
Neuronal Oxidative Stress Promotes α-Synuclein Aggregation In Vivo
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Breeding and Husbandry
2.2. Experimental Design
2.3. Behavioral Assessments
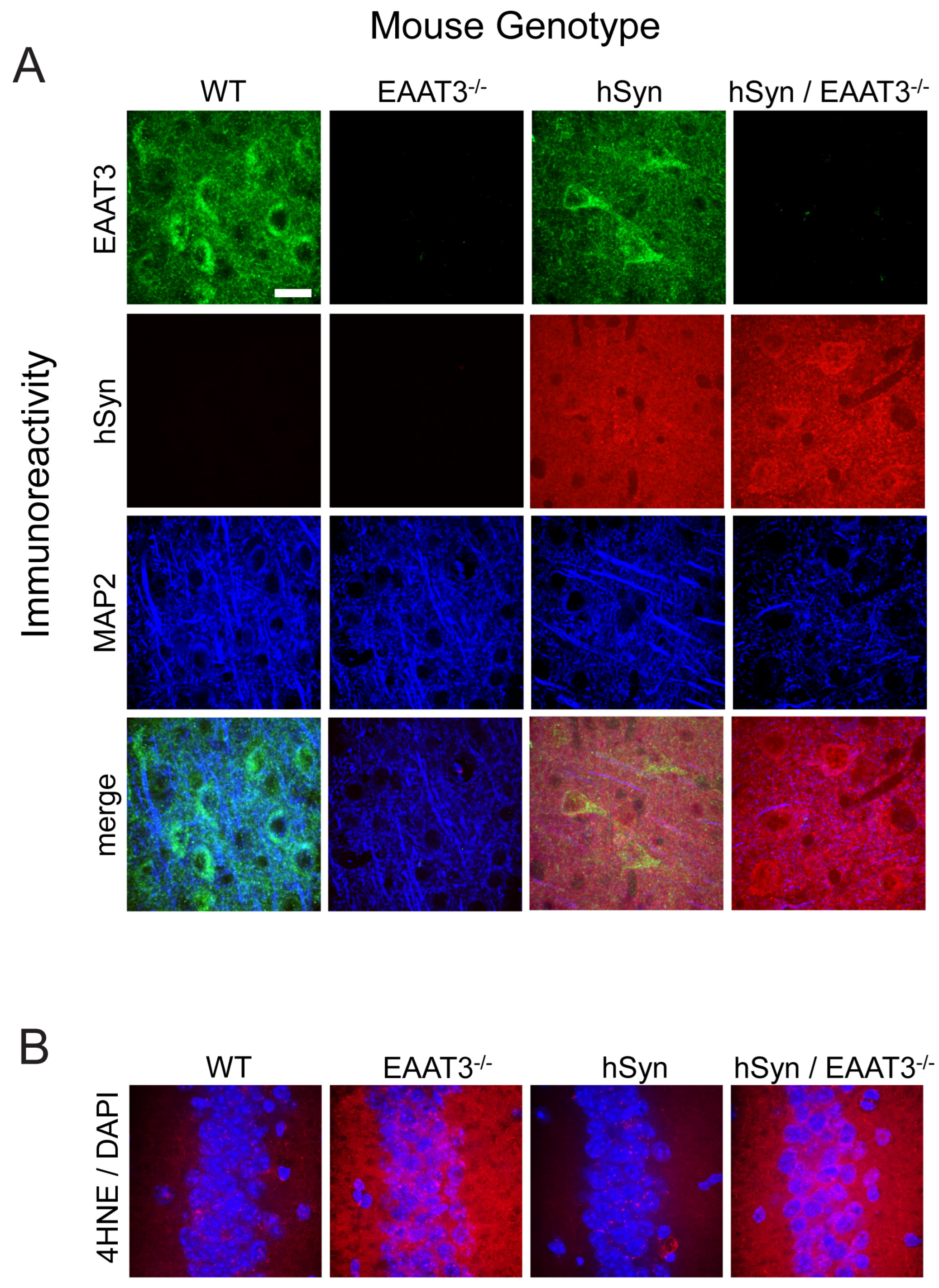
2.4. Immunohistochemistry and Image Analysis
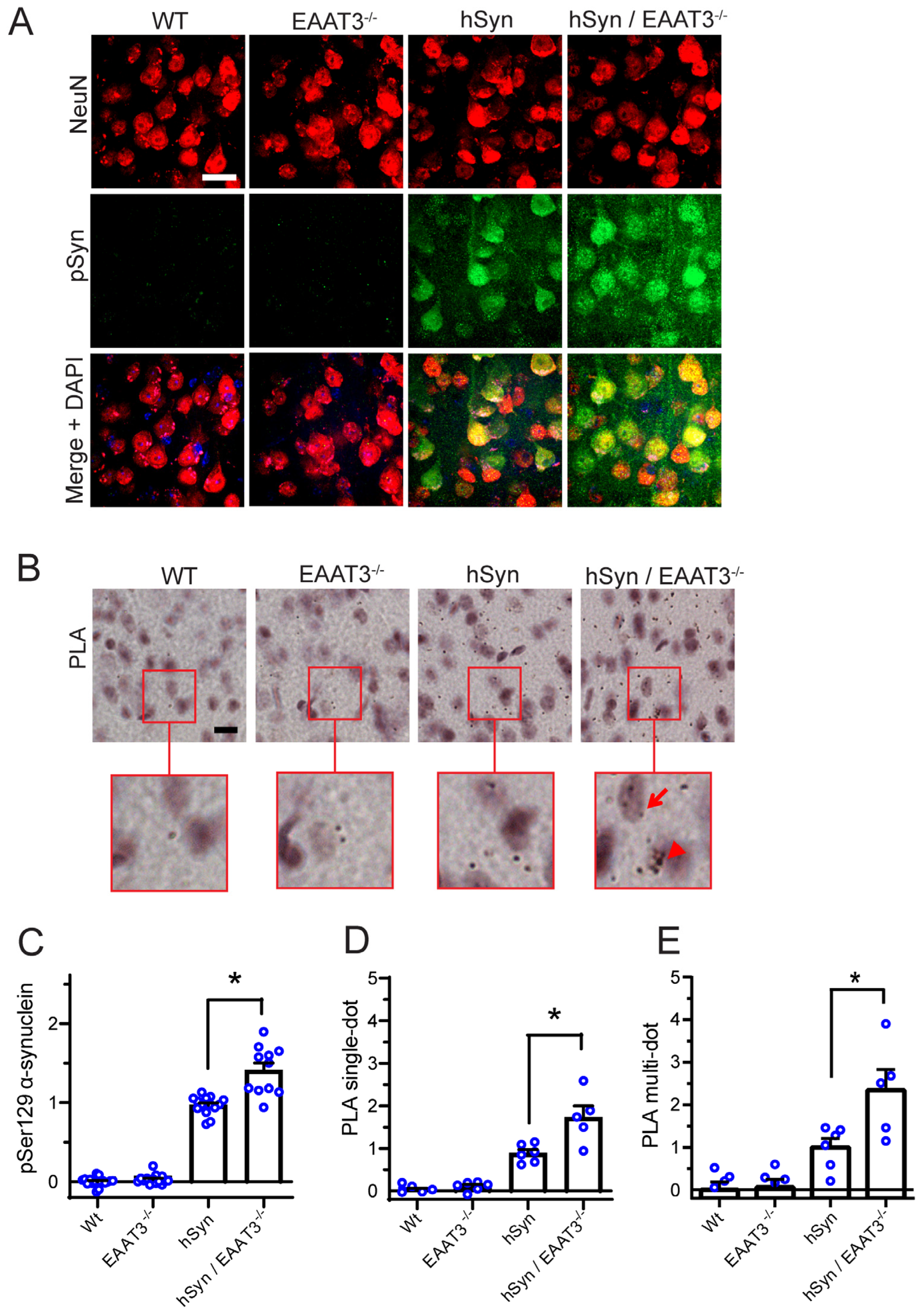
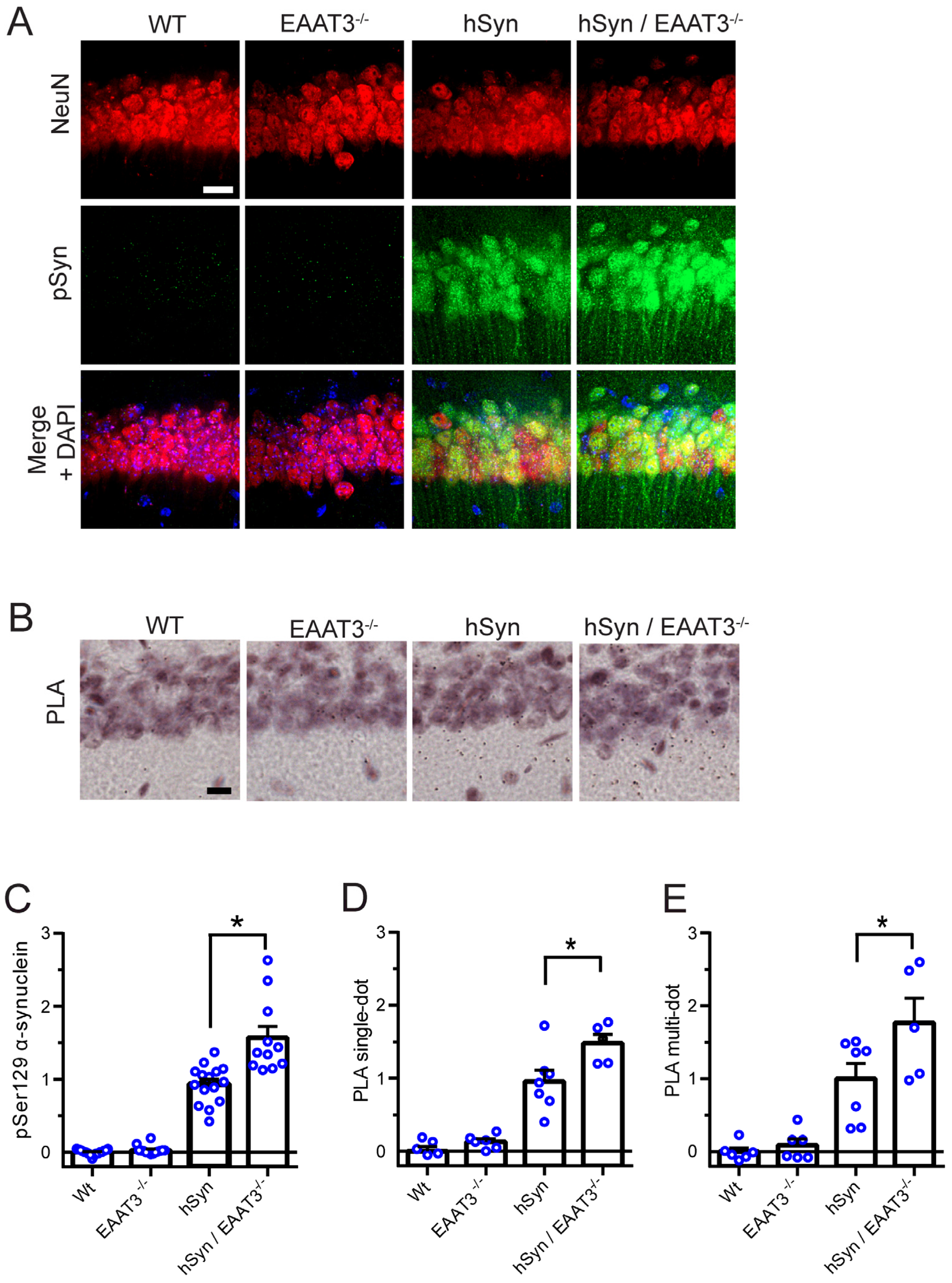
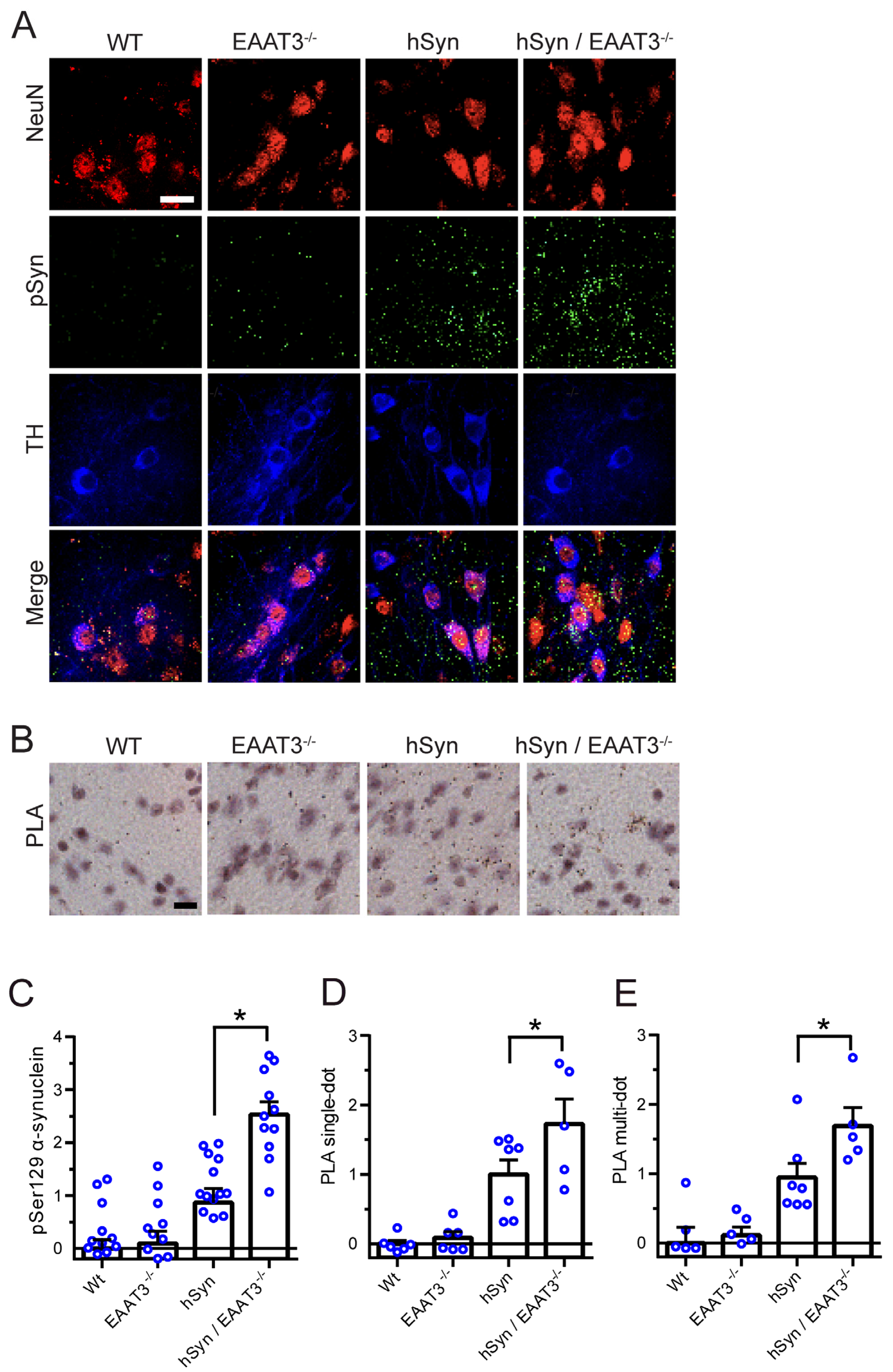
2.5. Proximity Ligation Assay
2.6. Western Blots
2.7. Statistics
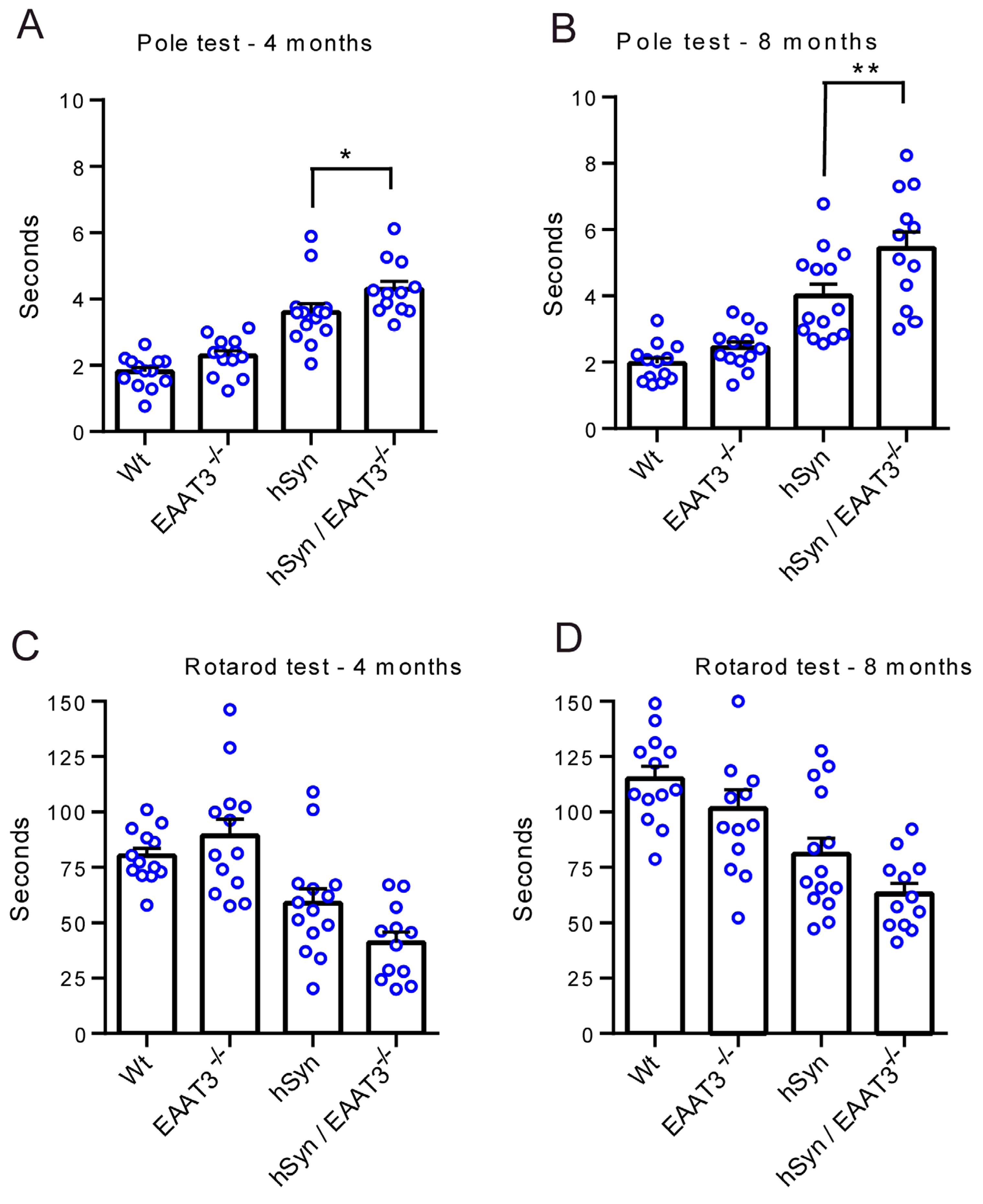
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luth, E.S.; Bartels, T.; Dettmer, U.; Kim, N.C.; Selkoe, D.J. Purification of alpha-synuclein from human brain reveals an instability of endogenous multimers as the protein approaches purity. Biochemistry 2015, 54, 279–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dettmer, U.; Newman, A.J.; von Saucken, V.E.; Bartels, T.; Selkoe, D. KTKEGV repeat motifs are key mediators of normal alpha-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 9596–9601. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, K.J.; Taylor, J.K.; Osterberg, V.R.; Churchill, M.J.; Pollock, E.; Moore, C.; Meshul, C.K.; Unni, V.K. Presynaptic alpha-synuclein aggregation in a mouse model of Parkinson’s disease. J. Neurosci. 2014, 34, 2037–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Norris, E.H.; Giasson, B.I. Role of oxidative damage in protein aggregation associated with Parkinson’s disease and related disorders. Antioxid. Redox Signal. 2005, 7, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Paxinou, E.; Chen, Q.; Weisse, M.; Giasson, B.I.; Norris, E.H.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.; Ischiropoulos, H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 2001, 21, 8053–8061. [Google Scholar] [CrossRef]
