
Neuroprotection against Aminochrome Neurotoxicity: Glutathione Transferase M2-2 and DT-Diaphorase

 , , and
, , and
Abstract
:
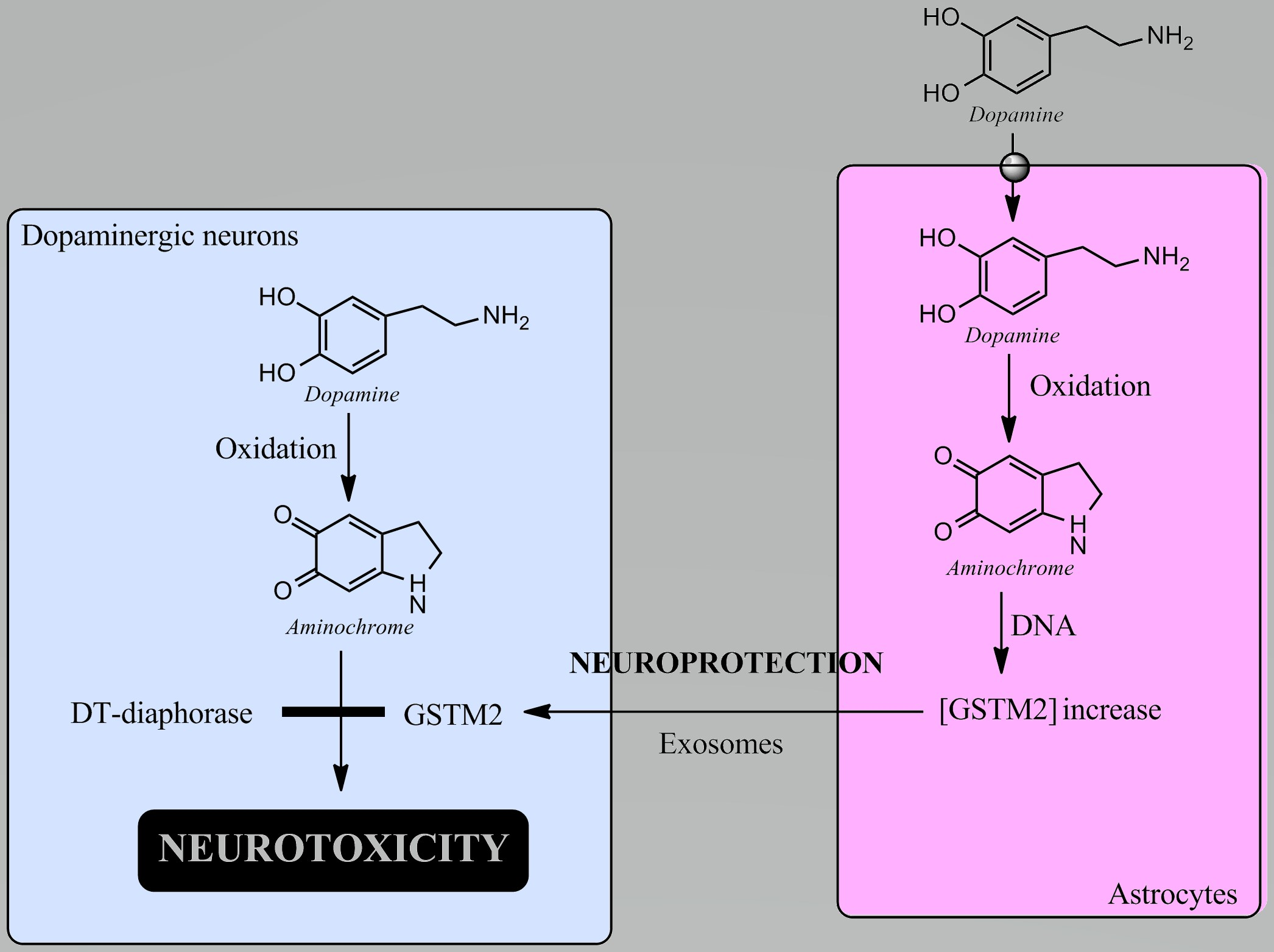{kind=link}
{kind=link}
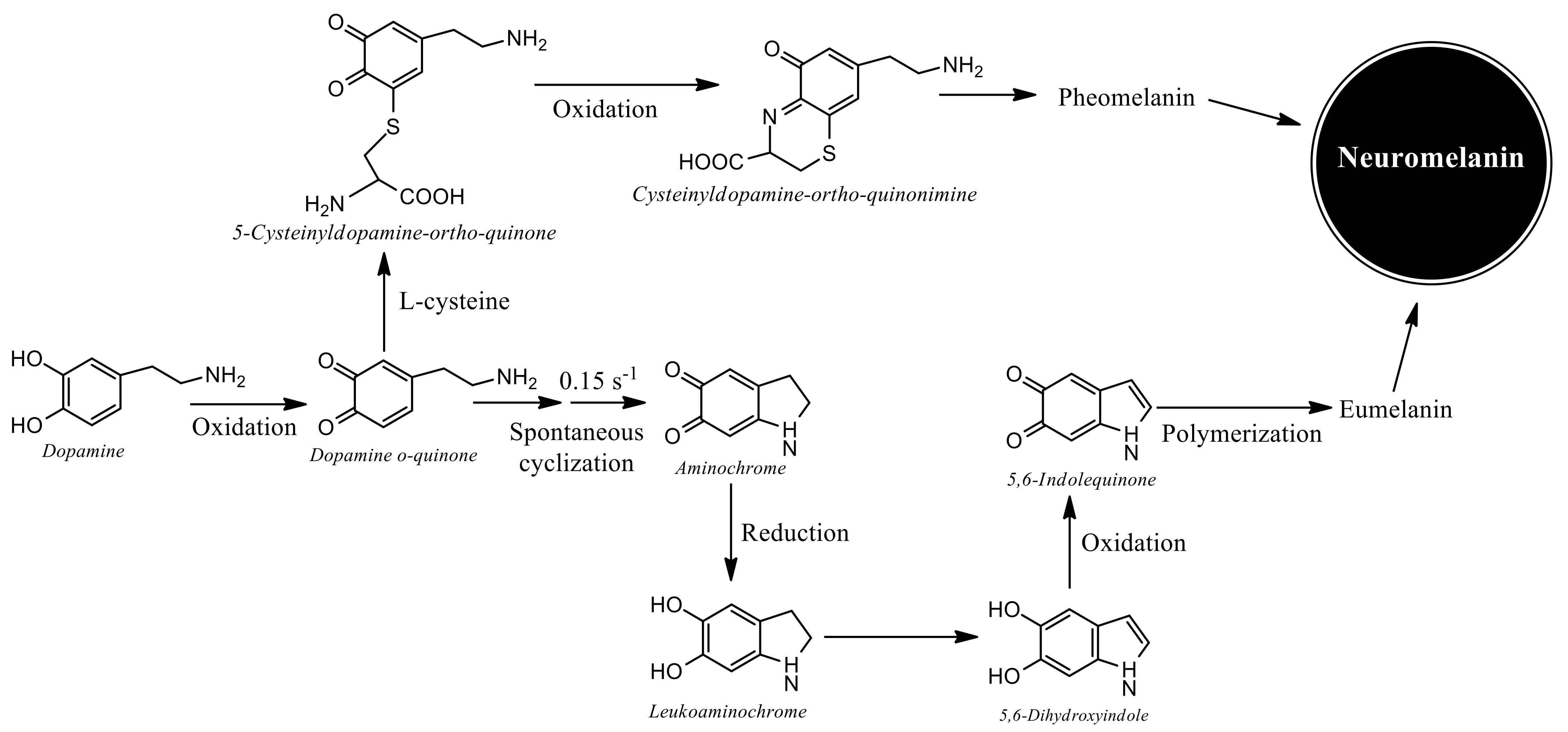{kind=link}
{kind=link}
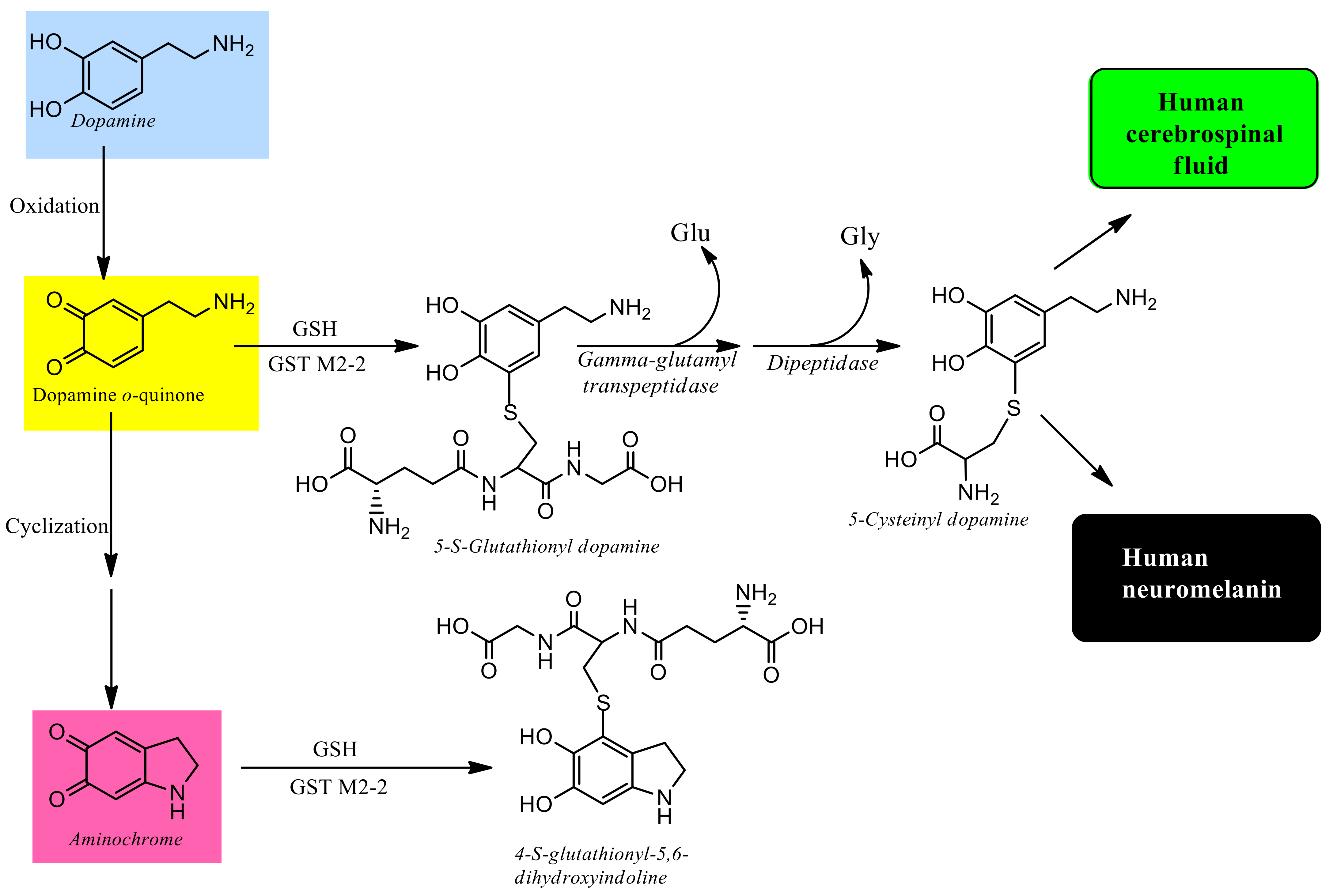{kind=link}
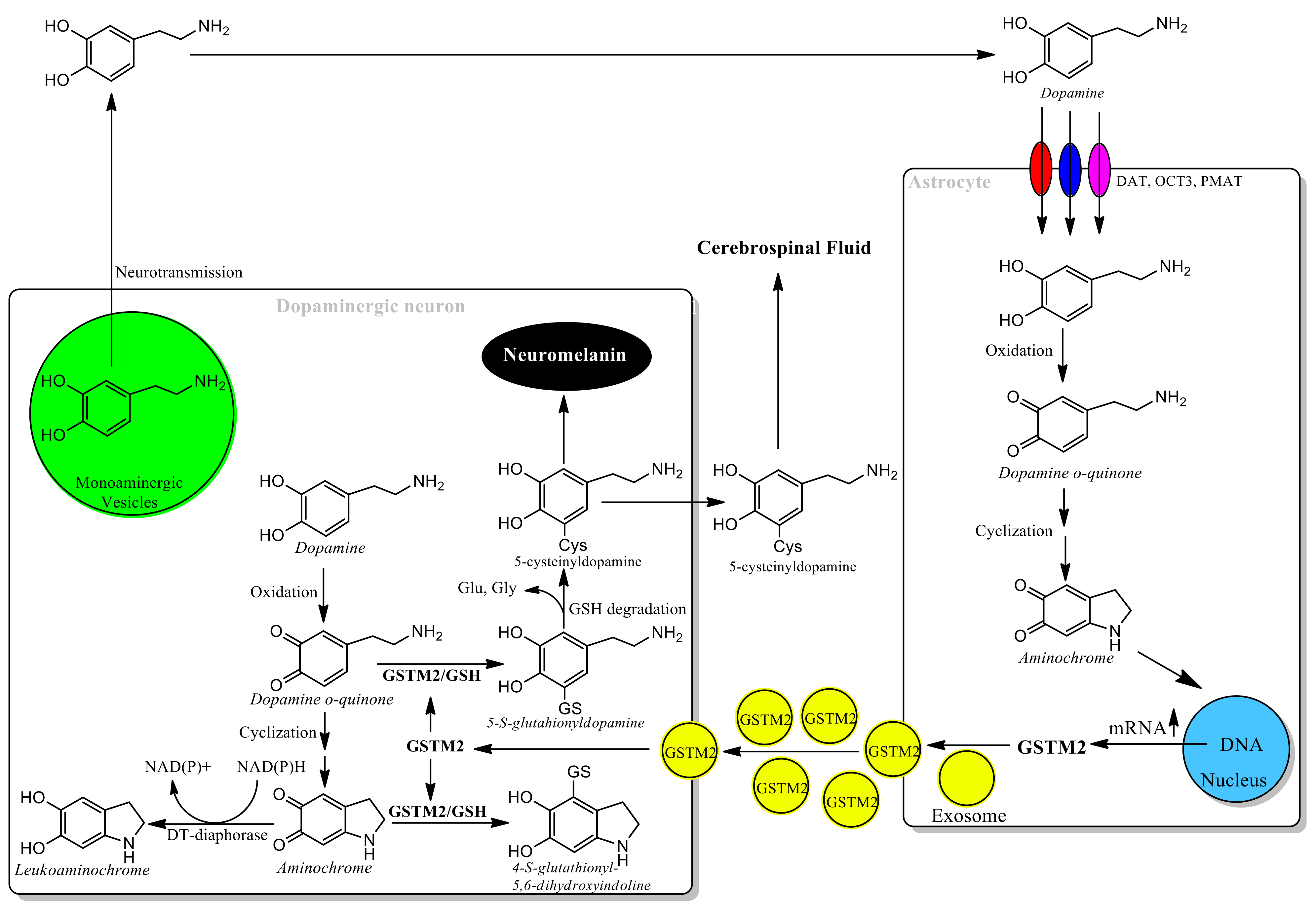{kind=link}
1. Glutathione as Antioxidant
2. Glutathione Transferase
3. Parkinson’s Disease Neurodegeneration
3.1. Alpha-Synuclein Oligomers
3.2. 3,4-Dihydroxyphenylacetaldehyde (DOPAL)
3.3. Aminochrome: The Neuromelanin Precursor
3.4. DT-Diaphorase
3.5. Glutathione Transferase M2-2
4. Astrocytes Protects Dopaminergic Neurons
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Josephy, P.D.; Mannervik, B. Molecular Toxicology, 2nd ed.; Oxford University Press: New York, NY, USA, 2006; pp. 303–332. [Google Scholar]
- Volumes 3A and 3B: Glutathione—Chemical, Biochemical, and Medical Aspects. In Coenzymes and Cofactors; Dolphin, D.; Poulson, R.; Avramovic, O. (Eds.) John Wiley & Sons: New York, NY, USA, 1989. [Google Scholar]
- Mannervik, B.; Carlberg, I.; Larson, K. Glutathione: General Review of Mechanism of Action. In Coenzymes and Cofactors; Dolphin, D., Poulson, R., Avramovic, O., Eds.; John Wiley & Sons: New York, NY, USA, 1989; Volume 3A, pp. 475–516. [Google Scholar]
- Aoyama, K. Glutathione in the brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Moron, M.S.; De Pierre, J.W.; Mannervik, B. Levels of glutathione, glutathione reductase and glutathione S-transferase activities in rat lung and liver. Biochim. Biophys. Acta 1979, 582, 67–78. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Purification and characterization of the flavoenzyme glutathione reductase from rat liver. J. Biol. Chem. 1975, 250, 5475–5480. [Google Scholar] [CrossRef]
- Eriksson, B. On the synthesis and enzymatic reduction of the coenzyme A-glutathione mixed disulfide. Acta Chem. Scand. 1966, 20, 1178–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelsson, K.; Eriksson, S.; Mannervik, B. Purification and characterization of cytoplasmic thioltransferase (glutathione: Disulfide oxidoreductase) from rat liver. Biochemistry 1978, 17, 2978–2984. [Google Scholar] [CrossRef]
