
Citronellal Attenuates Oxidative Stress–Induced Mitochondrial Damage through TRPM2/NHE1 Pathway and Effectively Inhibits Endothelial Dysfunction in Type 2 Diabetes Mellitus

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals and Treatments
2.3. Cell Culture and Treatment
2.4. Fundus Photography
2.5. Vascular Morphology Observation
2.6. Test of Endothelium-Dependent Relaxation (EDR)
2.7. Detection of Diastolic Function of Mesenteric Artery
2.8. Oxidative Stress-Level Assays
2.9. Electron Microscope
2.10. Mitochondrial Membrane Potential Analysis
2.11. Immunofluorescence Analysis
2.12. Immunohistochemistry Analysis
2.13. Fluorescence Determination of Ca2+ in Cell Suspension
2.14. Western Blot
2.15. Statistics
3. Results
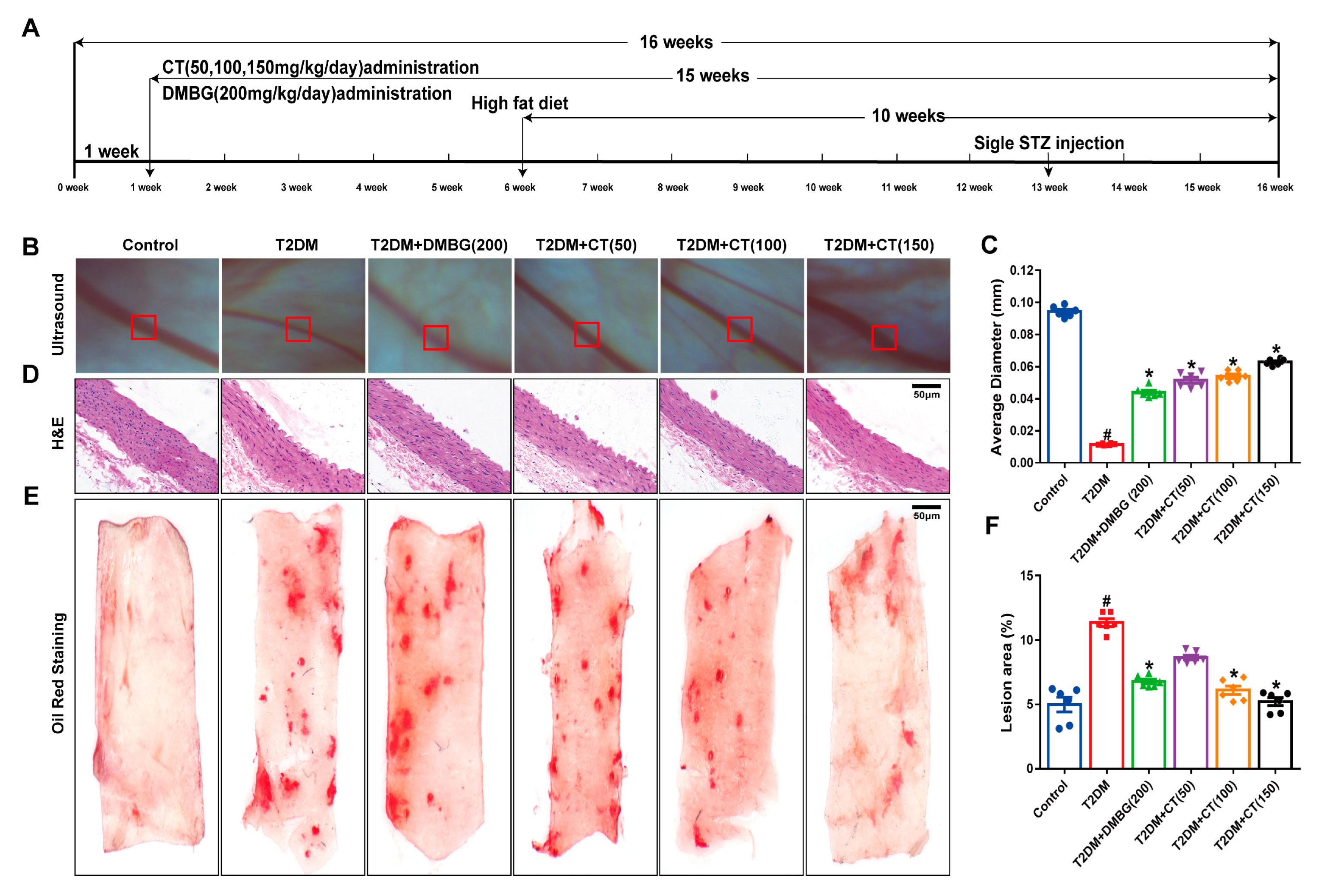
3.1. CT Improved Arterial Stenosis and Lipid Deposition in T2DM Rats
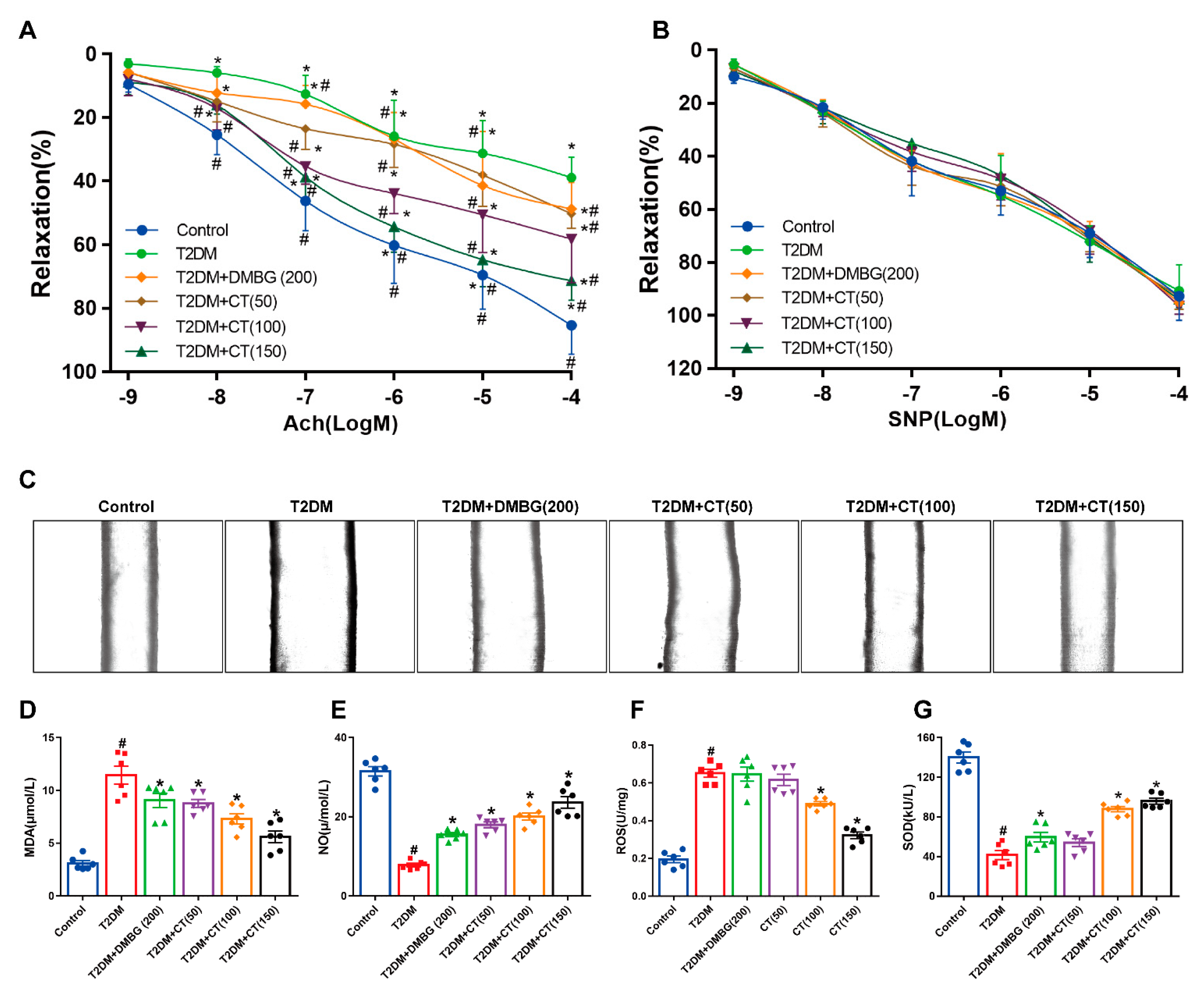
3.2. CT Alleviated the Endothelial Dysfunction and Oxidative Stress in T2DM Rats
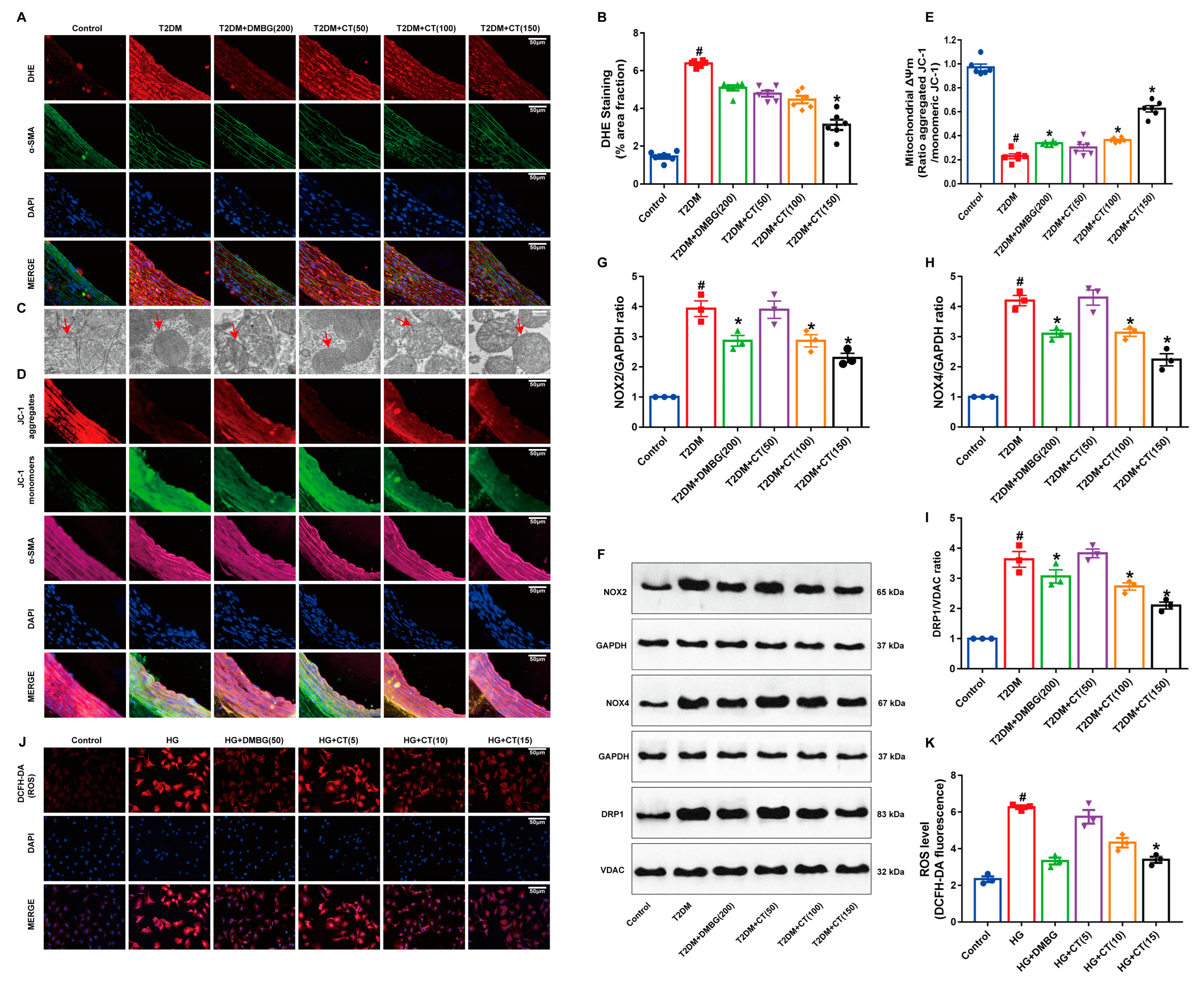
3.3. CT Attenuated Oxidative Stress-Induced Mitochondrial Damage in Carotid Arteries of T2DM Rats
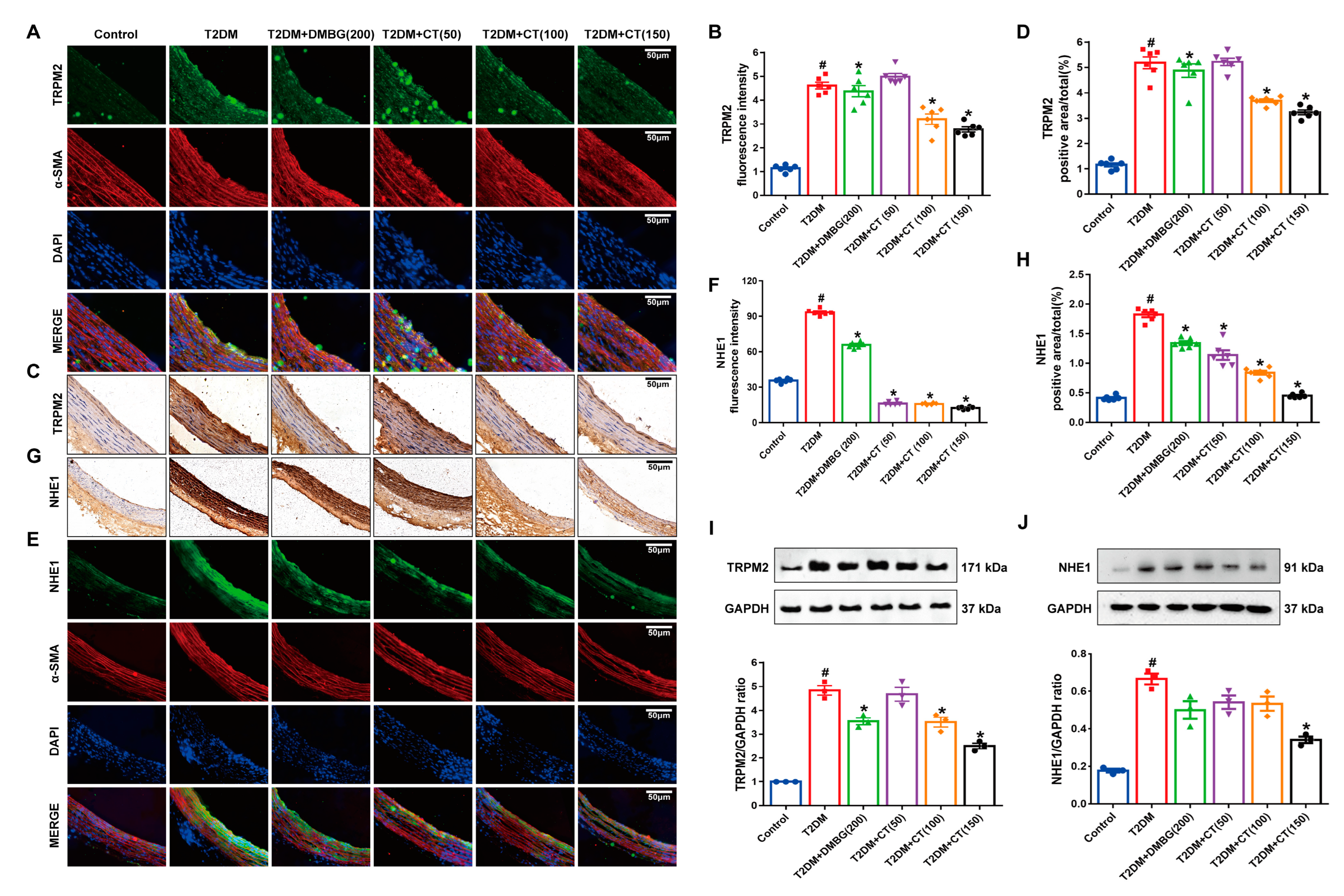
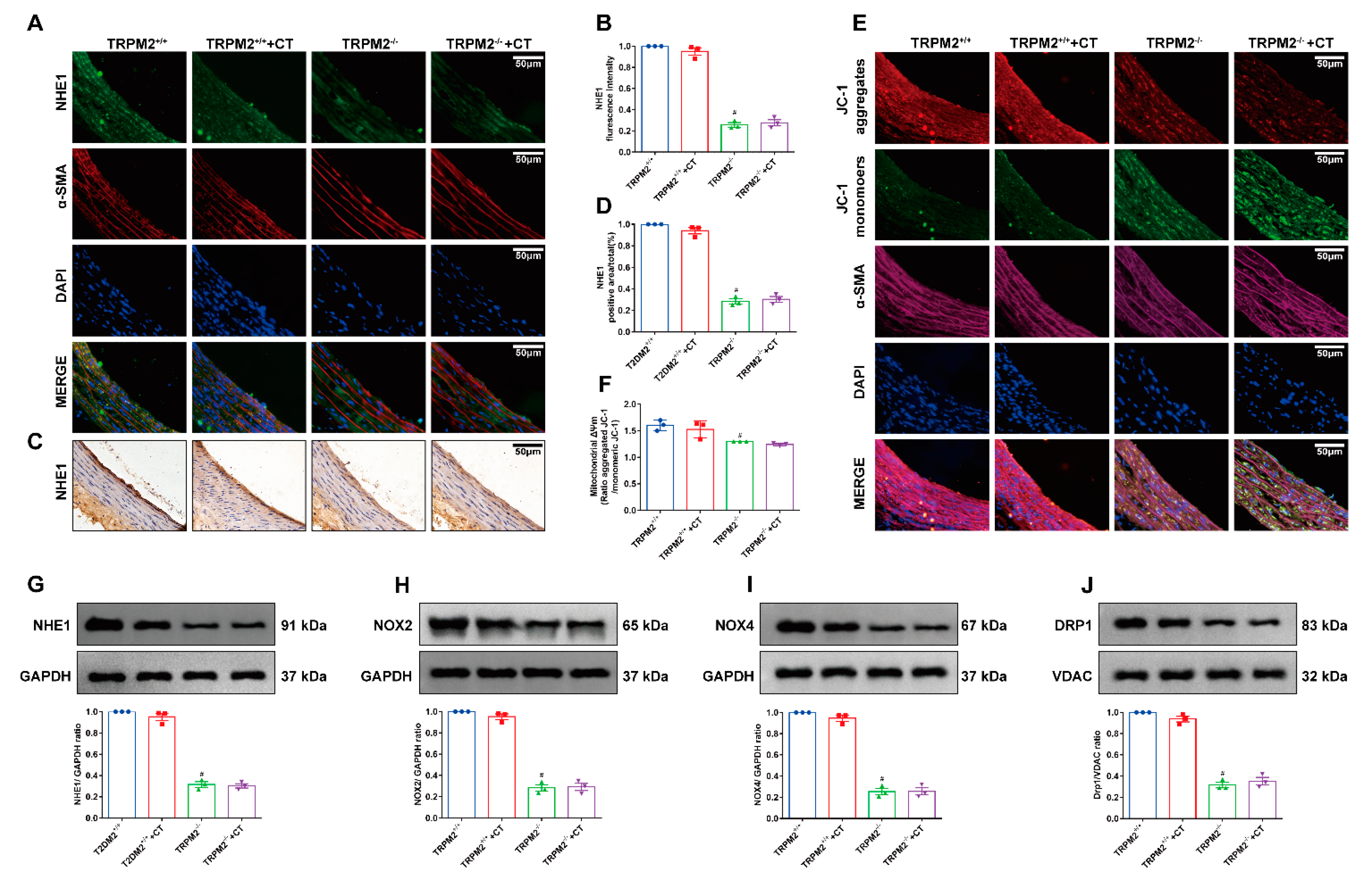
3.4. CT Alleviated the ED by Inhibiting the TRPM2/NHE1 Pathway in Carotid Arteries of T2DM Rats
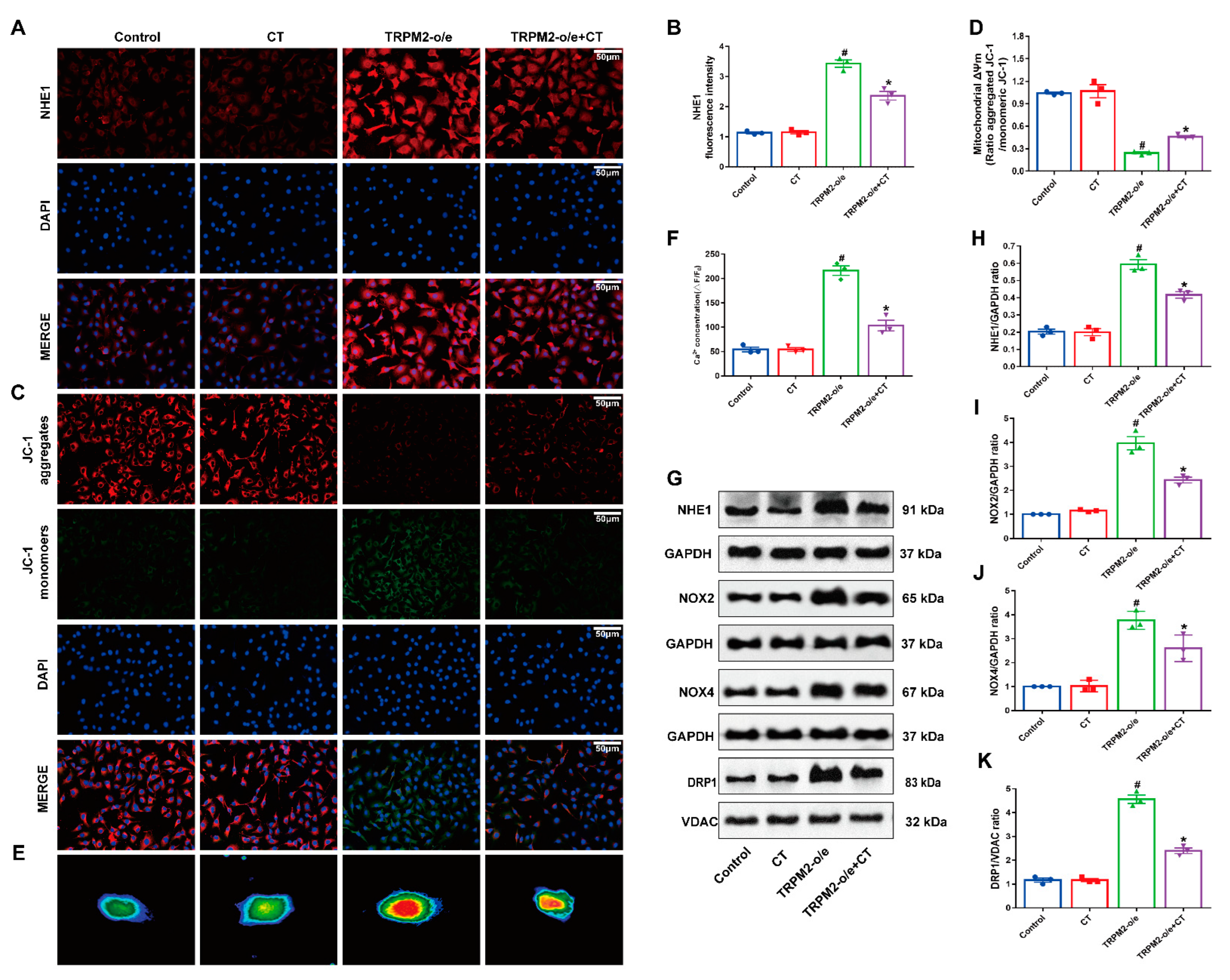
3.5. CT Inhibited the Up-Regulation of NHE1 and Mitochondrial Damage in HUVECs with Overexpression of TRPM2
3.6. CT Inhibited the Expression of NHE1 via TRPM–Dependent 2 Signaling Pathway
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, N.; Qiu, Y.; Liu, Y.; Liu, T.; Xue, X.; Song, P.; Xu, J.; Fu, Y.; Sun, R.; Yin, Y.; et al. Floralozone protects endothelial function in atherosclerosis by ameliorating NHE1. Acta Biochim. Biophys. Sin. 2021, 53, 1310–1320. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.X.; Guo, C.; Ou, W.S.; Jing, Y.; Niu, H.F.; Song, P.; Li, Q.Z.; Liu, Z.; Xu, J.; Li, P.; et al. Citronellal prevents endothelial dysfunction and atherosclerosis in rats. J. Cell Biochem. 