Mda, L.; Martos, L.S.; Mendes, M.E.; Gomes, N.E.; Rigato, O. TLR signaling pathway in patients with sepsis. Shock 2008, 30 (Suppl. S1), 73–77. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wei, H. Immune Intervention in Sepsis. Front. Pharmacol. 2021, 12, 718089. [Google Scholar] [CrossRef]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [Green Version]
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D., 2nd; Kreisel, D.; Krupnick, A.S.; et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011, 306, 2594–2605. [Google Scholar] [CrossRef]
- Zhuo, Y.; Li, D.; Cui, L.; Li, C.; Zhang, S.; Zhang, Q.; Zhang, L.; Wang, X.; Yang, L. Treatment with 3,4-dihydroxyphenylethyl alcohol glycoside ameliorates sepsis-induced ALI in mice by reducing inflammation and regulating M1 polarization. Biomed. Pharmacother. 2019, 116, 109012. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, T.; Jiang, L.; Gao, J.; Yu, D.; Ge, Y.; Lin, S. MCP-induced protein 1 attenuates sepsis-induced acute lung injury by modulating macrophage polarization via the JNK/c-Myc pathway. Int. Immunopharmacol. 2019, 75, 105741. [Google Scholar] [CrossRef]
- Shu, B.; Feng, Y.; Gui, Y.; Lu, Q.; Wei, W.; Xue, X.; Sun, X.; He, W.; Yang, J.; Dai, C. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-κB signaling suppression. Cell. Signal. 2018, 42, 249–258. [Google Scholar] [CrossRef]
- Tang, H.; Liang, Y.B.; Chen, Z.B.; Du, L.L.; Zeng, L.J.; Wu, J.G.; Yang, W.; Liang, H.P.; Ma, Z.F. Soluble Egg Antigen Activates M2 Macrophages via the STAT6 and PI3K Pathways, and Schistosoma Japonicum Alternatively Activates Macrophage Polarization to Improve the Survival Rate of Septic Mice. J. Cell. Biochem. 2017, 118, 4230–4239. [Google Scholar] [CrossRef]
- Tong, Y.; Yu, Z.; Chen, Z.; Zhang, R.; Ding, X.; Yang, X.; Niu, X.; Li, M.; Zhang, L.; Billiar, T.R.; et al. The HIV protease inhibitor Saquinavir attenuates sepsis-induced acute lung injury and promotes M2 macrophage polarization via targeting matrix metalloproteinase-9. Cell Death Dis. 2021, 12, 67. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Brewington, R.; Chatterji, M.; Zoubine, M.; Kinasewitz, G.T.; Peer, G.T.; Chang, A.C.; Taylor, F.B., Jr.; Shnyra, A. Infection-induced modulation of m1 and m2 phenotypes in circulating monocytes: Role in immune monitoring and early prognosis of sepsis. Shock 2004, 22, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Mena, H.A.; Olexen, C.M.; Ortiz Wilczyñski, J.M.; Negrotto, S.; Errasti, A.E.; Gómez, R.M.; Jenne, C.N.; Carrera Silva, E.A.; Schattner, M. Platelets Promote Macrophage Polarization toward Pro-inflammatory Phenotype and Increase Survival of Septic Mice. Cell Rep. 2019, 28, 896–908.e5. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. T cells and their immunometabolism: A novel way to understanding sepsis immunopathogenesis and future therapeutics. Eur. J. Cell Biol. 2018, 97, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Ruan, W.S.; Feng, M.X.; Xu, J.; Xu, Y.G.; Song, C.Y.; Lin, L.Y.; Li, L.; Lu, Y.Q. Early Activation of Myeloid-Derived Suppressor Cells Participate in Sepsis-Induced Immune Suppression via PD-L1/PD-1 Axis. Front. Immunol. 2020, 11, 1299. [Google Scholar] [CrossRef]
- Sang, Y.; Miller, L.C.; Blecha, F. Macrophage Polarization in Virus-Host Interactions. J. Clin. Cell. Immunol. 2015, 6, 311. [Google Scholar] [CrossRef] [Green Version]
- Gracia-Hernandez, M.; Sotomayor, E.M.; Villagra, A. Targeting Macrophages as a Therapeutic Option in Coronavirus Disease. Front. Pharmacol. 2020, 11, 577571. [Google Scholar]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. Macrophage Phenotype in Kidney Injury and Repair. Kidney Dis. 2015, 1, 138–146. [Google Scholar] [CrossRef]
- Lu, J.; Cao, Q.; Zheng, D.; Sun, Y.; Wang, C.; Yu, X.; Wang, Y.; Lee, V.W.; Zheng, G.; Tan, T.K.; et al. Discrete functions of M2a and M2c macrophage subsets determine their relative efficacy in treating chronic kidney disease. Kidney Int. 2013, 84, 745–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yu, W.; He, M.; Yuan, F. The Effects of M1/M2 Macrophages on the mRNA Expression Profile of Diabetic Glomerular Endothelial Cells. Nephron 2021, 145, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Li, B.; Rao, S.; Yeo, E.J.; Hudson, T.E.; Nowlin, B.T.; Pei, H.; Chen, L.; Zheng, J.J.; Carroll, T.J.; et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chen, J.; Xu, J.; Xie, J.; Harris, D.C.H.; Zheng, G. The Role of Macrophages in Kidney Fibrosis. Front. Physiol. 2021, 12, 705838. [Google Scholar] [CrossRef]