- Eriksson, S.A.; Mannervik, B. The reduction of the L-cysteine-glutathione mixed disulfide in rat liver. Involvement of an enzyme catalyzing thiol-disulfide interchange. FEBS Lett. 1970, 7, 26–28. [Google Scholar] [CrossRef] [Green Version]
- Mannervik, B.; Axelsson, K.; Sundewall, A.-C.; Holmgren, A. Relative contributions of thioltransferase- and thioredoxin-dependent systems in reduction of low-molecular-mass and protein disulphides. Biochem. J. 1983, 213, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Mannervik, B. The isoenzymes of glutathione transferase. Adv. Enzymol. Rel. Areas Mol. Biol. 1985, 57, 357–417. [Google Scholar]
- Bjørklund, G.; Peana, M.; Maes, M.; Dadar, M.; Severin, B. The glutathione system in Parkinson’s disease and its progression. Neurosci. Biobehav. Rev. 2021, 120, 470–478. [Google Scholar] [CrossRef]
- Perry, T.L.; Godin, D.V.; Hansen, S. Parkinson’s disease: A disorder due to nigral glutathione deficiency? Neurosci. Lett. 1982, 33, 305–310. [Google Scholar] [CrossRef]
- Hauser, R.A.; Lyons, K.E.; McClain, T.; Carter, S.; Perlmutter, D. Randomized, double-blind, pilot evaluation of intravenous glutathione in Parkinson’s disease. Mov. Disord. 2009, 24, 983. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Lau, R.C.; Shankland, E.G.; Wilbur, T.K.; Padowski, J.M. Phase IIb Study of Intranasal Glutathione in Parkinson’s Disease. J. Parkinsons. Dis. 2017, 7, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, R.C.; Domingues, H.S.; Salgado, A.J.; Teixeira, F.G. From regenerative strategies to pharmacological approaches: Can we fine-tune treatment for Parkinson’s disease? Neural. Regen Res. 2022, 17, 933–936. [Google Scholar] [PubMed]
- Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 9592. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I. Glutathione and related molecules in Parkinsonism. Int. J. Mol. Sci. 2021, 22, 8689. [Google Scholar] [CrossRef]
- Mannervik, B. Five decades with glutathione and the GSTome. J. Biol. Chem. 2012, 287, 6072–6083. [Google Scholar] [CrossRef] [Green Version]
- Mannervik, B.; Board, P.G.; Hayes, J.D.; Listowsky, I.; Pearson, W.R. Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 2005, 401, 1–8. [Google Scholar]
- Berhane, K.; Widersten, M.; Engström, Å.; Kozarich, J.W.; Mannervik, B. Detoxication of base propenals and other a,b-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proc. Natl. Acad. Sci. USA 1994, 91, 1480–1484. [Google Scholar] [CrossRef] [Green Version]