- Krishnan, S.; Chi, E.Y.; Wood, S.J.; Kendrick, B.S.; Li, C.; Garzon-Rodriguez, W.; Wypych, J.; Randolph, T.W.; Narhi, L.O.; Biere, A.L.; et al. Oxidative dimer formation is the critical rate-limiting step for Parkinson’s disease alpha-synuclein fibrillogenesis. Biochemistry 2003, 42, 829–837. [Google Scholar] [CrossRef]
- Leong, S.L.; Pham, C.L.; Galatis, D.; Fodero-Tavoletti, M.T.; Perez, K.; Hill, A.F.; Masters, C.L.; Ali, F.E.; Barnham, K.J.; Cappai, R. Formation of dopamine-mediated alpha-synuclein-soluble oligomers requires methionine oxidation. Free Radic. Biol. Med. 2009, 46, 1328–1337. [Google Scholar] [CrossRef]
- Cook, C.; Stetler, C.; Petrucelli, L. Disruption of protein quality control in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009423. [Google Scholar] [CrossRef]
- Munoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Park. Dis. 2012, 2012, 920953. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Castelao, B.; Goethals, M.; Vandekerckhove, J.; Castano, J.G. Mechanism of cleavage of alpha-synuclein by the 20S proteasome and modulation of its degradation by the RedOx state of the N-terminal methionines. Biochim. Biophys. Acta 2014, 1843, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Rodriguez-Rocha, H.; Bohovych, I.; Griggs, A.M.; Zavala-Flores, L.; Reyes-Reyes, E.M.; Seravalli, J.; Stanciu, L.A.; Lee, J.; Rochet, J.; et al. Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol. Dis. 2015, 81, 76–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice lacking ADPRT and poly (ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Alnakhala, H.; Terry-Kantor, E.; Newman, A.; Liu, L.; Imberdis, T.; Fanning, S.; Nuber, S.; Ramalingam, N.; Selkoe, D.; et al. Pathogenic Mechanisms of Cytosolic and Membrane-Enriched alpha-Synuclein Converge on Fatty Acid Homeostasis. J. Neurosci. 2022, 42, 2116–2130. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef] [Green Version]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly (ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Won, S.J.; Wang, J.; Fong, R.; Butler, N.J.M.; Moss, A.; Wong, C.; Pan, J.; Sanchez, J.; Huynh, A.; et al. Alpha-synuclein aggregates induce c-Abl activation and dopaminergic neuronal loss by a feed-forward redox stress mechanism. Prog. Neurobiol. 2021, 202, 102070. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Fauvet, B.; Gysbers, A.; Dikiy, I.; Oueslati, A.; Georgeon, S.; Lamontanara, A.J.; Bisquertt, A.; Eliezer, D.; Masliah, E.; et al. c-Abl phosphorylates alpha-synuclein and regulates its degradation: Implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Hum. Mol. Genet. 2014, 23, 2858–2879. [Google Scholar] [CrossRef]
- Watabe, M.; Aoyama, K.; Nakaki, T. Regulation of glutathione synthesis via interaction between glutamate transport-associated protein 3-18 (GTRAP3-18) and excitatory amino acid carrier-1 (EAAC1) at plasma membrane. Mol. Pharmacol. 2007, 72, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, A.R.; Szydlowska, K.; Nizinska, K.; Asaro, A.; van Vliet, E.A.; Popp, O.; Dittmar, G.; Fritsche-Guenther, R.; Kirwan, J.A.; Nykjaer, A.; et al. SorCS2 Controls Functional Expression of Amino Acid Transporter EAAT3 and Protects Neurons from Oxidative Stress and Epilepsy-Induced Pathology. Cell Rep. 2019, 26, 2792–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, R.C.; Cittolin-Santos, G.F.; Kim, J.E.; Won, S.J.; Brennan-Minnella, A.M.; Katz, M.; Glass, G.A.; Swanson, R.A. Neuronal Glutathione Content and Antioxidant Capacity can be Normalized In Situ by N-acetyl Cysteine Concentrations Attained in Human Cerebrospinal Fluid. Neurotherapeutics 2016, 13, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Rockenstein, E.; Mallory, M.; Hashimoto, M.; Song, D.; Shults, C.W.; Lang, I.; Masliah, E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res. 2002, 68, 568–578. [Google Scholar] [CrossRef]
