
N-Acetylcysteine Inhibits Platelet Function through the Regeneration of the Non-Oxidative Form of Albumin



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Blood Collection and Plasma Preparation
2.2. Plasma Protein Depletion
2.3. Platelet Aggregation Assays
2.4. Measurement of Thromboxane B2 and 12-Hydroxyeicosatetraenoic Acid
2.5. Measurement of Glutathione
2.6. Measurement of Cysteinylated form of Albumin
2.7. Albumin Cysteinylation
2.8. Platelet Adhesion Assays
2.9. Measurements of Intracellular Calcium Mobilization
2.10. Determination of Reactive Oxygen Species Generation
2.11. Statistical Analysis
3. Results
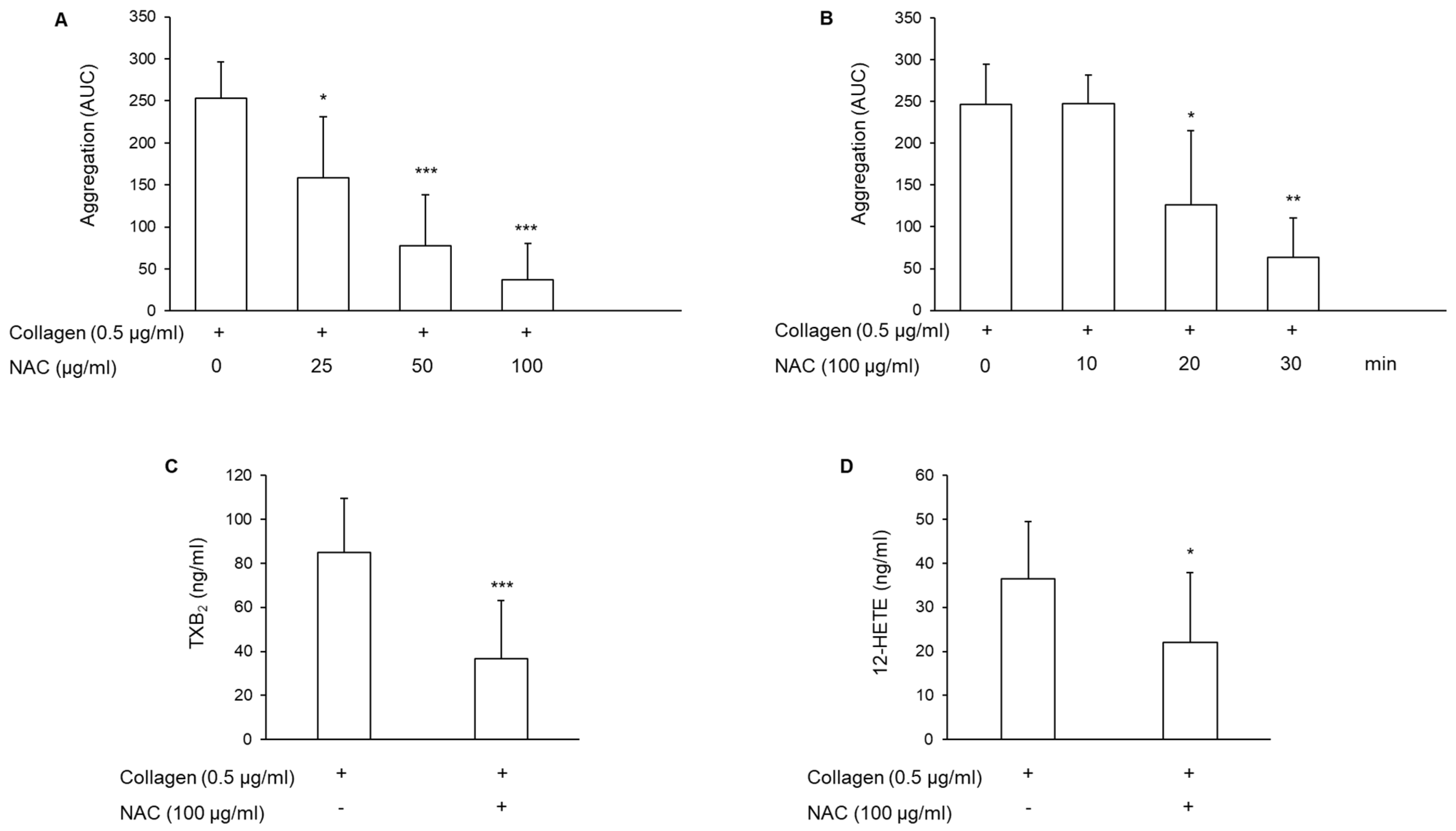
3.1. Effect of N-Acetylcysteine on Platelet Aggregation and Arachidonic Acid Metabolite Generation
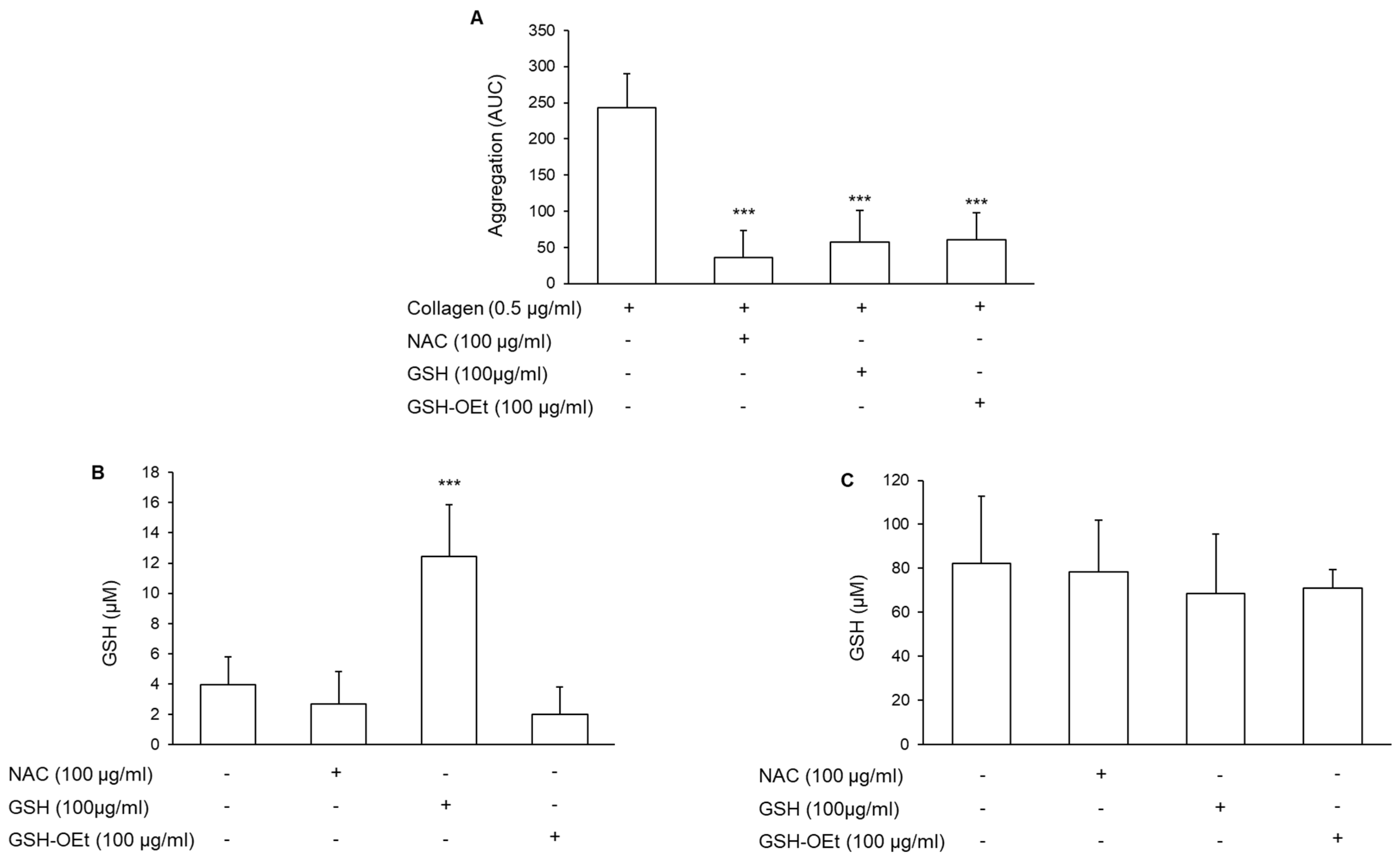
3.2. Involvement of the Intracellular Glutathione in the Antiplatelet Effect of N-Acetylcysteine
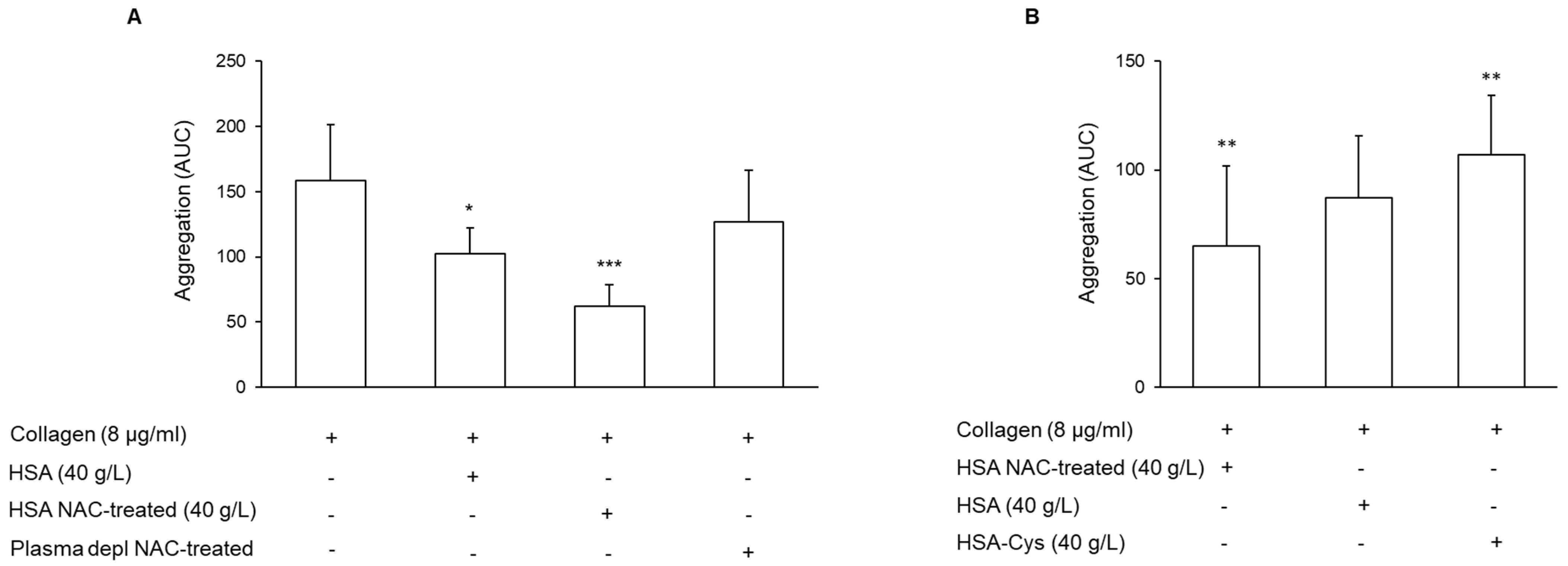
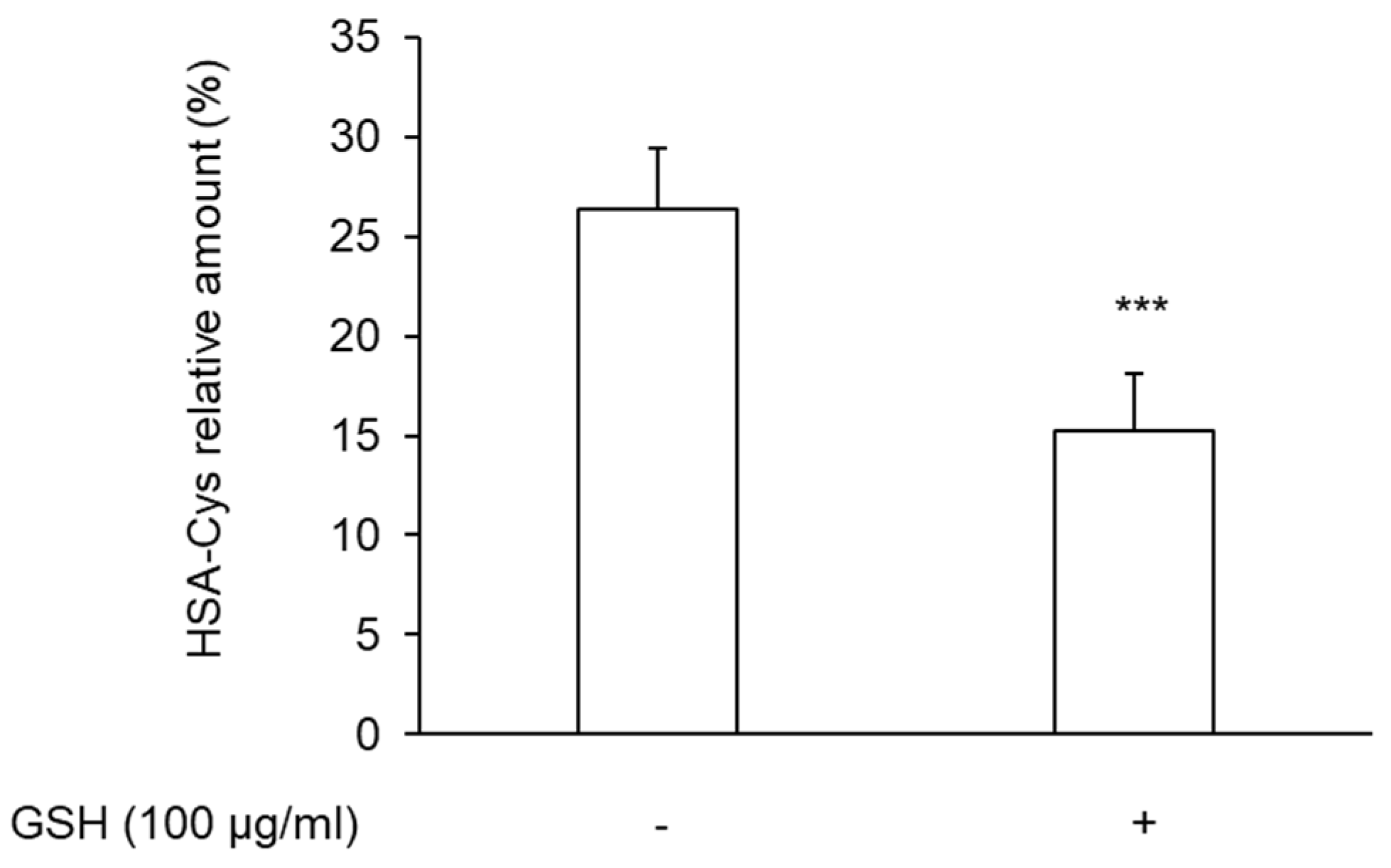
3.3. Effect of N-Acetylcysteine on the Restoration of HMA
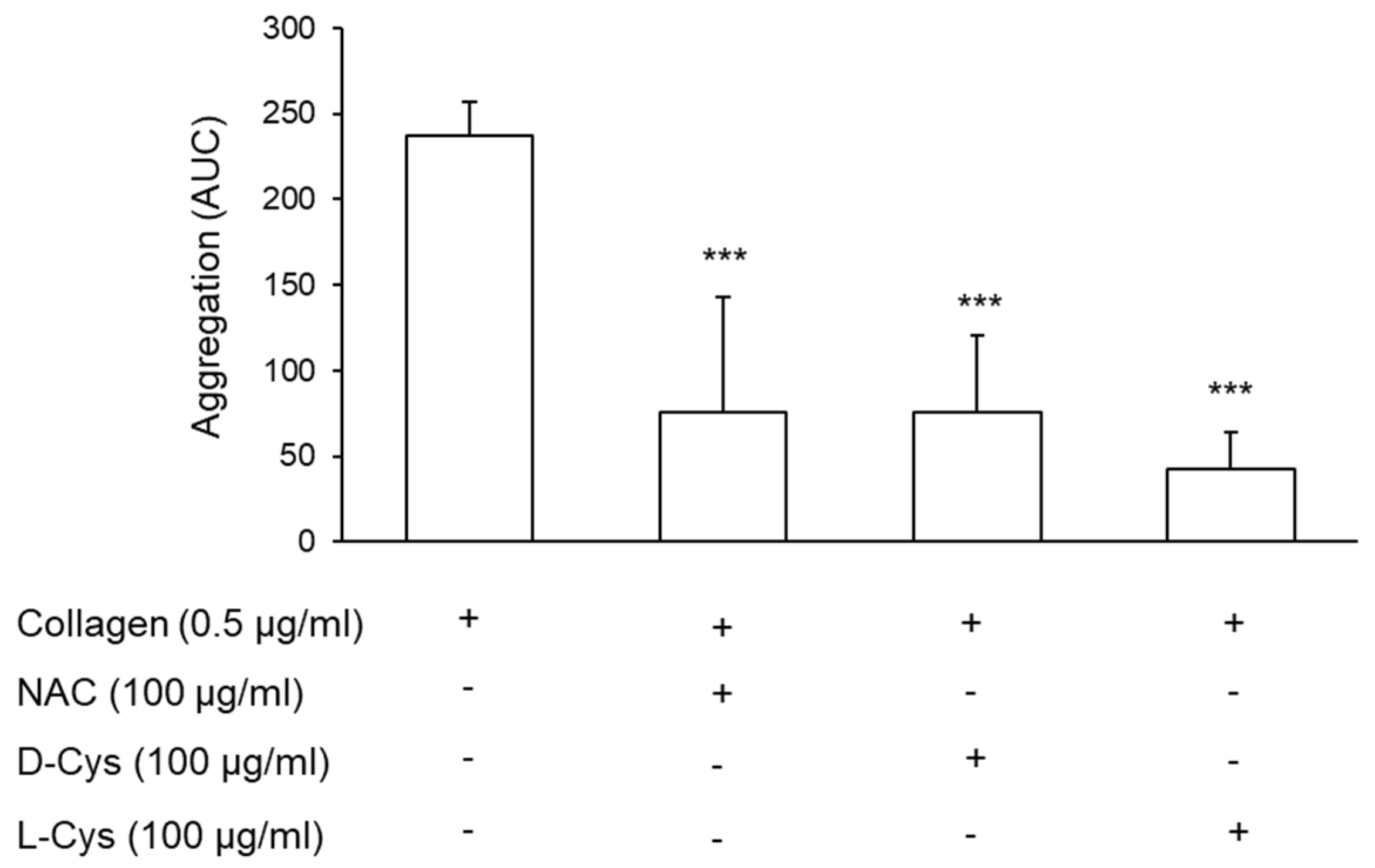
3.4. Effect of Cysteine on Platelet Aggregation
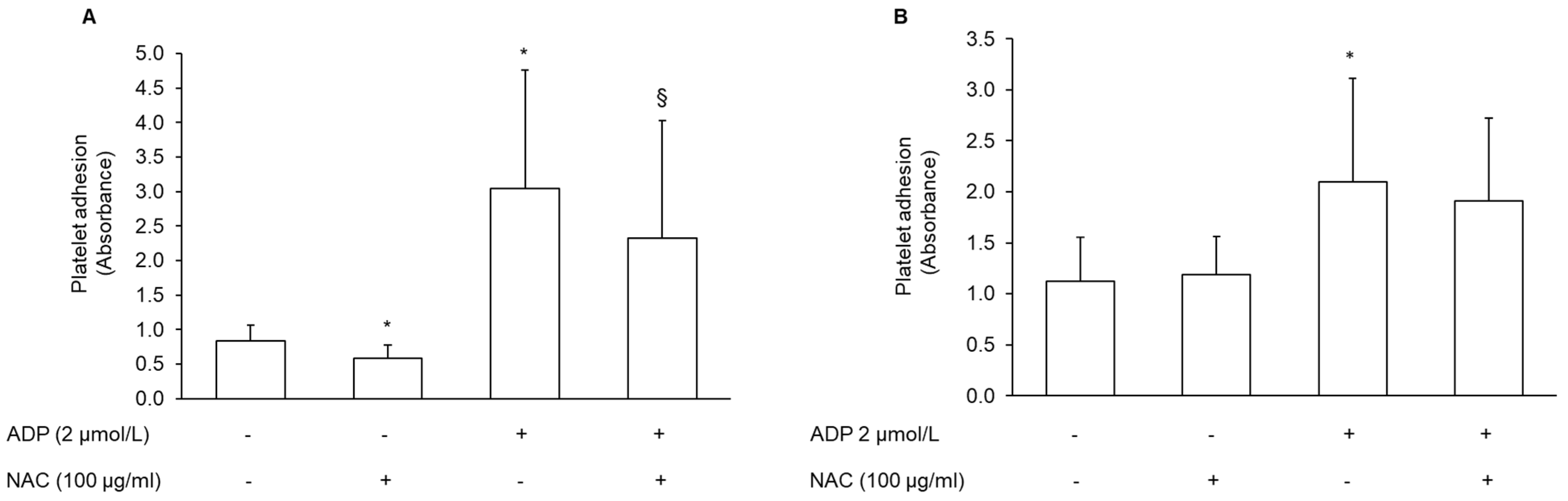
3.5. Effect of NAC-Pretreated PRP on Platelet Adhesion
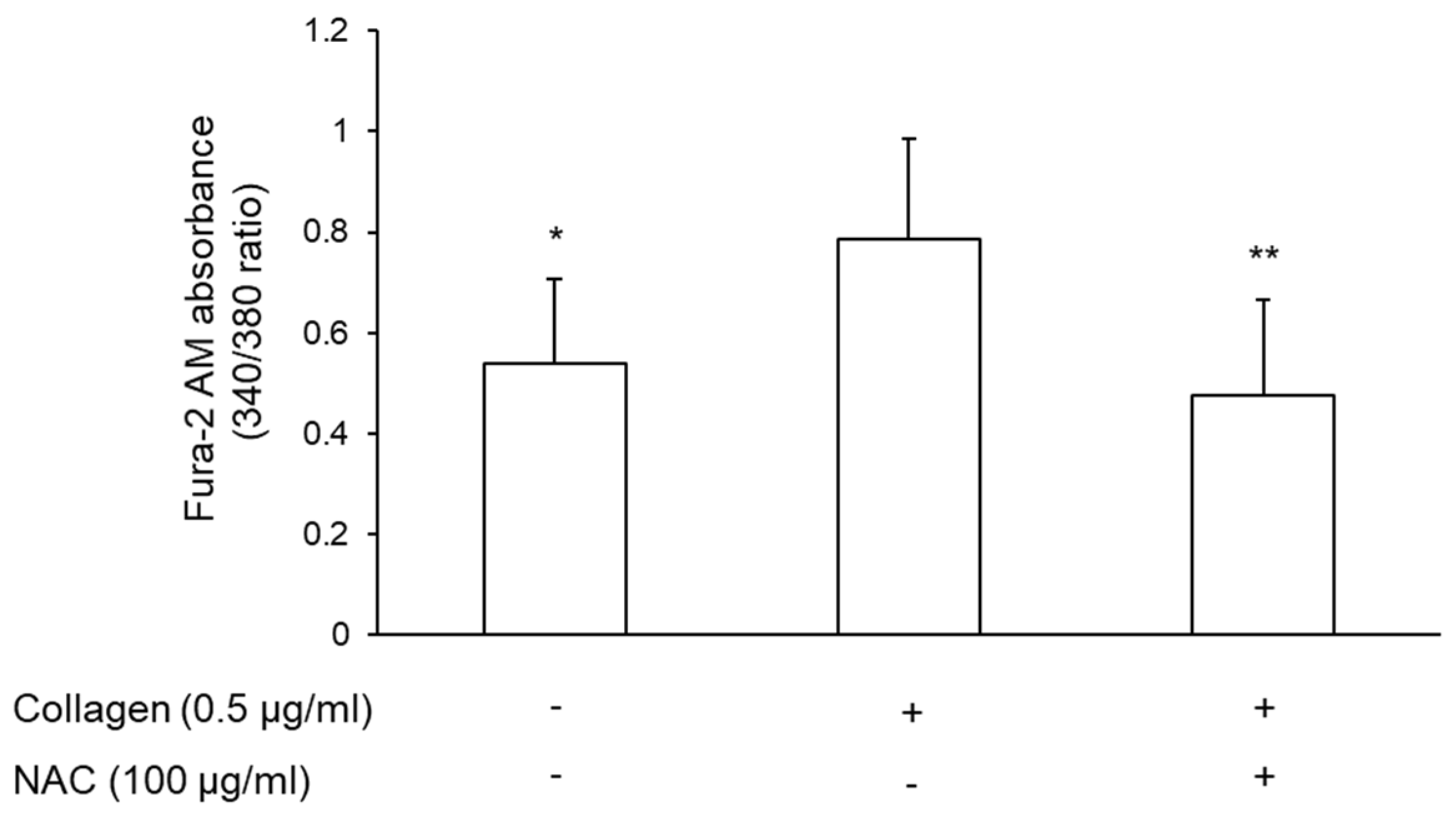
3.6. Effect of NAC-Pretreated Plasma on Intracellular Calcium Mobilization
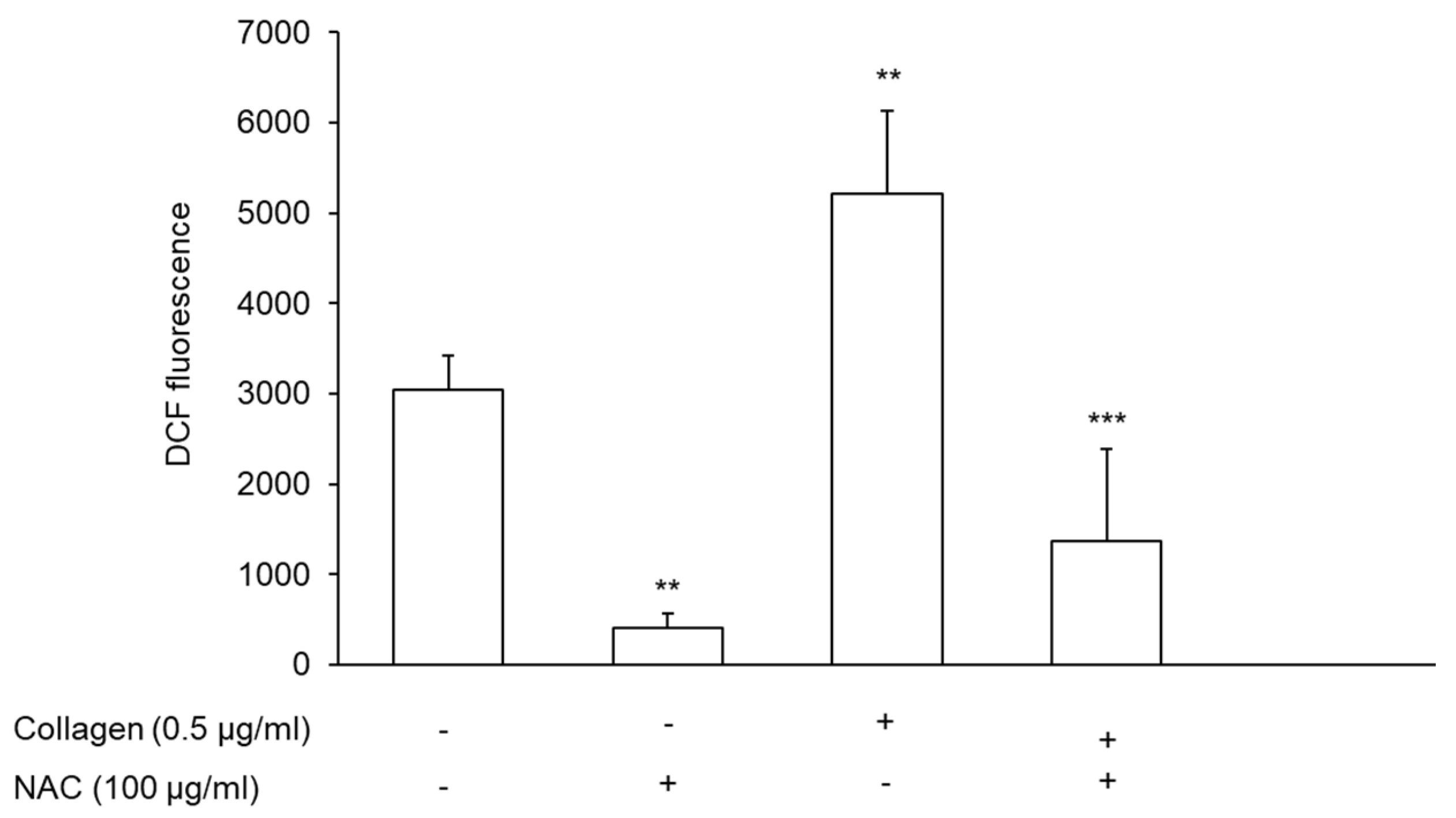
3.7. Effect of NAC-Pretreated PRP on Reactive Oxygen Species Generation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calzetta, L.; Matera, M.G.; Rogliani, P.; Cazzola, M. Multifaceted activity of N-acetyl-l-cysteine in chronic obstructive pulmonary disease. Expert Rev. Respir. Med. 2018, 12, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Prescott, L.F.; Illingworth, R.N.; Critchley, J.A.; Stewart, M.J.; Adam, R.D.; Proudfoot, A.T. Intravenous N-acetylcystine: The treatment of choice for paracetamol poisoning. Br. Med. J. 1979, 2, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenório, M.; Graciliano, N.; Moura, F.; Oliveira, A.; Goulart, M. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Berk, M.; Campochiaro, P.A.; Jaeschke, H.; Marenzi, G.; Richeldi, L.; Wen, F.Q.; Nicoletti, F.; Calverley, P.M.A. The Multifaceted Therapeutic Role of N-Acetylcysteine (NAC) in Disorders Characterized by Oxidative Stress. Curr. Neuropharmacol. 2021, 19, 1202–1224. [Google Scholar] [CrossRef] [PubMed]
- De Lizarrondo, S.M.; Gakuba, C.; Herbig, B.A.; Repesse, Y.; Ali, C.; Denis, C.; Lenting, P.; Touzé, E.; Diamond, S.L.; Vivien, D.; et al. Potent Thrombolytic Effect of N-Acetylcysteine on Arterial Thrombi. Circulation 2017, 136, 646–660. [Google Scholar] [CrossRef]
- Gibson, K.R.; Winterburn, T.J.; Barrett, F.; Sharma, S.; MacRury, S.M.; Megson, I.L. Therapeutic potential of N-acetylcysteine as an antiplatelet agent in patients with type-2 diabetes. Cardiovasc. Diabetol. 2011, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yee Aw, T.; Stokes, K.Y. N-acetylcysteine attenuates systemic platelet activation and cerebral vessel thrombosis in diabetes. Redox Biol. 2018, 14, 218–228. [Google Scholar] [CrossRef]
- Dludla, P.V.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Nyambuya, T.M.; Marcheggiani, F.; Cirilli, I.; Ziqubu, K.; Shabalala, S.C.; Johnson, R.; Louw, J.; et al. N-acetyl Cysteine Targets Hepatic Lipid Accumulation to Curb Oxidative Stress and Inflammation in NAFLD: A Comprehensive Analysis of the Literature. Antioxidants 2020, 9, 1283. [Google Scholar] [CrossRef]