2019, 120, 3790–3800. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, I.; Nuti, R.; Picchioni, T.; Dotta, F.; Palazzuoli, A. Molecular Dysfunction and Phenotypic Derangement in Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 3264. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial Dysfunction and Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 851941. [Google Scholar] [CrossRef] [PubMed]
- Grismaldo, A.; Sobrevia, L.; Morales, L. Role of platelet-derived growth factor c on endothelial dysfunction in cardiovascular diseases. Biochim. Et Biophys. Acta. Gen. Subj. 2022, 1866, 130188. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, N.; Chen, X.; Chen, H.; Yang, X.; Liu, C. Curcumin protects islet cells from glucolipotoxicity by inhibiting oxidative stress and NADPH oxidase activity both in vitro and in vivo. Islets 2019, 11, 152–164. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef]
- Ait-Aissa, K.; Koval, O.M.; Lindsey, N.R.; Grumbach, I.M. Mitochondrial Ca2+ Uptake Drives Endothelial Injury By Radiation Therapy. Arter. Thromb Vasc Biol 2022, 42, 1121–1136. [Google Scholar] [CrossRef]
- Ding, R.; Yin, Y.L.; Jiang, L.H. Reactive Oxygen Species-Induced TRPM2-Mediated Ca2+ Signalling in Endothelial Cells. Antioxidants 2021, 10, 718. [Google Scholar] [CrossRef]
- Hecquet, C.M.; Zhang, M.; Mittal, M.; Vogel, S.M.; Di, A.; Gao, X.; Bonini, M.G.; Malik, A.B. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ. Res. 2014, 114, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Zahra, A.; Jia, M.; Wang, Q.; Wang, Y.; Campbell, S.L.; Wu, J. Na+/H+ Exchanger 1, a Potential Therapeutic Drug Target for Cardiac Hypertrophy and Heart Failure. Pharmaceuticals 2022, 15, 875. [Google Scholar] [CrossRef] [PubMed]