- Chesselet, M.F.; Richter, F.; Zhu, C.; Magen, I.; Watson, M.B.; Subramaniam, S.R. A progressive mouse model of Parkinson’s disease: The Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 2012, 9, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Roshanbin, S.; Aniszewska, A.; Gumucio, A.; Masliah, E.; Erlandsson, A.; Bergstrom, J.; Ingelsson, M.; Ekmark-Lewen, S. Age-related increase of alpha-synuclein oligomers is associated with motor disturbances in L61 transgenic mice. Neurobiol. Aging 2021, 101, 207–220. [Google Scholar] [CrossRef]
- Gabrielyan, L.; Liang, H.; Minalyan, A.; Hatami, A.; John, V.; Wang, L. Behavioral Deficits and Brain alpha-Synuclein and Phosphorylated Serine-129 alpha-Synuclein in Male and Female Mice Overexpressing Human alpha-Synuclein. J. Alzheimers Dis. 2021, 79, 875–893. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. J. Physiol. 2020, 598, 3793–3801. [Google Scholar] [CrossRef]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; Swanson, R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1-/- mouse. Ann. Neurol. 2011, 69, 509–520. [Google Scholar] [CrossRef]
- Fu, Y.; Yuan, Y.; Halliday, G.; Rusznak, Z.; Watson, C.; Paxinos, G. A cytoarchitectonic and chemoarchitectonic analysis of the dopamine cell groups in the substantia nigra, ventral tegmental area, and retrorubral field in the mouse. Brain Struct. Funct. 2012, 217, 591–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 2015, 138, 1642–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeney, M.T.; Hoffman, E.K.; Farmer, K.; Bodle, C.R.; Fazzari, M.; Zharikov, A.; Castro, S.L.; Hu, X.; Mortimer, A.; Kofler, J.K.; et al. NADPH oxidase 2 activity in Parkinson’s disease. Neurobiol. Dis. 2022, 170, 105754. [Google Scholar] [CrossRef]