- Baez, S.; Segura-Aguilar, J.; Widersten, M.; Johansson, A.-S.; Mannervik, B. Glutathione transferases catalyse the detoxication of oxidized metabolites (o-quinones) of catecholamines and may serve as an antioxidant system preventing degenerative cellular processes. Biochem. J. 1997, 324, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Huenchuguala, S.; Sjödin, B.; Mannervik, B.; Segura-Aguilar, J. Novel Alpha-Synuclein Oligomers Formed with the Aminochrome-Glutathione Conjugate Are Not Neurotoxic. Neurotox. Res. 2019, 35, 432–440. [Google Scholar] [CrossRef]
- Cuevas, C.; Huenchuguala, S.; Muñoz, P.; Villa, M.; Paris, I.; Mannervik, B.; Segura-Aguilar, J. Glutathione transferase-M2–2 secreted from glioblastoma cell protects SH-SY5Y cells from aminochrome neurotoxicity. Neurotox. Res. 2015, 27, 217–228. [Google Scholar] [CrossRef]
- Dirr, H.; Reinemer, P.; Huber, R. X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function. Eur. J. Biochem. 1994, 220, 645–661. [Google Scholar] [CrossRef]
- Mannervik, B.; Jensson, H. Binary combinations of four protein subunits with different catalytic specificities explain the relationship between six basic glutathione S-transferases in rat liver cytosol. J. Biol. Chem. 1982, 257, 9909–9912. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Board, P.G.; Suzuki, T.; Shaw, D.C. Human muscle glutathione S-transferase (GST-4) shows close homology to human liver GST-1. Biochim. Biophys. Acta 1988, 953, 214–217. [Google Scholar] [CrossRef]
- Rowe, J.D.; Nieves, E.; Listowsky, I. Subunit diversity and tissue distribution of human glutathione S-transferases: Interpretations based on electrospray ionization-MS and peptide sequence-specific antisera. Biochem. J. 1997, 325, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J.; Baez, S.; Widersten, M.; Welch, C.J.; Mannervik, B. Human class Mu glutathione transferases, in particular isoenzyme M2–2, catalyze detoxication of the dopamine metabolite aminochrome. J. Biol. Chem. 1997, 272, 5727–5731. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J. Parkinson’s disease. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 1–168. [Google Scholar]