- Quintavalle, C.; Donnarumma, E.; Fiore, D.; Briguori, C.; Condorelli, G. Therapeutic strategies to prevent contrast-induced acute kidney injury. Curr. Opin. Cardiol. 2013, 28, 676–682. [Google Scholar] [CrossRef]
- Liu, X.-H.; Xu, C.-Y.; Fan, G.-H. Efficacy of N-acetylcysteine in preventing atrial fibrillation after cardiac surgery: A meta-analysis of published randomized controlled trials. BMC Cardiovasc. Disord. 2014, 14, 52. [Google Scholar] [CrossRef] [Green Version]
- Deepmala; Slattery, J.; Kumar, N.; Delhey, L.; Berk, M.; Dean, O.; Spielholz, C.; Frye, R. Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. Neurosci. Biobehav. Rev. 2015, 55, 294–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waring, W.S. Criteria for acetylcysteine treatment and clinical outcomes after paracetamol poisoning. Expert Rev. Clin. Pharmacol. 2012, 5, 311–318. [Google Scholar] [CrossRef]
- Gosselin, S.; Hoffman, R.; Juurlink, D.N.; Whyte, I.; Yarema, M.; Caro, J. Treating acetaminophen overdose: Thresholds, costs and uncertainties. Clin. Toxicol. 2013, 51, 130–133. [Google Scholar] [CrossRef]
- Grinberg, L.; Fibach, E.; Amer, J.; Atlas, D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic. Biol. Med. 2005, 38, 136–145. [Google Scholar] [CrossRef]
- Turell, L.; Radi, R.; Alvarez, B. The thiol pool in human plasma: The central contribution of albumin to redox processes. Free Radic. Biol. Med. 2013, 65, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Colombo, G.; Clerici, M.; Giustarini, D.; Rossi, R.; Milzani, A.D.G.; Dalle-Donne, I. Redox Albuminomics: Oxidized Albumin in Human Diseases. Antioxid. Redox Signal. 2012, 17, 1515–1527. [Google Scholar] [CrossRef]