- Delic, V.; Chandra, S.; Abdelmotilib, H.; Maltbie, T.; Wang, S.; Kem, D.; Scott, H.J.; Underwood, R.N.; Liu, Z.; Volpicelli-Daley, L.A.; et al. Sensitivity and specificity of phospho-Ser129 alpha-synuclein monoclonal antibodies. J. Comp. Neurol. 2018, 526, 1978–1990. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Bandres-Ciga, S.; Saez-Atienzar, S.; Singleton, A.B. Genetic risk factors in Parkinson’s disease. Cell Tissue Res. 2018, 373, 9–20. [Google Scholar] [CrossRef]
- Ball, N.; Teo, W.P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Koller, W.; Vetere-Overfield, B.; Gray, C.; Alexander, C.; Chin, T.; Dolezal, J.; Hassanein, R.; Tanner, C. Environmental risk factors in Parkinson’s disease. Neurology 1990, 40, 1218–1221. [Google Scholar] [CrossRef]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox. Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.; Kholodilov, N.; Vila, M.; Trillat, A.C.; Goodchild, R.; Larsen, K.E.; Staal, R.; Tieu, K.; Schmitz, Y.; Yuan, C.A.; et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 2002, 99, 14524–14529. [Google Scholar] [CrossRef] [Green Version]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef]
- Andersen, C.; Gronnemose, A.L.; Pedersen, J.N.; Nowak, J.S.; Christiansen, G.; Nielsen, J.; Mulder, F.A.A.; Otzen, D.E.; Jorgensen, T.J.D. Lipid Peroxidation Products HNE and ONE Promote and Stabilize Alpha-Synuclein Oligomers by Chemical Modifications. Biochemistry 2021, 60, 3644–3658. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Hu, D.; Han, S.; Reaney, S.H.; Di Monte, D.A.; Fink, A.L. Effect of 4-hydroxy-2-nonenal modification on alpha-synuclein aggregation. J. Biol. Chem. 2007, 282, 5862–5870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, E.J.; Ho, D.H.; Park, E.; Jung, J.W.; Cho, K.; Hong, J.H.; Lee, H.J.; Kim, K.P.; Lee, S.J. Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of alpha-synuclein. Antioxid. Redox Signal. 2013, 18, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornai, F.; Schluter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef] [Green Version]
- Rocha, S.M.; Bantle, C.M.; Aboellail, T.; Chatterjee, D.; Smeyne, R.J.; Tjalkens, R.B. Rotenone induces regionally distinct alpha-synuclein protein aggregation and activation of glia prior to loss of dopaminergic neurons in C57Bl/6 mice. Neurobiol. Dis. 2022, 167, 105685. [Google Scholar] [CrossRef]
- Sherer, T.B.; Kim, J.H.; Betarbet, R.; Greenamyre, J.T. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 2003, 179, 9–16. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- McCormack, A.L.; Mak, S.K.; Shenasa, M.; Langston, W.J.; Forno, L.S.; Di Monte, D.A. Pathologic modifications of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated squirrel monkeys. J. Neuropathol. Exp. Neurol. 2008, 67, 793–802. [Google Scholar] [CrossRef]
- Scudamore, O.; Ciossek, T. Increased Oxidative Stress Exacerbates alpha-Synuclein Aggregation In Vivo. J. Neuropathol. Exp. Neurol. 2018, 77, 443–453. [Google Scholar] [CrossRef]
- Ishihara, Y.; Takemoto, T.; Itoh, K.; Ishida, A.; Yamazaki, T. Dual Role of Superoxide Dismutase 2 Induced in Activated Microglia: Oxidative stress tolerance and convergence of inflammatory responses. J. Biol. Chem. 2015, 290, 22805–22817. [Google Scholar] [CrossRef]
- Nieoullon, A.; Canolle, B.; Masmejean, F.; Guillet, B.; Pisano, P.; Lortet, S. The neuronal excitatory amino acid transporter EAAC1/EAAT3: Does it represent a major actor at the brain excitatory synapse? J. Neurochem. 2006, 98, 1007–1018. [Google Scholar] [CrossRef]