- Chen, Y.C.; Chen, R.S.; Weng, Y.H.; Huang, Y.Z.; Chen, C.C.; Hung, J.; Lin, Y.Y. The severity progression of non-motor symptoms in Parkinson’s disease: A 6-year longitudinal study in Taiwanese patients. Sci. Rep. 2021, 11, 14781. [Google Scholar]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Wang, X.Y.; Han, Y.Y.; Li, G.; Zhang, B. Association between autonomic dysfunction and olfactory dysfunction in Parkinson’s disease in southern Chinese. BMC Neurol. 2019, 19, 17. [Google Scholar] [CrossRef] [PubMed]
- Williams, A. MPTP parkinsonism. Br. Med. J. 1984, 289, 1401–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, R.M. Neurobehavioral deficits and parkinsonism in occupations with manganese exposure: A review of methodological issues in the epidemiological literature. Saf. Health Work 2013, 4, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caviedes, P.; Segura-Aguilar, J. The price of development in Chile: Overcoming environmental hazards produced by heavy industrial exploitation. Neuroreport 2001, 12, A25. [Google Scholar] [CrossRef]
- Rehman, K.; Irshad, K.; Kamal, S.; Imran, I.; Akash, M.S.H. Exposure of Environmental Contaminants and Development of Neurological Disorders. Crit. Rev. Eukaryot. Gene Expr. 2021, 31, 35–53. [Google Scholar] [CrossRef]
- Savica, R.; Boeve, B.F.; Mielke, M.M. When do α-Synucleinopathies Start? An Epidemiological Timeline: A Review. JAMA Neurol. 2018, 75, 503–509. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. A timeline for Parkinson’s disease. Parkinsonism Relat. Disord. 2010, 16, 79–84. [Google Scholar] [CrossRef]
- Ray, B.; Bhat, A.; Mahalakshmi, A.M.; Tuladhar, S.; Bishir, M.; Mohan, S.K.; Veeraraghavan, V.P.; Chandra, R.; Essa, M.M.; Chidambaram, S.B.; et al. Mitochondrial and Organellar Crosstalk in Parkinson’s Disease. ASN Neuro 2021, 13, 17590914211028364. [Google Scholar] [CrossRef]
- Lizama, B.N.; Chu, C.T. Neuronal autophagy and mitophagy in Parkinson’s disease. Mol. Aspects Med. 2021, 12, 100972. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, J.H.; Joe, E.H.; Jou, I. Small heterodimer partner (SHP) aggravates ER stress in Parkinson’s disease-linked LRRK2 mutant astrocyte by regulating XBP1 SUMOylation. J. Biomed. Sci. 2021, 28, 51. [Google Scholar] [CrossRef]
- Yang, H. LncRNA MALAT1 potentiates inflammation disorder in Parkinson’s disease. Int. J. Immunogenet. 2021, 48, 419–428. [Google Scholar] [CrossRef]
