
Ageing and Low-Level Chronic Inflammation: The Role of the Biological Clock
 ,
,  ,
,  , , and
, , and
Abstract
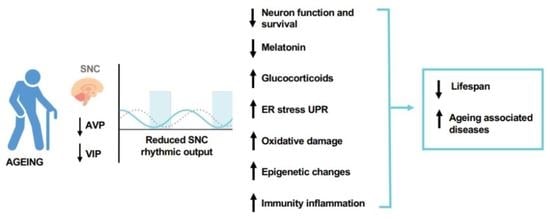
:
{kind=link}
{kind=link}
1. Introduction
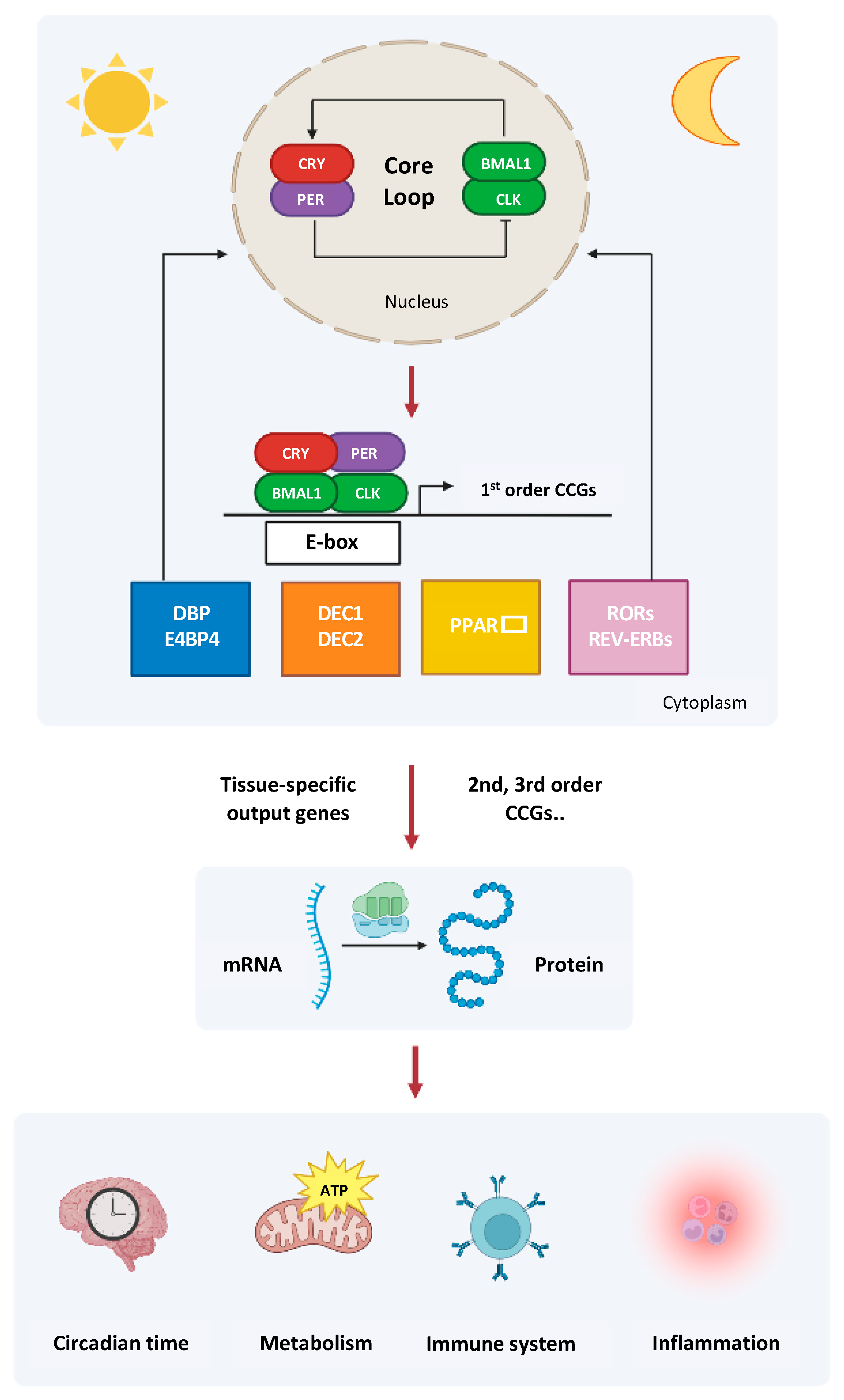
2. The Circadian Clock Circuitry
3. The Role of Biological Clock Derangement in the Ageing Process
4. The Biological Clock and the Innate Immune System
5. The Biological Clock and the Adaptive Immune System
6. Ageing, Inflammation, and the Immune Response
7. Endoplasmic Reticulum Stress and UPR during Ageing
8. UPR Mediators and Inflammation
9. Ageing and NLRP3 Inflammasome Formation
10. The Circadian Epigenome and the Epigenetic Clock in Ageing
11. “Circadian Medicine”: A New Anti-Ageing Therapy
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 12, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 2019, 9, 790. [Google Scholar] [CrossRef]
- Hergenhan, S.; Holtkamp, S.; Scheiermann, C. Molecular Interactions Between Components of the Circadian Clock and the Immune System. J. Mol. Biol. 2020, 432, 3700–3713. [Google Scholar] [CrossRef]
- Logan, R.W.; Sarkar, D.K. Circadian nature of immune function. Mol. Cell Endocrinol. 2012, 349, 82–90. [Google Scholar] [CrossRef]
- Man, K.; Loudon, A.; Chawla, A. Immunity around the clock. Science 2016, 354, 999–1003. [Google Scholar] [CrossRef] [Green Version]
- Scheiermann, C.; Gibbs, J.; Ince, L.; Loudon, A. Clocking into immunity. Nat. Rev. Immunol. 2018, 18, 423–437. [Google Scholar] [CrossRef]
- Mazzoccoli, G.; Sothern, R.B.; De Cata, A.; Giuliani, F.; Fontana, A.; Copetti, M.; Pellegrini, F.; Tarquini, R. A timetable of 24-hour patterns for human lymphocyte subpopulations. J. Biol. Regul. Homeost. Agents 2011, 25, 387–395. [Google Scholar]
- Méndez-Ferrer, S.; Chow, A.; Merad, M.; Frenette, P.S. Circadian rhythms influence hematopoietic stem cells. Curr. Opin. Hematol. 2009, 16, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, S.; Lange, T.; Born, J. Selective mobilization of cytotoxic leukocytes by epinephrine. J. Immunol. 2010, 184, 503–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebisawa, T.; Numazawa, K.; Shimada, H.; Izutsu, H.; Sasaki, T.; Kato, N.; Tokunaga, K.; Mori, A.; Honma, K.; Honma, S.; et al. Self-sustained circadian rhythm in cultured human mononuclear cells isolated from peripheral blood. Neurosci. Res. 2010, 66, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; De Cata, A.; Greco, A.; Carughi, S.; Giuliani, F.; Tarquini, R. Circadian rhythmicity of lymphocyte subpopulations and relationship with neuro-endocrine system. J. Biol. Regul. Homeost. Agents 2010, 24, 341–350. [Google Scholar]