- Reyes, R.C.; Brennan, A.M.; Shen, Y.; Baldwin, Y.; Swanson, R.A. Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J. Neurosci. 2012, 32, 12973–12978. [Google Scholar] [CrossRef] [Green Version]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warr, O.; Takahashi, M.; Attwell, D. Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J. Physiol. 1999, 514, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Seubert, P.; Rockenstein, E.; Patrick, C.; Trejo, M.; Ubhi, K.; Ettle, B.; Ghassemiam, M.; Barbour, R.; Schenk, D.; et al. Axonopathy in an alpha-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of C-terminal-truncated alpha-synuclein. Am. J. Pathol. 2013, 182, 940–953. [Google Scholar] [CrossRef] [Green Version]
- Rockenstein, E.; Nuber, S.; Overk, C.R.; Ubhi, K.; Mante, M.; Patrick, C.; Adame, A.; Trejo-Morales, M.; Gerez, J.; Picotti, P.; et al. Accumulation of oligomer-prone alpha-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 2014, 137, 1496–1513. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Chen, W.; Junn, E.; Im, J.Y.; Grosso, H.; Sonsalla, P.K.; Feng, X.; Ray, N.; Fernandez, J.R.; Chao, Y.; et al. Enhanced phosphatase activity attenuates alpha-synucleinopathy in a mouse model. J. Neurosci. 2011, 31, 6963–6971. [Google Scholar] [CrossRef] [Green Version]
- Elbaz, A.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Peterson, B.J.; Ahlskog, J.E.; Schaid, D.J.; Rocca, W.A. Risk tables for parkinsonism and Parkinson’s disease. J. Clin. Epidemiol. 2002, 55, 25–31. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Genotype | Mean Weight | Number Allocated | Mortality |
|---|---|---|---|
| Wild-type (EAAT3+/+, XY) | 28.3 ± 0.8 | 18 | 1 |
| hSyn (EAAT3+/+, XhSynY) | 26.8 ± 0.9 | 20 | 1 |
| EAAT3−/− (EAAT3−/−, XY) | 26.7 ± 0.7 | 17 | 2 |
| hSyn/EAAT3−/− (EAAT3−/−, XhSynY) | 22.5 ± 0.8 | 16 | 4 |
| Antibody | Source | ID | Host | Dilution |
|---|---|---|---|---|