- Herrera, A.; Muñoz, P.; Steinbusch, H.W.M.; Segura-Aguilar, J. Are Dopamine Oxidation Metabolites Involved in the Loss of Dopaminergic Neurons in the Nigrostriatal System in Parkinson’s Disease? ACS Chem. Neurosci. 2017, 8, 702–711. [Google Scholar] [CrossRef]
- Franco, R.; Rivas-Santisteban, R.; Navarro, G.; Pinna, A.; Reyes-Resina, I. Genes Implicated in Familial Parkinson’s Disease Provide a Dual Picture of Nigral Dopaminergic Neurodegeneration with Mitochondria Taking Center Stage. Int. J. Mol. Sci. 2021, 22, 4643. [Google Scholar] [CrossRef]
- Chu, Y.T.; Tai, C.H.; Lin, C.H.; Wu, R.M. Updates on the Genetics of Parkinson’s Disease: Clinical Implications and Future Treatment. Acta Neurol. Taiwan 2021, 30, 83–93. [Google Scholar]
- Segura-Aguilar, J. Preclinical models based on genetic mutations associated with the familial form of Parkinson’s disease. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 259–263. [Google Scholar]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J. Preclinical models based on endogenous neurotoxins. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 213–227. [Google Scholar]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef]
- Fornai, F.; Lenzi, P.; Gesi, M.; Ferrucci, M.; Lazzeri, G.; Natale, G.; Ruggieri, S.; Paparelli, A. Recent knowledge on molecular components of Lewy bodies discloses future therapeutic strategies in Parkinson’s disease. Curr. Drug Targets CNS Neurol. Disord. 2003, 2, 149–152. [Google Scholar] [CrossRef]
- Kunath, T.; Natalwala, A.; Chan, C.; Chen, Y.; Stecher, B.; Taylor, M.; Khan, S.; Muqit, M.M.K. Are PARKIN patients ideal candidates for dopaminergic cell replacement therapies? Eur. J. Neurosci. 2019, 49, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Gaig, C.; Martí, M.J.; Ezquerra, M.; Cardozo, A.; Rey, M.J.; Tolosa, E. G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. BMJ Case Rep. 2009, 78, 626–628. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J. Can we conclude a potential therapeutic action for Parkinson’s disease by using postmortem tissue and a preclinical model based on an exogenous neurotoxin? Cell Death Dis. 2018, 9, 748. [Google Scholar] [CrossRef]