- van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Wang, C.; Ma, C.; Gong, L.; Guo, Y.; Fu, K.; Zhang, Y.; Zhou, H.; Li, Y. Macrophage Polarization and Its Role in Liver Disease. Front. Immunol. 2021, 12, 803037. [Google Scholar] [CrossRef]
- Tomar, S.; Zumbrun, E.E.; Nagarkatti, M.; Nagarkatti, P.S. Protective role of cannabinoid receptor 2 activation in galactosamine/lipopolysaccharide-induced acute liver failure through regulation of macrophage polarization and microRNAs. J. Pharmacol. Exp. Ther. 2015, 353, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, Y.; Ding, H.; Shi, X.; Ren, H. Mesenchymal stem cell-secreted prostaglandin E(2) ameliorates acute liver failure via attenuation of cell death and regulation of macrophage polarization. Stem Cell Res. Ther. 2021, 12, 15. [Google Scholar] [CrossRef]
- Xie, J.; Wu, X.; Zhou, Q.; Yang, Y.; Tian, Y.; Huang, C.; Meng, X.; Li, J. PICK1 confers anti-inflammatory effects in acute liver injury via suppressing M1 macrophage polarization. Biochimie 2016, 127, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rada, P.; Pardo, V.; Mobasher, M.A.; García-Martínez, I.; Ruiz, L.; González-Rodríguez, Á.; Sanchez-Ramos, C.; Muntané, J.; Alemany, S.; James, L.P.; et al. SIRT1 Controls Acetaminophen Hepatotoxicity by Modulating Inflammation and Oxidative Stress. Antioxid. Redox Signal. 2018, 28, 1187–1208. [Google Scholar] [CrossRef]
- Wang, Q.; Wei, S.; Zhou, H.; Shen, G.; Gan, X.; Zhou, S.; Qiu, J.; Shi, C.; Lu, L. Hyperglycemia exacerbates acetaminophen-induced acute liver injury by promoting liver-resident macrophage proinflammatory response via AMPK/PI3K/AKT-mediated oxidative stress. Cell Death Discov. 2019, 5, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.; Lou, N.; Jiao, J.; Guo, F.; Xiang, H.; Shang, D. Macrophages in pancreatitis: Mechanisms and therapeutic potential. Biomed. Pharmacother. 2020, 131, 110693. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.F.; Rohban, R.; Esni, F. Macrophages in the pancreas: Villains by circumstances, not necessarily by actions. Immun. Inflamm. Dis. 2020, 8, 807–824. [Google Scholar] [CrossRef]
- Sakai, Y.; Masamune, A.; Satoh, A.; Nishihira, J.; Yamagiwa, T.; Shimosegawa, T. Macrophage migration inhibitory factor is a critical mediator of severe acute pancreatitis. Gastroenterology 2003, 124, 725–736. [Google Scholar] [CrossRef]
- de Oliveira, C.; Khatua, B.; Noel, P.; Kostenko, S.; Bag, A.; Balakrishnan, B.; Patel, K.S.; Guerra, A.A.; Martinez, M.N.; Trivedi, S.; et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J. Clin. Investig. 2020, 130, 1931–1947. [Google Scholar] [CrossRef]
- Bonjoch, L.; Gea-Sorlí, S.; Closa, D. Lipids generated during acute pancreatitis increase inflammatory status of macrophages by interfering with their M2 polarization. Pancreatology 2015, 15, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, L.; Shi, J.; He, R.; Yang, W.; Habtezion, A.; Niu, N.; Lu, P.; Xue, J. Macrophage phenotypic switch orchestrates the inflammation and repair/regeneration following acute pancreatitis injury. EBioMedicine 2020, 58, 102920. [Google Scholar] [CrossRef]
- Murtaugh, L.C.; Keefe, M.D. Regeneration and repair of the exocrine pancreas. Annu. Rev. Physiol. 2015, 77, 229–249. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Döppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Morrell, E.D.; Bhatraju, P.K.; Mikacenic, C.R.; Radella, F., 2nd; Manicone, A.M.; Stapleton, R.D.; Wurfel, M.M.; Gharib, S.A. Alveolar Macrophage Transcriptional Programs Are Associated with Outcomes in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2019, 200, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, J.; Shuai, W.; Meng, J.; Feng, J.; Han, Z. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflamm. Res. 2020, 69, 883–895. [Google Scholar] [CrossRef]