- Gazmuri, R.J.; Radhakrishnan, J.; Ayoub, I.M. Sodium-Hydrogen Exchanger Isoform-1 Inhibition: A Promising Pharmacological Intervention for Resuscitation from Cardiac Arrest. Molecules 2019, 24, 1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, T.; Shi, Y.; Xiong, Z.G.; Sun, D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: New therapeutic targets for stroke? Prog. Neurobiol. 2014, 115, 189–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dini, S.; Chen, Q.; Fatemi, F.; Asri, Y. Phytochemical and biological activities of some Iranian medicinal plants. Pharm. Biol. 2022, 60, 664–689. [Google Scholar] [CrossRef]
- Paul, A.K.; Jahan, R.; Paul, A.; Mahboob, T.; Bondhon, T.A.; Jannat, K.; Hasan, A.; Nissapatorn, V.; Wilairatana, P.; de Lourdes Pereira, M.; et al. The Role of Medicinal and Aromatic Plants against Obesity and Arthritis: A Review. Nutrients 2022, 14, 985. [Google Scholar] [CrossRef]
- Saini, R.K.; Ranjit, A.; Sharma, K.; Prasad, P.; Shang, X.; Gowda, K.G.M.; Keum, Y.S. Bioactive Compounds of Citrus Fruits: A Review of Composition and Health Benefits of Carotenoids, Flavonoids, Limonoids, and Terpenes. Antioxidants 2022, 11, 239. [Google Scholar] [CrossRef]
- Yoshida, N.; Koizumi, M.; Adachi, I.; Kawakami, J. Inhibition of P-glycoprotein-mediated transport by terpenoids contained in herbal medicines and natural products. Food Chem. Toxicol. 2006, 44, 2033–2039. [Google Scholar] [CrossRef]
- Qiu, Y.; Chao, C.Y.; Jiang, L.; Zhang, J.; Niu, Q.Q.; Guo, Y.Q.; Song, Y.T.; Li, P.; Zhu, M.L.; Yin, Y.L. Citronellal alleviate macro- and micro-vascular damage in high fat diet/streptozotocin-Induced diabetic rats via a S1P/S1P1 dependent signaling pathway. Eur. J. Pharm. 2022, 920, 174796. [Google Scholar] [CrossRef]
- Liao, L.; Gong, L.; Zhou, M.; Xue, X.; Li, Y.; Peng, C. Leonurine Ameliorates Oxidative Stress and Insufficient Angiogenesis by Regulating the PI3K/Akt-eNOS Signaling Pathway in H2O2-Induced HUVECs. Oxidative Med. Cell. Longev. 2021, 2021, 9919466. [Google Scholar] [CrossRef]