- Watanabe, H.; Imafuku, T.; Otagiri, M.; Maruyama, T. Clinical Implications Associated with the Posttranslational Modification–Induced Functional Impairment of Albumin in Oxidative Stress–Related Diseases. J. Pharm. Sci. 2017, 106, 2195–2203. [Google Scholar] [CrossRef] [Green Version]
- Arques, S. Serum albumin and cardiovascular disease: State-of-the-art review. Ann. Cardiol. Angeiol. 2020, 69, 192–200. [Google Scholar] [CrossRef]
- Jørgensen, K.A.; Stoffersen, E. On the inhibitory effect of albumin on platelet aggregation. Thromb. Res. 1980, 17, 13–18. [Google Scholar] [CrossRef]
- Silver, M.; Smith, J.; Ingerman, C.; Kocsis, J. Arachidonic acid-induced human platelet aggregation and prostaglandin formation. Prostaglandins 1973, 4, 863–875. [Google Scholar] [CrossRef]
- Lam, F.W.; Cruz, M.A.; Leung, H.-C.E.; Parikh, K.S.; Smith, C.W.; Rumbaut, R.E. Histone induced platelet aggregation is inhibited by normal albumin. Thromb. Res. 2013, 132, 69–76. [Google Scholar] [CrossRef]
- Pasterk, L.; Lemesch, S.; Leber, B.; Trieb, M.; Curcic, S.; Stadlbauer, V.; Schuligoi, R.; Schicho, R.; Heinemann, A.; Marsche, G. Oxidized plasma albumin promotes platelet-endothelial crosstalk and endothelial tissue factor expression. Sci. Rep. 2016, 6, 22104. [Google Scholar] [CrossRef]
- Bhat, A.; Das, S.; Yadav, G.; Chaudhary, S.; Vyas, A.K.; Islam, M.; Gupta, A.C.; Bajpai, M.; Maiwall, R.; Maras, J.S.; et al. Hyperoxidized Albumin Modulates Platelets and Promotes Inflammation Through CD36 Receptor in Severe Alcoholic Hepatitis. Hepatol. Commun. 2020, 4, 50–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coenen, D.M.; Heinzmann, A.C.; Karel, M.F.; Cosemans, J.M.; Koenen, R.R. The multifaceted contribution of platelets in the emergence and aftermath of acute cardiovascular events. Atherosclerosis 2021, 319, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.; Mendelsohn, M.E.; Amarante, P.; Smick, D.; Andon, N.; Davies, P.F.; Cooke, J.P.; Loscalzo, J. N-acetylcysteine potentiates platelet inhibition by endothelium-derived relaxing factor. Circ. Res. 1989, 65, 789–795. [Google Scholar] [CrossRef] [Green Version]
- Anfossi, G.; Russo, I.; Massucco, P.; Mattiello, L.; Balbo, A.; Cavalot, F.; Trovati, M. Studies on Inhibition of Human Platelet Function by Sodium Nitroprusside. Kinetic Evaluation of the Effect on Aggregation and Cyclic Nucleotide Content. Thromb. Res. 2001, 102, 319–330. [Google Scholar] [CrossRef]
- Niemi, T.T.; Munsterhjelm, E.; Pöyhiä, R.; Hynninen, M.S.; Salmenperä, M.T. The effect of N-acetylcysteine on blood coagulation and platelet function in patients undergoing open repair of abdominal aortic aneurysm. Blood Coagul. Fibrinolysis 2006, 17, 29–34. [Google Scholar] [CrossRef]
- Nikbakht, M.; Ahmadi, F.; Vaseghi, G.; Talasaz, A.H.; Eshraghi, A.; Naderi, J.; Daneshmehr, M.A. The Role of N-Acetylcysteine in Platelet Aggregation and Reperfusion Injury in Recent Years. Curr. Clin. Pharmacol. 2017, 12, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Squellerio, I.; Porro, B.; Songia, P.; Veglia, F.; Caruso, D.; Tremoli, E.; Cavalca, V. Liquid chromatography–tandem mass spectrometry for simultaneous measurement of thromboxane B2 and 12(S)-hydroxyeicosatetraenoic acid in serum. J. Pharm. Biomed. Anal. 2014, 96, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Squellerio, I.; Caruso, D.; Porro, B.; Veglia, F.; Tremoli, E.; Cavalca, V. Direct glutathione quantification in human blood by LC–MS/MS: Comparison with HPLC with electrochemical detection. J. Pharm. Biomed. Anal. 2012, 71, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Baron, G.; Brioschi, M.; Longoni, M.; Butti, R.; Valvassori, E.; Tremoli, E.; Carini, M.; Agostoni, P.; Vistoli, G.; et al. N-Acetyl-Cysteine Regenerates Albumin Cys34 by a Thiol-Disulfide Breaking Mechanism: An Explanation of Its Extracellular Antioxidant Activity. Antioxidants 2020, 9, 367. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz-Quiles, J.; Casulleras, M.; Oettl, K.; Titos, E.; Flores-Costa, R.; Duran-Güell, M.; López-Vicario, C.; Pavesi, M.; Stauber, R.E.; Arroyo, V.; et al. Oxidized Albumin Triggers a Cytokine Storm in Leukocytes Through P38 Mitogen-Activated Protein Kinase: Role in Systemic Inflammation in Decompensated Cirrhosis. Hepatology 2018, 68, 1937–1952. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef] [PubMed]
- Brioschi, M.; Gianazza, E.; Mallia, A.; Zoanni, B.; Altomare, A.; Fernandez, A.M.; Agostoni, P.; Aldini, G.; Banfi, C. S-Thiolation Targets Albumin in Heart Failure. Antioxidants 2020, 9, 763. [Google Scholar] [CrossRef] [PubMed]
- Davlouros, P.; Xanthopoulou, I.; Mparampoutis, N.; Giannopoulos, G.; Deftereos, S.; Alexopoulos, D. Role of Calcium in Platelet Activation: Novel Insights and Pharmacological Implications. Med. Chem. 2016, 12, 131–138. [Google Scholar] [CrossRef]
- Violi, F.; Pignatelli, P. Platelet Oxidative Stress and Thrombosis. Thromb. Res. 2012, 129, 378–381. [Google Scholar] [CrossRef]
- Gresele, P.; Deckmyn, H.; Huybrechts, E.; Vermylen, J. Serum albumin enhances the impairment of platelet aggregation with thromboxane synthase inhibition by increasing the formation of prostaglandin D2. Biochem. Pharmacol. 1984, 33, 2083–2088. [Google Scholar] [CrossRef]