- Fang, W.; Cai, S.X.; Wang, C.L.; Sun, X.X.; Li, K.; Yan, X.W.; Sun, Y.B.; Sun, X.Z.; Gu, C.K.; Dai, M.Y.; et al. Modulation of mitogen-activated protein kinase attenuates sepsis-induced acute lung injury in acute respiratory distress syndrome rats. Mol. Med. Rep. 2017, 16, 9652–9658. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Chun, W.; Lee, H.J.; Min, J.H.; Kim, S.M.; Seo, J.Y.; Ahn, K.S.; Oh, S.R. The Role of Macrophages in the Development of Acute and Chronic Inflammatory Lung Diseases. Cells 2021, 10, 897. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M. What differentiates normal lung repair and fibrosis? Inflammation, resolution of repair, and fibrosis. Proc. Am. Thorac. Soc. 2008, 5, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Fu, X.; Chen, X.; Han, X.; Dong, P. M2 macrophages induce EMT through the TGF-β/Smad2 signaling pathway. Cell Biol. Int. 2017, 41, 960–968. [Google Scholar] [CrossRef]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Weyand, C.M.; Shen, Y.; Goronzy, J.J. Redox-sensitive signaling in inflammatory T cells and in autoimmune disease. Free Radic. Biol. Med. 2018, 125, 36–43. [Google Scholar] [CrossRef]
- Canton, M.; Sánchez-Rodríguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 2021, 12, 734229. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassi, R.; Burgoyne, J.R.; DeNicola, G.F.; Rudyk, O.; DeSantis, V.; Charles, R.L.; Eaton, P.; Marber, M.S. Redox-dependent dimerization of p38α mitogen-activated protein kinase with mitogen-activated protein kinase kinase. J. Biol. Chem. 2017, 292, 16161–16173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surh, Y.J.; Kundu, J.K.; Na, H.K.; Lee, J.S. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J. Nutr. 2005, 135, 2993S–3001S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z.G. NADPH Oxidases Are Essential for Macrophage Differentiation. J. Biol. Chem. 2016, 291, 20030–20041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Nauseef, W.M. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 2019, 60, 130–140. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy: An emerging immunological paradigm. J. Immunol. 2012, 189, 15–20. [Google Scholar] [CrossRef]
- Martinez, J.; Malireddi, R.K.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Weigert, A.; von Knethen, A.; Fuhrmann, D.; Dehne, N.; Brüne, B. Redox-signals and macrophage biology. Mol. Aspects Med. 2018, 63, 70–87. [Google Scholar] [CrossRef]
- Allan, E.R.; Tailor, P.; Balce, D.R.; Pirzadeh, P.; McKenna, N.T.; Renaux, B.; Warren, A.L.; Jirik, F.R.; Yates, R.M. NADPH oxidase modifies patterns of MHC class II-restricted epitopic repertoires through redox control of antigen processing. J. Immunol. 2014, 192, 4989–5001. [Google Scholar] [CrossRef] [Green Version]
- Dhiman, M.; Garg, N.J. P47phox−/− mice are compromised in expansion and activation of CD8+ T cells and susceptible to Trypanosoma cruzi infection. PLoS Pathog. 2014, 10, e1004516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Hoffmann, F.W.; Fay, J.D.; Hashimoto, A.C.; Chapagain, M.L.; Kaufusi, P.H.; Hoffmann, P.R. Stimulation of unprimed macrophages with immune complexes triggers a low output of nitric oxide by calcium-dependent neuronal nitric-oxide synthase. J. Biol. Chem. 2012, 287, 4492–4502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibbs, J.B., Jr.; Taintor, R.R.; Vavrin, Z. Macrophage cytotoxicity: Role for L-arginine deiminase and imino nitrogen oxidation to nitrite. Science 1987, 235, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [Green Version]
- Flores-Costa, R.; Duran-Güell, M.; Casulleras, M.; López-Vicario, C.; Alcaraz-Quiles, J.; Diaz, A.; Lozano, J.J.; Titos, E.; Hall, K.; Sarno, R.; et al. Stimulation of soluble guanylate cyclase exerts antiinflammatory actions in the liver through a VASP/NF-κB/NLRP3 inflammasome circuit. Proc. Natl. Acad. Sci. USA 2020, 117, 28263–28274. [Google Scholar] [CrossRef]
- Coffey, M.J.; Phare, S.M.; Luo, M.; Peters-Golden, M. Guanylyl cyclase and protein kinase G mediate nitric oxide suppression of 5-lipoxygenase metabolism in rat alveolar macrophages. Biochim. Biophys. Acta 2008, 1781, 299–305. [Google Scholar] [CrossRef]
- Brown, G.C.; Cooper, C.E. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994, 356, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Flashman, E.; Mecinović, J.; Kramer, H.B.; Kessler, B.M.; Frapart, Y.M.; Boucher, J.L.; Clifton, I.J.; McDonough, M.A.; Schofield, C.J. Studies on the reaction of nitric oxide with the hypoxia-inducible factor prolyl hydroxylase domain 2 (EGLN1). J. Mol. Biol. 2011, 410, 268–279. [Google Scholar] [CrossRef]