| NeuN | Millipore, Temecula, CA, USA | MAB377 | Mouse | 1:1000 |
| pSyn (S129) | Abcam, Cambridge, MA, USA | Ab51253 | Rabbit | 1:1000 |
| TH | Abcam, Cambridge, MA, USA | Ab76442 | Chicken | 1:1000 |
| Synuclein (syn211) | Abcam, Cambridge, MA, USA | Ab80627 | Mouse | 1:750 |
| EAAT3 | Cell Signaling, Boston, MA, USA | 14501s | Rabbit | 1:1000 |
| 4HNE Actin | Alpha Diagnostic, San Antonio, TX, USA Sigma-Aldrich, St. Louis, MO, USA | HNE11-S A2066 | Rabbit Rabbit | 1:1000 1:100 |
| anti-mouse IgG 594 | Thermo Fisher Scientific, Waltham, MA, USA | A21203 | Donkey | 1:1000 |
| anti-rabbit IgG 488 | Thermo Fisher Scientific, Waltham, MA, USA | A21206 | Donkey | 1:1000 |
| anti-chicken IgG 405 | Thermo Fisher Scientific, Waltham, MA, USA | A175675 | Goat | 1:1000 |
| anti-rabbit IgG, HRP-linked F(ab’)2 | Amersham Biosciences, UK | NA934v | Donkey | 1:5000 |
| anti-mouse IgG, HRP-linked F(ab’)2 | Amersham Biosciences, UK | NA931v | Sheep | 1:5000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, S.J.; Fong, R.; Butler, N.; Sanchez, J.; Zhang, Y.; Wong, C.; Tambou Nzoutchoum, O.; Huynh, A.; Pan, J.; Swanson, R.A. Neuronal Oxidative Stress Promotes α-Synuclein Aggregation In Vivo. Antioxidants 2022, 11, 2466. https://doi.org/10.3390/antiox11122466
Won SJ, Fong R, Butler N, Sanchez J, Zhang Y, Wong C, Tambou Nzoutchoum O, Huynh A, Pan J, Swanson RA. Neuronal Oxidative Stress Promotes α-Synuclein Aggregation In Vivo. Antioxidants. 2022; 11(12):2466. https://doi.org/10.3390/antiox11122466
Chicago/Turabian StyleWon, Seok Joon, Rebecca Fong, Nicholas Butler, Jennifer Sanchez, Yiguan Zhang, Candance Wong, Olive Tambou Nzoutchoum, Annie Huynh, June Pan, and Raymond A. Swanson. 2022. "Neuronal Oxidative Stress Promotes α-Synuclein Aggregation In Vivo" Antioxidants 11, no. 12: 2466. https://doi.org/10.3390/antiox11122466