- Li, P.; Yin, Y.L.; Guo, T.; Sun, X.Y.; Ma, H.; Zhu, M.L.; Zhao, F.R.; Xu, P.; Chen, Y.; Wan, G.R.; et al. Inhibition of Aberrant MicroRNA-133a Expression in Endothelial Cells by Statin Prevents Endothelial Dysfunction by Targeting GTP Cyclohydrolase 1 in Vivo. Circulation 2016, 134, 1752–1765. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, T.; Peng, Q.S.; Yue, S.W.; Wang, S.X. Berberine via suppression of transient receptor potential vanilloid 4 channel improves vascular stiffness in mice. J. Cell Mol. Med. 2015, 19, 2607–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.; Hu, J.; Zhao, J.; Yang, J.; Huang, N.; Song, P.; Xu, J.; Zhang, M.; Li, P.; Yin, Y. Experimental study of blood pressure and its impact on spontaneous hypertension in rats with Xin Mai Jia. Biomed. Pharmacother. 2019, 112, 108689. [Google Scholar] [CrossRef] [PubMed]
- Walker, O.S.; Ragos, R.; Gurm, H.; Lapierre, M.; May, L.L.; Raha, S. Delta-9-tetrahydrocannabinol disrupts mitochondrial function and attenuates syncytialization in human placental BeWo cells. Physiol. Rep. 2020, 8, e14476. [Google Scholar] [CrossRef]
- Ye, J.; Yin, Y.; Yin, Y.; Zhang, H.; Wan, H.; Wang, L.; Zuo, Y.; Gao, D.; Li, M.; Li, J.; et al. Tau-induced upregulation of C/EBPβ-TRPC1-SOCE signaling aggravates tauopathies: A vicious cycle in Alzheimer neurodegeneration. Aging Cell 2020, 19, e13209. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, C.; Zhang, M.; Liang, B.; Zhu, H.; Lee, J.; Viollet, B.; Xia, L.; Zhang, Y.; Zou, M.H. Activation of AMP-activated protein kinase α2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat. Med. 2012, 18, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yu, J.; Ke, F.; Lan, M.; Li, D.; Tan, K.; Ling, J.; Wang, Y.; Wu, K.; Li, D. Curcumin Alleviates Diabetic Retinopathy in Experimental Diabetic Rats. Ophthalmic Res. 2018, 60, 43–54. [Google Scholar] [CrossRef]
- Cao, J.; Zhao, C.; Gong, L.; Cheng, X.; Yang, J.; Zhu, M.; Lv, X. MiR-181 Enhances Proliferative and Migratory Potentials of Retinal Endothelial Cells in Diabetic Retinopathy by Targeting KLF6. Curr. Eye Res. 2022, 47, 882–888. [Google Scholar] [CrossRef]