- Brüne, B. Nitric oxide: NO apoptosis or turning it ON? Cell Death Differ. 2003, 10, 864–869. [Google Scholar] [CrossRef]
- Wiese, M.; Gerlach, R.G.; Popp, I.; Matuszak, J.; Mahapatro, M.; Castiglione, K.; Chakravortty, D.; Willam, C.; Hensel, M.; Bogdan, C.; et al. Hypoxia-mediated impairment of the mitochondrial respiratory chain inhibits the bactericidal activity of macrophages. Infect. Immun. 2012, 80, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Gan, P.; Gao, Z.; Zhao, X.; Qi, G. Surfactin inducing mitochondria-dependent ROS to activate MAPKs, NF-κB and inflammasomes in macrophages for adjuvant activity. Sci. Rep. 2016, 6, 39303. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lu, H.; Liu, Q.; Huang, G.; Lim, C.P.; Zhang, L.; Hao, A.; Cao, X. Function of GRIM-19, a mitochondrial respiratory chain complex I protein, in innate immunity. J. Biol. Chem. 2012, 287, 27227–27235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redente, E.F.; Dwyer-Nield, L.D.; Merrick, D.T.; Raina, K.; Agarwal, R.; Pao, W.; Rice, P.L.; Shroyer, K.R.; Malkinson, A.M. Tumor progression stage and anatomical site regulate tumor-associated macrophage and bone marrow-derived monocyte polarization. Am. J. Pathol. 2010, 176, 2972–2985. [Google Scholar] [CrossRef]
- Balce, D.R.; Li, B.; Allan, E.R.; Rybicka, J.M.; Krohn, R.M.; Yates, R.M. Alternative activation of macrophages by IL-4 enhances the proteolytic capacity of their phagosomes through synergistic mechanisms. Blood 2011, 118, 4199–4208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Knethen, A.; Soller, M.; Tzieply, N.; Weigert, A.; Johann, A.M.; Jennewein, C.; Köhl, R.; Brüne, B. PPARgamma1 attenuates cytosol to membrane translocation of PKCalpha to desensitize monocytes/macrophages. J. Cell Biol. 2007, 176, 681–694. [Google Scholar] [CrossRef]
- Bourdonnay, E.; Serezani, C.H.; Aronoff, D.M.; Peters-Golden, M. Regulation of alveolar macrophage p40phox: Hierarchy of activating kinases and their inhibition by PGE. J. Leukoc. Biol. 2012, 92, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Kuchler, L.; Giegerich, A.K.; Sha, L.K.; Knape, T.; Wong, M.S.; Schröder, K.; Brandes, R.P.; Heide, H.; Wittig, I.; Brüne, B.; et al. SYNCRIP-dependent Nox2 mRNA destabilization impairs ROS formation in M2-polarized macrophages. Antioxid. Redox. Signal. 2014, 21, 2483–2497. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barra, V.; Kuhn, A.M.; von Knethen, A.; Weigert, A.; Brüne, B. Apoptotic cell-derived factors induce arginase II expression in murine macrophages by activating ERK5/CREB. Cell. Mol. Life Sci. 2011, 68, 1815–1827. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Liu, Q.; Orandle, M.S.; Sadiq-Ali, S.; Koontz, S.M.; Choi, U.; Torres-Velez, F.J.; Jackson, S.H. P47(phox) Directs Murine Macrophage Cell Fate Decisions. Am. J. Pathol. 2012, 180, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Harvey, C.J.; Thimmulappa, R.K.; Sethi, S.; Kong, X.; Yarmus, L.; Brown, R.H.; Feller-Kopman, D.; Wise, R.; Biswal, S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 2011, 3, 78ra32. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Thimmulappa, R.; Kombairaju, P.; Biswal, S. NADPH oxidase-dependent reactive oxygen species mediate amplified TLR4 signaling and sepsis-induced mortality in Nrf2-deficient mice. J. Immunol. 2010, 185, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2008, 39, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, A.M.; Tzieply, N.; Schmidt, M.V.; von Knethen, A.; Namgaladze, D.; Yamamoto, M.; Brüne, B. Antioxidant signaling via Nrf2 counteracts lipopolysaccharide-mediated inflammatory responses in foam cell macrophages. Free Radic. Biol. Med. 2011, 50, 1382–1391. [Google Scholar] [CrossRef]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef]
- He, C.; Ryan, A.J.; Murthy, S.; Carter, A.B. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J. Biol. Chem. 2013, 288, 20745–20757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Galván-Peña, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar]
- Kieler, M.; Hofmann, M.; Schabbauer, G. More than just protein building blocks: How amino acids and related metabolic pathways fuel macrophage polarization. FEBS J. 2021, 288, 3694–3714. [Google Scholar] [CrossRef]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef]