- Bollinger, T.; Leutz, A.; Leliavski, A.; Skrum, L.; Kovac, J.; Bonacina, L.; Benedict, C.; Lange, T.; Westermann, J.; Oster, H.; et al. Circadian clocks in mouse and human CD4+ T cells. PLoS ONE 2011, 6, e29801. [Google Scholar] [CrossRef]
- Keller, M.; Mazuch, J.; Abraham, U.; Eom, G.D.; Herzog, E.D.; Volk, H.D.; Kramer, A.; Maier, B. A circadian clock in macrophages controls inflammatory immune responses. Proc. Natl. Acad. Sci. USA 2009, 106, 21407–21412. [Google Scholar] [CrossRef] [Green Version]
- Mazzoccoli, G.; Sothern, R.B.; Greco, G.; Pazienza, V.; Vinciguerra, M.; Liu, S.; Cai, Y. Time-Related Dynamics of Variation in Core Clock Gene Expression Levels in Tissues Relevant to the Immune System. Int. J. Immunopathol. Pharmacol. 2011, 24, 869–879. [Google Scholar] [CrossRef]
- Pourcet, B.; Duez, H. Circadian Control of Inflammasome Pathways: Implications for Circadian Medicine. Front. Immunol. 2020, 11, 1630. [Google Scholar] [CrossRef]
- Albrecht, U. Timing to perfection: The biology of central and peripheral circadian clocks. Neuron 2012, 74, 246–260. [Google Scholar] [CrossRef] [Green Version]
- Koike, N.; Yoo, S.H.; Huang, H.C.; Kumar, V.; Lee, C.; Kim, T.K.; Takahashi, J.S. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 2012, 338, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, K.; Shirai, H.; Ishida, N. CLOCK is involved in the circadian transactivation of peroxisome-proliferator-activated receptor alpha (PPARalpha) in mice. Biochem. J. 2005, 386, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Canaple, L.; Rambaud, J.; Dkhissi-Benyahya, O.; Rayet, B.; Tan, N.S.; Michalik, L.; Delaunay, F.; Wahli, W.; Laudet, V. Reciprocal regulation of brain and muscle Arnt-like protein 1 and peroxisome proliferator-activated receptor alpha defines a novel positive feedback loop in the rodent liver circadian clock. Mol. Endocrinol. 2006, 20, 1715–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozek, K.; Relógio, A.; Kielbasa, S.M.; Heine, M.; Dame, C.; Kramer, A.; Herzel, H. Regulation of clock-controlled genes in mammals. PLoS ONE 2009, 4, e4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Male, V.; Nisoli, I.; Gascoyne, D.M.; Brady, H.J. E4BP4: An unexpected player in the immune response. Trends Immunol. 2012, 33, 98–102. [Google Scholar] [CrossRef]