- Mussbacher, M.; Schossleitner, K.; Kral-Pointner, J.B.; Salzmann, M.; Schrammel, A.; Schmid, J.A. More than Just a Monolayer: The Multifaceted Role of Endothelial Cells in the Pathophysiology of Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 483–492. [Google Scholar] [CrossRef]
- Gao, L.; Mann, G.E. Vascular NAD(P)H oxidase activation in diabetes: A double-edged sword in redox signalling. Cardiovasc Res. 2009, 82, 9–20. [Google Scholar] [CrossRef]
- Shi, Y.; Vanhoutte, P.M. Reactive oxygen-derived free radicals are key to the endothelial dysfunction of diabetes. J. Diabetes 2009, 1, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.H.; Ke, Z.Q.; Wu, S.; Yang, X.S.; Chen, Q.J.; Huang, S.T.; Liu, C. Evaluation of the antioxidant and endothelial protective effects of Lysimachia christinae Hance (Jin Qian Cao) extract fractions. BMC Complement. Altern. Med. 2018, 18, 128. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, J.; Song, P.; Viollet, B.; Zou, M.H. In vivo activation of AMP-activated protein kinase attenuates diabetes-enhanced degradation of GTP cyclohydrolase I. Diabetes 2009, 58, 1893–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Wei, M.; Ni, J.; Sun, F.; Sun, L.; Wang, J.; Yu, T.; Wang, K.; Lv, P.; Wang, Y.; et al. Tianzhi granule improves cognition and BPSD of vascular dementia: A randomized controlled trial. J. Transl. Med. 2020, 18, 76. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Thapa, S.; Mahawar, H.; Kumar, D.; Geat, N.; Singh, S.K. Prospecting potential of endophytes for modulation of biosynthesis of therapeutic bioactive secondary metabolites and plant growth promotion of medicinal and aromatic plants. Antonie Van Leeuwenhoek 2022, 115, 699–730. [Google Scholar] [CrossRef]
- Madonna, R.; De Caterina, R. Aquaporin-1 and sodium-hydrogen exchangers as pharmacological targets in diabetic atherosclerosis. Curr. Drug Targets 2015, 16, 361–365. [Google Scholar] [CrossRef]