- Kim, S.B.; Chi, H.S.; Park, J.S.; Hong, C.D.; Yang, W.S. Effect of increasing serum albumin on plasma D-dimer, von Willebrand factor, and platelet aggregation in CAPD patients. Am. J. Kidney Dis. 1999, 33, 312–317. [Google Scholar] [CrossRef]
- Johansson, M.; Eriksson, A.C.; Östgren, C.J.; Whiss, P.A. Platelet adhesion in type 2 diabetes: Impact of plasma albumin and mean platelet volume. Thromb. J. 2021, 19, 40. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.R.; Neilson, I.L.; Barrett, F.; Winterburn, T.J.; Sharma, S.; MacRury, S.M.; Megson, I.L. Evaluation of the Antioxidant Properties of N-acetylcysteine in Human Platelets: Prerequisite for Bioconversion to Glutathione for Antioxidant and Antiplatelet Activity. J. Cardiovasc. Pharmacol. 2009, 54, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Holinstat, M. 12-Lipoxygenase: A Potential Target for Novel Anti-Platelet Therapeutics. Cardiovasc. Hematol. Agents Med. Chem. 2011, 9, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Setty, B.N.; Werner, M.H.; Hannun, Y.A.; Stuart, M.J. 15-Hydroxyeicosatetraenoic acid-mediated potentiation of thrombin-induced platelet functions occurs via enhanced production of phosphoinositide-derived second messengers--sn-1,2-diacylglycerol and inositol-1,4,5-trisphosphate. Blood 1992, 80, 2765–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch-Decker, S.; Both, T.; Mülhopt, S.; Paur, H.-R.; Weiss, C.; Diabaté, S. Regulation of the arachidonic acid mobilization in macrophages by combustion-derived particles. Part. Fibre Toxicol. 2011, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Jamasbi, J.; Ayabe, K.; Goto, S.; Nieswandt, B.; Peter, K.; Siess, W. Platelet receptors as therapeutic targets: Past, present and future. Thromb. Haemost. 2017, 117, 1249–1257. [Google Scholar] [CrossRef]