- Koo, S.J.; Szczesny, B.; Wan, X.; Putluri, N.; Garg, N.J. Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma cruzi in Macrophages. Front. Immunol. 2018, 9, 202. [Google Scholar] [CrossRef] [Green Version]
- Meng, T.; Yu, J.; Lei, Z.; Wu, J.; Wang, S.; Bo, Q.; Zhang, X.; Ma, Z.; Yu, J. Propofol reduces lipopolysaccharide-induced, NADPH oxidase (NOX 2) mediated TNF- α and IL-6 production in macrophages. Clin. Dev. Immunol. 2013, 2013, 325481. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.P.; Henry, R.J.; Villapol, S.; Stoica, B.A.; Kumar, A.; Burns, M.P.; Faden, A.I.; Loane, D.J. NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: Implications for traumatic brain injury. J. Neuroinflammation 2017, 14, 65. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Lee, J.H.; Choi, S.H.; Kim, S.; Almazan, F.; Witztum, J.L.; Miller, Y.I. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: Toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase. Circ. Res. 2009, 104, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Jeong, J.M.; Kim, S.J.; Seo, W.; Kim, M.H.; Choi, W.M.; Yoo, W.; Lee, J.H.; Shim, Y.R.; Yi, H.S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat. Commun. 2017, 8, 2247. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.V.; Poulsen, N.B.; Linneberg Matthiesen, C.; Vilhardt, F. Novel and Converging Ways of NOX2 and SOD3 in Trafficking and Redox Signaling in Macrophages. Antioxidants 2021, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Qin, J.; Lan, L.; Zhang, H.; Liu, F.; Wu, Z.; Ni, H.; Wang, Y. PTEN inhibits macrophage polarization from M1 to M2 through CCL2 and VEGF-A reduction and NHERF-1 synergism. Cancer Biol. Ther. 2015, 16, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Park, J.; Han, S.J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox. Biol. 2020, 34, 101553. [Google Scholar] [CrossRef]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Horie, T.; Ono, K.; Nagao, K.; Nishi, H.; Kinoshita, M.; Kawamura, T.; Wada, H.; Shimatsu, A.; Kita, T.; Hasegawa, K. Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J. Cell. Physiol. 2008, 215, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.; Kim, L.C.; Song, W.; Edwards, D.N.; Cook, R.S.; Chen, J. Disruption of the Scaffolding Function of mLST8 Selectively Inhibits mTORC2 Assembly and Function and Suppresses mTORC2-Dependent Tumor Growth In Vivo. Cancer Res. 2019, 79, 3178–3184. [Google Scholar] [CrossRef]
- Griffiths, H.R.; Gao, D.; Pararasa, C. Redox regulation in metabolic programming and inflammation. Redox. Biol. 2017, 12, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Bertolio, R.; Napoletano, F.; Mano, M.; Maurer-Stroh, S.; Fantuz, M.; Zannini, A.; Bicciato, S.; Sorrentino, G.; Del Sal, G. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 2019, 10, 1326. [Google Scholar] [CrossRef] [Green Version]
- Cerychova, R.; Pavlinkova, G. HIF-1, Metabolism, and Diabetes in the Embryonic and Adult Heart. Front. Endocrinol. 2018, 9, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piantadosi, C.A. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poderoso, J.J.; Carreras, M.C.; Lisdero, C.; Riobo, N.; Schopfer, F.; Boveris, A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch. Biochem. Biophys. 1996, 328, 85–92. [Google Scholar] [CrossRef]
- Pearce, L.L.; Epperly, M.W.; Greenberger, J.S.; Pitt, B.R.; Peterson, J. Identification of respiratory complexes I and III as mitochondrial sites of damage following exposure to ionizing radiation and nitric oxide. Nitric Oxide 2001, 5, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Rodriguez, M.; Castro, L.; Telleri, R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch. Biochem. Biophys. 1994, 308, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230.e7. [Google Scholar] [CrossRef] [Green Version]
- Castro, L.; Tórtora, V.; Mansilla, S.; Radi, R. Aconitases: Non-redox Iron-Sulfur Proteins Sensitive to Reactive Species. Acc. Chem. Res. 2019, 52, 2609–2619. [Google Scholar] [CrossRef]