- Sun, L.; Yau, H.Y.; Wong, W.Y.; Li, R.A.; Huang, Y.; Yao, X. Role of TRPM2 in H(2)O(2)-induced cell apoptosis in endothelial cells. PLoS ONE 2012, 7, e43186. [Google Scholar] [CrossRef]
- Osmanlıoğlu, H.; Yıldırım, M.K.; Akyuva, Y.; Yıldızhan, K.; Nazıroğlu, M. Morphine Induces Apoptosis, Inflammation, and Mitochondrial Oxidative Stress via Activation of TRPM2 Channel and Nitric Oxide Signaling Pathways in the Hippocampus. Mol. Neurobiol. 2020, 57, 3376–3389. [Google Scholar] [CrossRef]
- Zhang, W.; Hirschler-Laszkiewicz, I.; Tong, Q.; Conrad, K.; Sun, S.C.; Penn, L.; Barber, D.L.; Stahl, R.; Carey, D.J.; Cheung, J.Y.; et al. TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage. Am. J. Physiol. Cell Physiol. 2006, 290, C1146–C1159. [Google Scholar] [CrossRef]
- Zhu, M.L.; Wang, G.; Wang, H.; Guo, Y.M.; Song, P.; Xu, J.; Li, P.; Wang, S.; Yang, L. Amorphous nano-selenium quantum dots improve endothelial dysfunction in rats and prevent atherosclerosis in mice through Na+/H+ exchanger 1 inhibition. Vasc. Pharm. 2019, 115, 26–32. [Google Scholar] [CrossRef]
- Cui, G.M.; Zhao, Y.X.; Zhang, N.N.; Liu, Z.S.; Sun, W.C.; Peng, Q.S. Amiloride attenuates lipopolysaccharide-accelerated atherosclerosis via inhibition of NHE1-dependent endothelial cell apoptosis. Acta Pharm. Sin. 2013, 34, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.F.; Chen, S.; Feng, J.D.; Zhang, M.Y.; Liu, X.X. Probucol via inhibition of NHE1 attenuates LPS-accelerated atherosclerosis and promotes plaque stability in vivo. Exp. Mol. Pathol. 2014, 96, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Benincasa, G.; Coscioni, E.; Napoli, C. Cardiovascular risk factors and molecular routes underlying endothelial dysfunction: Novel opportunities for primary prevention. Biochem. Pharmacol. 2022, 202, 115108. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, P.; Zhao, Z.H.; Zhang, Y.; Ma, Z.M.; Wang, S.X. Vitamin B6 Prevents Endothelial Dysfunction, Insulin Resistance, and Hepatic Lipid Accumulation in Apoe (-/-) Mice Fed with High-Fat Diet. J. Diabetes Res. 2016, 2016, 1748065. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting NADPH oxidases in vascular pharmacology. Vasc. Pharmacol. 2012, 56, 216–231. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Li, S.; Yu, K. DJ-1 alleviates high glucose-induced endothelial cells injury via PI3K/Akt-eNOS signaling pathway. Mol. Med. Rep. 2018, 17, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. TEM 2014, 25, 452–463. [Google Scholar] [CrossRef]
- de Marañón, A.M.; Canet, F.; Abad-Jiménez, Z.; Jover, A.; Morillas, C.; Rocha, M.; Victor, V.M. Does Metformin Modulate Mitochondrial Dynamics and Function in Type 2 Diabetic Patients? Antioxid. Redox Signal. 2021, 35, 377–385. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Cao, A.; Li, C.; Wang, L.; Wu, Q.; Li, X.; Lv, X.; Zhu, J.; Chun, H.; et al. Crocin Improves Endothelial Mitochondrial Dysfunction via GPx1/ROS/KCa3.1 Signal Axis in Diabetes. Front. Cell Dev. Biol. 2021, 9, 651434. [Google Scholar] [CrossRef]
- Rahadian, A.; Fukuda, D.; Salim, H.M.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; Sata, M. Canagliflozin Prevents Diabetes-Induced Vascular Dysfunction in ApoE-Deficient Mice. J. Atheroscler. Thromb. 2020, 27, 1141–1151. [Google Scholar] [CrossRef]
- Zhang, Y.; Mao, X.D.; Cao, A.L.; Chu, S.; Li, Z.J.; Wang, Y.M.; Peng, W.; Wang, L.; Wang, H. Astragaloside IV prevents endothelial dysfunction by improving oxidative stress in streptozotocin-induced diabetic mouse aortas. Exp. Med. 2021, 22, 1197. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Han, F.; Xuan, L.; Lv, Y.; Gong, L.; Yan, Y.; Wan, Z.; Guo, L.; Liu, H.; Xu, B.; et al. Clusterin ameliorates endothelial dysfunction in diabetes by suppressing mitochondrial fragmentation. Free Radic. Biol. Med. 2019, 145, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, W.; Zabrzyński, J.; Gagat, M.; Grzanka, A. The Role of TRPM2 in Endothelial Function and Dysfunction. Int. J. Mol. Sci. 2021, 22, 7635. [Google Scholar] [CrossRef]