- Muñoz, P.; Cardenas, S.; Huenchuguala, S.; Briceño, A.; Couve, E.; Paris, I.; Segura-Aguilar, J. DT-Diaphorase Prevents Aminochrome-Induced Alpha-Synuclein Oligomer Formation and Neurotoxicity. Toxicol. Sci. 2015, 145, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Cascella, R.; Chen, S.W.; Bigi, A.; Camino, J.D.; Xu, C.K.; Dobson, C.M.; Chiti, F.; Cremades, N.; Cecchi, C. The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells. Nat. Commun. 2021, 12, 1814. [Google Scholar] [CrossRef]
- Grünblatt, E.; Ruder, J.; Monoranu, C.M.; Riederer, P.; Youdim, M.B.; Mandel, S.A. Differential Alterations in Metabolism and Proteolysis-Related Proteins in Human Parkinson’s Disease Substantia Nigra. Neurotox Res. 2018, 33, 560–568. [Google Scholar] [CrossRef]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S. The catecholaldehyde hypothesis: Where MAO fits in. J. Neural. Transm. 2020, 127, 169–177. [Google Scholar] [CrossRef]
- Coelho-Cerqueira, E.; de Araújo, C.C.C.; Follmer, C. Formation of large oligomers of DOPAL-modified α-synuclein is modulated by the oxidation of methionine residues located at C-terminal domain. Biochem. Biophys. Res. Commun. 2019, 509, 367–372. [Google Scholar] [CrossRef]
- Grünblatt, E.; Mandel, S.; Jacob-Hirsch, J.; Zeligson, S.; Amariglo, N.; Rechavi, G.; Li, J.; Ravid, R.; Roggendorf, W.; Riederer, P.; et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiqui-tin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesi-cle trafficking genes. J. Neural. Transm. 2004, 111, 1543–1573. [Google Scholar] [CrossRef]
- Liu, G.; Yu, J.; Ding, J.; Xie, C.; Sun, L.; Rudenko, I.; Zheng, W.; Sastry, N.; Luo, J.; Rudow, G.; et al. Aldehyde dehy-drogenase 1 defines and protects a nigrostriatal dopaminergic neuron subpopulation. J. Clin. Investig. 2014, 124, 3032–3046. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J. Dopamine oxidation to neuromelanin and neurotoxic metabolites. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 213–223. [Google Scholar]
- Liang, C.L.; Nelson, O.; Yazdani, U.; Pasbakhsh, P.; German, D.C. Inverse relationship between the contents of neuromelanin pigment and the vesicular monoamine transporter-2: Human midbrain dopamine neurons. J. Comp. Neurol. 2004, 473, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar J; Mannervik B; Inzunza J; Varshney M; Nalvarte I; Muñoz P; Astrocytes protect dopaminergic neurons against aminochrome neurotoxicity. Neural Regen Res 2022, 17, 1861–1866.