- Castro, L.A.; Robalinho, R.L.; Cayota, A.; Meneghini, R.; Radi, R. Nitric oxide and peroxynitrite-dependent aconitase inactivation and iron-regulatory protein-1 activation in mammalian fibroblasts. Arch. Biochem. Biophys. 1998, 359, 215–224. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Meiser, J.; Krämer, L.; Sapcariu, S.C.; Battello, N.; Ghelfi, J.; D’Herouel, A.F.; Skupin, A.; Hiller, K. Pro-inflammatory Macrophages Sustain Pyruvate Oxidation through Pyruvate Dehydrogenase for the Synthesis of Itaconate and to Enable Cytokine Expression. J. Biol. Chem. 2016, 291, 3932–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox. Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Prados, J.C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD(+) in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, J.N.; Auwerx, J. Transcriptional targets of sirtuins in the coordination of mammalian physiology. Curr. Opin. Cell Biol. 2008, 20, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.I.; Guarente, L. It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Lee, S.W.; Lee, S.Y.; Hong, K.W.; Bae, S.S.; Kim, K.; Kim, C.D. SIRT1/Adenosine Monophosphate-Activated Protein Kinase α Signaling Enhances Macrophage Polarization to an Anti-inflammatory Phenotype in Rheumatoid Arthritis. Front. Immunol. 2017, 8, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Lu, Y.; Martinez, J.; Bi, Y.; Lian, G.; Wang, T.; Milasta, S.; Wang, J.; Yang, M.; Liu, G.; et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc. Natl. Acad. Sci. USA 2016, 113, 1564–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizaki, T.; Schenk, S.; Imamura, T.; Babendure, J.L.; Sonoda, N.; Bae, E.J.; Oh, D.Y.; Lu, M.; Milne, J.C.; Westphal, C.; et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E419–E428. [Google Scholar] [CrossRef] [Green Version]
- Fabbiano, S.; Suárez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Stevanovic Dokic, A.; Colin, D.J.; Trajkovski, M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fu, W.; Chen, J.; Olashaw, N.; Zhang, X.; Nicosia, S.V.; Bhalla, K.; Bai, W. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat. Cell Biol. 2007, 9, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 2008, 135, 907–918. [Google Scholar] [CrossRef] [Green Version]
- de Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Dalla Man, C.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: Potential biochemical mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Heinonen, T.; Ciarlo, E.; Rigoni, E.; Regina, J.; Le Roy, D.; Roger, T. Dual Deletion of the Sirtuins SIRT2 and SIRT3 Impacts on Metabolism and Inflammatory Responses of Macrophages and Protects From Endotoxemia. Front. Immunol. 2019, 10, 2713. [Google Scholar] [CrossRef]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traba, J.; Geiger, S.S.; Kwarteng-Siaw, M.; Han, K.; Ra, O.H.; Siegel, R.M.; Gius, D.; Sack, M.N. Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3-mediated activation of superoxide dismutase. J. Biol. Chem. 2017, 292, 12153–12164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox. Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Dominy, J.E., Jr.; Lee, Y.; Jedrychowski, M.P.; Chim, H.; Jurczak, M.J.; Camporez, J.P.; Ruan, H.B.; Feldman, J.; Pierce, K.; Mostoslavsky, R.; et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 2012, 48, 900–913. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Gupta, V.; Carroll, K.S.; Liebler, D.C. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat. Commun. 2014, 5, 4776. [Google Scholar] [CrossRef] [Green Version]
- Lo Sasso, G.; Menzies, K.J.; Mottis, A.; Piersigilli, A.; Perino, A.; Yamamoto, H.; Schoonjans, K.; Auwerx, J. SIRT2 deficiency modulates macrophage polarization and susceptibility to experimental colitis. PLoS ONE 2014, 9, e103573. [Google Scholar] [CrossRef]
- Kurundkar, D.; Kurundkar, A.R.; Bone, N.B.; Becker, E.J., Jr.; Liu, W.; Chacko, B.; Darley-Usmar, V.; Zmijewski, J.W.; Thannickal, V.J. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight 2019, 4, e120722. [Google Scholar] [CrossRef]
- Xi, J.; Chen, Y.; Jing, J.; Zhang, Y.; Liang, C.; Hao, Z.; Zhang, L. Sirtuin 3 suppresses the formation of renal calcium oxalate crystals through promoting M2 polarization of macrophages. J. Cell. Physiol. 2019, 234, 11463–11473. [Google Scholar] [CrossRef]