- Jiang, L.H.; Yang, W.; Zou, J.; Beech, D.J. TRPM2 channel properties, functions and therapeutic potentials. Expert Opin. Ther. Targets 2010, 14, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Malko, P.; Jiang, L.H. TRPM2 channel-mediated cell death: An important mechanism linking oxidative stress-inducing pathological factors to associated pathological conditions. Redox Biol. 2020, 37, 101755. [Google Scholar] [CrossRef] [PubMed]
- Abuarab, N.; Munsey, T.S.; Jiang, L.H.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017, 10, eaal4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, M.A.; Beltrán, A.R.; Araya, J.E.; Cornejo, M.; Toledo, F.; Fuentes, G.; Sobrevia, L. Involvement of Intracellular pH in Vascular Insulin Resistance. Curr. Vasc. Pharm. 2019, 17, 440–446. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Y.-L.; Wang, H.-H.; Gui, Z.-C.; Mi, S.; Guo, S.; Wang, Y.; Wang, Q.-Q.; Yue, R.-Z.; Lin, L.-B.; Fan, J.-X.; et al. Citronellal Attenuates Oxidative Stress–Induced Mitochondrial Damage through TRPM2/NHE1 Pathway and Effectively Inhibits Endothelial Dysfunction in Type 2 Diabetes Mellitus. Antioxidants 2022, 11, 2241. https://doi.org/10.3390/antiox11112241
Yin Y-L, Wang H-H, Gui Z-C, Mi S, Guo S, Wang Y, Wang Q-Q, Yue R-Z, Lin L-B, Fan J-X, et al. Citronellal Attenuates Oxidative Stress–Induced Mitochondrial Damage through TRPM2/NHE1 Pathway and Effectively Inhibits Endothelial Dysfunction in Type 2 Diabetes Mellitus. Antioxidants. 2022; 11(11):2241. https://doi.org/10.3390/antiox11112241
Chicago/Turabian StyleYin, Ya-Ling, Huan-Huan Wang, Zi-Chen Gui, Shan Mi, Shuang Guo, Yue Wang, Qian-Qian Wang, Rui-Zhu Yue, Lai-Biao Lin, Jia-Xin Fan, and et al. 2022. "Citronellal Attenuates Oxidative Stress–Induced Mitochondrial Damage through TRPM2/NHE1 Pathway and Effectively Inhibits Endothelial Dysfunction in Type 2 Diabetes Mellitus" Antioxidants 11, no. 11: 2241. https://doi.org/10.3390/antiox11112241