- Wakamatsu, K.; Fukushima, S.; Minagawa, A.; Omodaka, T.; Hida, T.; Hatta, N.; Takata, M.; Uhara, H.; Okuyama, R.; Ihn, H. Significance of 5-S-Cysteinyldopa as a Marker for Melanoma. Int. J. Mol. Sci. 2020, 21, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriagada, C.; Paris, I.; de las Matas, S.M.J.; Martinez-Alvarado, P.; Cardenas, S.; Castañeda, P.; Graumann, R.; Perez-Pastene, C.; Olea-Azar, C.; Couve, E.; et al. On the neurotoxicity mechanism of leukoaminochrome o-semiquinone radical derived from dopamine oxidation: Mitochondria damage, necrosis, and hydroxyl radical formation. Neurobiol. Dis. 2004, 16, 468–477. [Google Scholar] [CrossRef]
- Aguirre, P.; Urrutia, P.; Tapia, V.; Villa, M.; Paris, I.; Segura-Aguilar, J.; Núñez, M.T. The dopamine metabolite aminochrome inhibits mitochondrial complex I and modifies the expression of iron transporters DMT1 and FPN1. Biometals 2012, 25, 795–803. [Google Scholar] [CrossRef]
- Fuentes, P.; Paris, I.; Nassif, M.; Caviedes, P.; Segura-Aguilar, J. Inhibition of VMAT-2 and DT-diaphorase induce cell death in a substantia nigra-derived cell line—An experimental cell model for dopamine toxicity studies. Chem. Res. Toxicol. 2007, 20, 776–783. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Huenchuguala, S. Aminochrome Induces Irreversible Mitochondrial Dysfunction by Inducing Autophagy Dysfunction in Parkinson’s Disease. Front. Neurosci. 2018, 12, 106. [Google Scholar] [CrossRef] [Green Version]
- Huenchuguala, S.; Muñoz, P.; Segura-Aguilar, J. The Importance of Mitophagy in Maintaining Mitochondrial Function in U373MG Cells. Bafilomycin A1 Restores Aminochrome-Induced Mitochondrial Damage. ACS Chem. Neurosci. 2017, 8, 2247–2253. [Google Scholar] [CrossRef]
- Santos, C.C.; Araújo, F.M.; Ferreira, R.S.; Silva, V.B.; Silva, J.H.C.; Grangeiro, M.S.; Soares, É.N.; Pereira, É.P.L.; Souza, C.S.; Costa, S.L.; et al. Aminochrome induces microglia and astrocyte activation. Toxicol. In Vitro 2017, 42, 54–60. [Google Scholar] [CrossRef]
- De Araújo, F.M.; Ferreira, R.S.; Souza, C.S.; Dos Santos, C.C.; Rodrigues, T.L.R.S.; ESilva, J.H.C.; Gasparotto, J.; Gelain, D.P.; El-Bachá, R.S.; DCosta, M.F.; et al. Aminochrome decreases NGF, GDNF and induces neuroinflammation in organotypic midbrain slice cultures. Neurotoxicology 2018, 66, 98–106. [Google Scholar] [CrossRef]
- Xiong, R.; Siegel, D.; Ross, D. Quinone-induced protein handling changes: Implications for major protein handling systems in quinone-mediated toxicity. Toxicol. Appl. Pharmacol. 2014, 280, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Huenchuguala, S.; Muñoz, P.; Zavala, P.; Villa, M.; Cuevas, C.; Ahumada, U.; Graumann, R.; Nore, B.F.; Couve, E.; Mannervik, B.; et al. Glutathione transferase mu 2 protects glioblastoma cells against aminochrome toxicity by preventing autophagy and lysosome dysfunction. Autophagy 2014, 10, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Parkinsons Dis. 2012, 2012, 920953. [Google Scholar] [CrossRef]
- Meléndez, C.; Muñoz, P.; Segura-Aguilar, J. DT-Diaphorase Prevents Aminochrome-Induced Lysosome Dysfunction in SH-SY5Y Cells. Neurotox. Res. 2019, 35, 255–259. [Google Scholar] [CrossRef]
- Paris, I.; Perez-Pastene, C.; Cardenas, S.; Iturriaga-Vasquez, P.; Muñoz, P.; Couve, E.; Caviedes, P.; Segura-Aguilar, J. Aminochrome induces disruption of actin, alpha-, and beta-tubulin cytoskeleton networks in substantia-nigra-derived cell line. Neurotox. Res. 2010, 18, 82–92. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. Neuroprotective mechanisms against dopamine oxidation-dependent neurotoxicity. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 213–227. [Google Scholar]
- Schultzberg, M.; Segura-Aguilar, J.; Lind, C. Distribution of DT diaphorase in the rat brain: Biochemical and immunohistochemical studies. Neuroscience 1988, 27, 763–776. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Metodiewa, D.; Welch, C.J. Metabolic activation of dopamine o-quinones to o-semiquinones by NADPH cytochrome P450 reductase may play an important role in oxidative stress and apoptotic effects. Biochim. Biophys. Acta 1998, 1381, 1–6. [Google Scholar] [CrossRef]