- Song, M.Y.; Kim, S.H.; Ryoo, G.H.; Kim, M.K.; Cha, H.N.; Park, S.Y.; Hwang, H.P.; Yu, H.C.; Bae, E.J.; Park, B.H. Adipose sirtuin 6 drives macrophage polarization toward M2 through IL-4 production and maintains systemic insulin sensitivity in mice and humans. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Lee, Y.; Ka, S.O.; Cha, H.N.; Chae, Y.N.; Kim, M.K.; Park, S.Y.; Bae, E.J.; Park, B.H. Myeloid Sirtuin 6 Deficiency Causes Insulin Resistance in High-Fat Diet-Fed Mice by Eliciting Macrophage Polarization Toward an M1 Phenotype. Diabetes 2017, 66, 2659–2668. [Google Scholar] [CrossRef] [Green Version]
- Koo, J.H.; Jang, H.Y.; Lee, Y.; Moon, Y.J.; Bae, E.J.; Yun, S.K.; Park, B.H. Myeloid cell-specific sirtuin 6 deficiency delays wound healing in mice by modulating inflammation and macrophage phenotypes. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, F.; Lv, L.; Li, T.; Zhou, X.; Deng, C.X.; Guan, K.L.; Lei, Q.Y.; Xiong, Y. Oxidative stress activates SIRT2 to deacetylate and stimulate phosphoglycerate mutase. Cancer Res. 2014, 74, 3630–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Buechler, N.L.; Long, D.L.; Furdui, C.M.; Yoza, B.K.; McCall, C.E.; Vachharajani, V. Cysteine thiol oxidation on SIRT2 regulates inflammation in obese mice with sepsis. Inflammation 2019, 42, 156–169. [Google Scholar] [CrossRef]
- Long, D.; Wu, H.; Tsang, A.W.; Poole, L.B.; Yoza, B.K.; Wang, X.; Vachharajani, V.; Furdui, C.M.; McCall, C.E. The Oxidative State of Cysteine Thiol 144 Regulates the SIRT6 Glucose Homeostat. Sci. Rep. 2017, 7, 11005. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Folgado, S.L.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Samarghandian, S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomed. Pharmacother. 2020, 127, 110234. [Google Scholar] [CrossRef]
- Chen, L.; Yang, S.; Zumbrun, E.E.; Guan, H.; Nagarkatti, P.S.; Nagarkatti, M. Resveratrol attenuates lipopolysaccharide-induced acute kidney injury by suppressing inflammation driven by macrophages. Mol. Nutr. Food Res. 2015, 59, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, Y.; Xu, Y.; Ruan, W.; Wang, H.; Zhang, Y.; Saavedra, J.M.; Zhang, L.; Huang, Z.; Pang, T. A Dual AMPK/Nrf2 Activator Reduces Brain Inflammation After Stroke by Enhancing Microglia M2 Polarization. Antioxid. Redox. Signal. 2018, 28, 141–163. [Google Scholar] [CrossRef]
- Tang, J.; Diao, P.; Shu, X.; Li, L.; Xiong, L. Quercetin and Quercitrin Attenuates the Inflammatory Response and Oxidative Stress in LPS-Induced RAW264.7 Cells: In Vitro Assessment and a Theoretical Model. BioMed Res. Int. 2019, 2019, 7039802. [Google Scholar] [PubMed] [Green Version]
- Cao, J.; Li, Q.; Shen, X.; Yao, Y.; Li, L.; Ma, H. Dehydroepiandrosterone attenuates LPS-induced inflammatory responses via activation of Nrf2 in RAW264.7 macrophages. Mol. Immunol. 2021, 131, 97–111. [Google Scholar] [CrossRef]
- Aesif, S.W.; Anathy, V.; Kuipers, I.; Guala, A.S.; Reiss, J.N.; Ho, Y.S.; Janssen-Heininger, Y.M. Ablation of glutaredoxin-1 attenuates lipopolysaccharide-induced lung inflammation and alveolar macrophage activation. Am. J. Respir. Cell Mol. Biol. 2011, 44, 491–499. [Google Scholar] [CrossRef] [Green Version]
- Gorelenkova Miller, O.; Cole, K.S.; Emerson, C.C.; Allimuthu, D.; Golczak, M.; Stewart, P.L.; Weerapana, E.; Adams, D.J.; Mieyal, J.J. Novel chloroacetamido compound CWR-J02 is an anti-inflammatory glutaredoxin-1 inhibitor. PLoS ONE 2017, 12, e0187991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Liu, Y.; Zhao, S.; Xu, W.; Li, Y.; Zhao, P.; Wang, D.; Cheng, H.; Ke, Y.; Zhang, X. Oxidative stress-induced FABP5 S-glutathionylation protects against acute lung injury by suppressing inflammation in macrophages. Nat. Commun. 2021, 12, 7094. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chen, S.; Liu, Q.; Ren, H.; Li, Y.; Pan, J.; Luo, Y.; Cai, T.; Liu, R.; Chen, J.; et al. Glutaredoxin 1 regulates macrophage polarization through mediating glutathionylation of STAT. Thorac. Cancer 2020, 11, 2966–2974. [Google Scholar] [CrossRef] [PubMed]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Lundberg, M.; Johansson, C.; Chandra, J.; Enoksson, M.; Jacobsson, G.; Ljung, J.; Johansson, M.; Holmgren, A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 2001, 276, 26269–26275. [Google Scholar] [CrossRef] [Green Version]