- Ding, Z.; Kim, S.; Dorsam, R.T.; Jin, J.; Kunapuli, S.P. Inactivation of the human P2Y12 receptor by thiol reagents requires interaction with both extracellular cysteine residues, Cys17 and Cys270. Blood 2003, 101, 3908–3914. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, Z.M.; Mendolicchio, G.L. Adhesion Mechanisms in Platelet Function. Circ. Res. 2007, 100, 1673–1685. [Google Scholar] [CrossRef]
- Moroi, M.; Jung, S.M. Integrin-mediated platelet adhesion. Front. Biosci. 1998, 3, d719–d728. [Google Scholar] [CrossRef] [Green Version]
- Bergmeier, W.; Hynes, R.O. Extracellular Matrix Proteins in Hemostasis and Thrombosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005132. [Google Scholar] [CrossRef]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. N-Acetylcysteine ethyl ester (NACET): A novel lipophilic cell-permeable cysteine derivative with an unusual pharmacokinetic feature and remarkable antioxidant potential. Biochem. Pharmacol. 2012, 84, 1522–1533. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eligini, S.; Porro, B.; Aldini, G.; Colli, S.; Banfi, C. N-Acetylcysteine Inhibits Platelet Function through the Regeneration of the Non-Oxidative Form of Albumin. Antioxidants 2022, 11, 445. https://doi.org/10.3390/antiox11030445
Eligini S, Porro B, Aldini G, Colli S, Banfi C. N-Acetylcysteine Inhibits Platelet Function through the Regeneration of the Non-Oxidative Form of Albumin. Antioxidants. 2022; 11(3):445. https://doi.org/10.3390/antiox11030445
Chicago/Turabian StyleEligini, Sonia, Benedetta Porro, Giancarlo Aldini, Susanna Colli, and Cristina Banfi. 2022. "N-Acetylcysteine Inhibits Platelet Function through the Regeneration of the Non-Oxidative Form of Albumin" Antioxidants 11, no. 3: 445. https://doi.org/10.3390/antiox11030445