- Lozano, J.; Muñoz, P.; Nore, B.F.; Ledoux, S.; Segura-Aguilar, J. Stable expression of short interfering RNA for DT-diaphorase induces neurotoxicity. Chem. Res. Toxicol. 2010, 23, 1492–1496. [Google Scholar] [CrossRef]
- Zafar, K.S.; Siegel, D.; Ross, D. A potential role for cyclized quinones derived from dopamine, DOPA, and 3,4-dihydroxyphenylacetic acid in proteasomal inhibition. Mol. Pharmacol. 2006, 70, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Herrera, A.; Muñoz, P.; Paris, I.; Díaz-Veliz, G.; Mora, S.; Inzunza, J.; Hultenby, K.; Cardenas, C.; Jaña, F.; Raisman-Vozari, R.; et al. Aminochrome induces dopaminergic neuronal dysfunction: A new animal model for Parkinson’s disease. Cell Mol. Life Sci. 2016, 73, 3583–3597. [Google Scholar] [CrossRef]
- Dagnino-Subiabre, A.; Cassels, B.K.; Baez, S.; Johansson, A.S.; Mannervik, B.; Segura-Aguilar, J. Glutathione transferase M2–2 catalyzes conjugation of dopamine and dopa o-quinones. Biochem. Biophys. Res. Commun. 2000, 274, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Carstam, R.; Brinck, C.; Hindemith-Augustsson, A.; Rorsman, H.; Rosengren, E. The neuromelanin of the human substantia nigra. Biochim. Biophys. Acta 1991, 1097, 152–160. [Google Scholar] [CrossRef]
- Cheng, F.C.; Kuo, J.S.; Chia, L.G.; Dryhurst, G. Elevated 5-S-cysteinyldopamine/homovanillic acid ratio and reduced homovanillic acid in cerebrospinal fluid: Possible markers for and potential insights into the pathoetiology of Parkinson’s disease. J. Neural. Transm. 1996, 103, 433–446. [Google Scholar] [CrossRef]
- Rosengren, E.; Linder-Eliasson, E.; Carlsson, A. Detection of 5-S-cysteinyldopamine in human brain. J. Neural. Transm. 1985, 63, 247–253. [Google Scholar] [CrossRef]
- Wang, X.F.; Cynader, M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem. 2000, 74, 1434–1442. [Google Scholar]
- Tarczyluk, M.A.; Nagel, D.A.; O’Neil, J.D.; Parri, H.R.; Tse, E.H.; Coleman, M.D.; Hill, E.J. Functional astrocyte-neuron lactate shuttle in a human stem cell-derived neuronal network. J. Cereb. Blood Flow Metab. 2013, 32, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A. Astrocytic Metabolism Focusing on Glutamate Homeostasis: A Short Review Dedicated to Vittorio Gallo. Neurochem. Res. 2020, 45, 522–525. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. A new mechanism for protection of dopaminergic neurons mediated by astrocytes. Neural. Regen Res. 2015, 10, 1225–1227. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. On the role of endogenous neurotoxins and neuroprotection in Parkinson’s disease. Neural. Regen Res. 2017, 12, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Valdes, R.; Armijo, A.; Muñoz, P.; Hultenby, K.; Hagg, A.; Inzunza, J.; Nalvarte, I.; Varshney, M.; Mannervik, B.; Segura-Aguilar, J. Cellular Trafficking of Glutathione Transferase M2–2 Between U373MG and SHSY-S7 Cells is Mediated by Exosomes. Neurotox. Res. 2021, 39, 182–190. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segura-Aguilar, J.; Muñoz, P.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Mannervik, B. Neuroprotection against Aminochrome Neurotoxicity: Glutathione Transferase M2-2 and DT-Diaphorase. Antioxidants 2022, 11, 296. https://doi.org/10.3390/antiox11020296
Segura-Aguilar J, Muñoz P, Inzunza J, Varshney M, Nalvarte I, Mannervik B. Neuroprotection against Aminochrome Neurotoxicity: Glutathione Transferase M2-2 and DT-Diaphorase. Antioxidants. 2022; 11(2):296. https://doi.org/10.3390/antiox11020296
Chicago/Turabian StyleSegura-Aguilar, Juan, Patricia Muñoz, Jose Inzunza, Mukesh Varshney, Ivan Nalvarte, and Bengt Mannervik. 2022. "Neuroprotection against Aminochrome Neurotoxicity: Glutathione Transferase M2-2 and DT-Diaphorase" Antioxidants 11, no. 2: 296. https://doi.org/10.3390/antiox11020296