- Hanschmann, E.M.; Berndt, C.; Hecker, C.; Garn, H.; Bertrams, W.; Lillig, C.H.; Hudemann, C. Glutaredoxin 2 Reduces Asthma-Like Acute Airway Inflammation in Mice. Front. Immunol. 2020, 11, 561724. [Google Scholar] [CrossRef]
- Coelho, C. Itaconate or how I learned to stop avoiding the study of immunometabolism. PLoS Pathog. 2022, 18, e1010361. [Google Scholar] [CrossRef]
- Li, R.; Zhang, P.; Wang, Y.; Tao, K. Itaconate: A Metabolite Regulates Inflammation Response and Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5404780. [Google Scholar] [CrossRef]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.H.; et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef]
- Swain, A.; Bambouskova, M.; Kim, H.; Andhey, P.S.; Duncan, D.; Auclair, K.; Chubukov, V.; Simons, D.M.; Roddy, T.P.; Stewart, K.M.; et al. Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat. Metab. 2020, 2, 594–602. [Google Scholar] [CrossRef]
- Runtsch, M.C.; Angiari, S.; Hooftman, A.; Wadhwa, R.; Zhang, Y.; Zheng, Y.; Spina, J.S.; Ruzek, M.C.; Argiriadi, M.A.; McGettrick, A.F.; et al. Itaconate and itaconate derivatives target JAK1 to suppress alternative activation of macrophages. Cell Metab. 2022, 34, 487–501.e8. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Ren, J.; Gao, D.S.; Dai, Y.; Yu, L. The Emerging Application of Itaconate: Promising Molecular Targets and Therapeutic Opportunities. Front. Chem. 2021, 9, 669308. [Google Scholar] [CrossRef] [PubMed]
- ElAzzouny, M.; Tom, C.T.; Evans, C.R.; Olson, L.L.; Tanga, M.J.; Gallagher, K.A.; Martin, B.R.; Burant, C.F. Dimethyl Itaconate Is Not Metabolized into Itaconate Intracellularly. J. Biol. Chem. 2017, 292, 4766–4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Zhang, S.; Jiao, Y.; Li, C.; Liang, X.; Jia, H.; Nie, Z.; Zhang, Y. Dimethyl Itaconate Alleviates the Inflammatory Responses of Macrophages in Sepsis. Inflammation 2021, 44, 549–557. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Zhang, H.; Xiao, J.; Yang, C.; Chen, W.; Wei, Z.; Chen, X.; Liu, J. 4-Octyl Itaconate Alleviates Lipopolysaccharide-Induced Acute Lung Injury in Mice by Inhibiting Oxidative Stress and Inflammation. Drug Des. Devel. Ther. 2020, 14, 5547–5558. [Google Scholar] [CrossRef]
- Sohail, A.; Iqbal, A.A.; Sahini, N.; Chen, F.; Tantawy, M.; Waqas, S.F.H.; Winterhoff, M.; Ebensen, T.; Schultz, K.; Geffers, R.; et al. Itaconate and derivatives reduce interferon responses and inflammation in influenza A virus infection. PLoS Pathog. 2022, 18, e1010219. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Y.; Zhang, Y.; Xu, F.; Chen, J.; Duan, L.; Zhang, T.; Wang, J.; Zhang, F. Breaking the vicious loop between inflammation, oxidative stress and coagulation, a novel anti-thrombus insight of nattokinase by inhibiting LPS-induced inflammation and oxidative stress. Redox. Biol. 2020, 32, 101500. [Google Scholar] [CrossRef]
- Zhang, W.B.; Yang, F.; Wang, Y.; Jiao, F.Z.; Zhang, H.Y.; Wang, L.W.; Gong, Z.J. Inhibition of HDAC6 attenuates LPS-induced inflammation in macrophages by regulating oxidative stress and suppressing the TLR4-MAPK/NF-κB pathways. Biomed. Pharmacother. 2019, 117, 109166. [Google Scholar] [CrossRef]
- Zhou, Y.; Gao, C.; Vong, C.T.; Tao, H.; Li, H.; Wang, S.; Wang, Y. Rhein regulates redox-mediated activation of NLRP3 inflammasomes in intestinal inflammation through macrophage-activated crosstalk. Br. J. Pharmacol. 2022, 179, 1978–1997. [Google Scholar] [CrossRef]
- Song, B.; Zhang, C.; Hu, W.; Guo, C.; Xia, Z.; Hu, W.; Qin, M.; Jiang, W.; Lv, J.; Xu, D.; et al. Nano-designed carbon monoxide donor SMA/CORM2 exhibits protective effect against acetaminophen induced liver injury through macrophage reprograming and promoting liver regeneration. J. Control. Release 2021, 331, 350–363. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez, S.; Rius-Pérez, S. Macrophage Polarization and Reprogramming in Acute Inflammation: A Redox Perspective. Antioxidants 2022, 11, 1394. https://doi.org/10.3390/antiox11071394
Pérez S, Rius-Pérez S. Macrophage Polarization and Reprogramming in Acute Inflammation: A Redox Perspective. Antioxidants. 2022; 11(7):1394. https://doi.org/10.3390/antiox11071394
Chicago/Turabian StylePérez, Salvador, and Sergio Rius-Pérez. 2022. "Macrophage Polarization and Reprogramming in Acute Inflammation: A Redox Perspective" Antioxidants 11, no. 7: 1394. https://doi.org/10.3390/antiox11071394