- Nakamura, T.J.; Takasu, N.N.; Nakamura, W. The suprachiasmatic nucleus: Age-related decline in biological rhythms. J. Physiol. Sci. 2016, 66, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Melhuish Beaupre, L.M.; Brown, G.M.; Gonçalves, V.F.; Kennedy, J.L. Melatonin’s neuroprotective role in mitochondria and its potential as a biomarker in aging, cognition and psychiatric disorders. Transl. Psychiatry 2021, 11, 339. [Google Scholar] [CrossRef] [PubMed]
- Hood, S.; Amir, S. The aging clock: Circadian rhythms and later life. J. Clin. Investig. 2017, 127, 437–446. [Google Scholar] [CrossRef]
- Chou, L.Y.; Ho, C.T.; Hung, S.C. Paracrine Senescence of Mesenchymal Stromal Cells Involves Inflammatory Cytokines and the NF-κB Pathway. Cells 2022, 11, 3324. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N. The Senescence Markers p16INK4A, p14ARF/p19ARF, and p21 in Organ Development and Homeostasis. Cells 2022, 11, 1966. [Google Scholar] [CrossRef]
- Kowalska, E.; Ripperger, J.A.; Hoegger, D.C.; Bruegger, P.; Buch, T.; Birchler, T.; Mueller, A.; Albrecht, U.; Contaldo, C.; Brown, S.A. NONO couples the circadian clock to the cell cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 1592–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashikawa, K.I.; Katamune, C.; Kusunose, N.; Matsunaga, N.; Koyanagi, S.; Ohdo, S. Dysfunction of the circadian transcriptional factor CLOCK in mice resists chemical carcinogen-induced tumorigenesis. Sci. Rep. 2017, 7, 9995. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Khapre, R.V.; Kondratova, A.A.; Patel, S.; Dubrovsky, Y.; Wrobel, M.; Antoch, M.P.; Kondratov, R.V. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging 2014, 6, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.I.; Guarente, L. It takes two to tango: NAD + and sirtuins in aging / longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [Green Version]
- Sadria, M.; Layton, A.T. Aging affects circadian clock and metabolism and modulates timing of medication. iScience 2021, 24, 102245. [Google Scholar] [CrossRef]
- Mezhnina, V.; Ebeigbe, O.P.; Poe, A.; Kondratov, R.V. Circadian Control of Mitochondria in Reactive Oxygen Species Homeostasis. Antioxid. Redox Signal. 2022, 37, 647–663. [Google Scholar] [CrossRef]
- Ferlazzo, N.; Andolina, G.; Cannata, A.; Costanzo, M.G.; Rizzo, V.; Currò, M.; Ientile, R.; Caccamo, D. Is Melatonin the Cornucopia of the 21st Century? Antioxidants 2020, 9, 1088. [Google Scholar] [CrossRef]
- Musiek, E.S.; Lim, M.M.; Yang, G.; Bauer, A.Q.; Qi, L.; Lee, Y.; Roh, J.H.; Ortiz-Gonzalez, X.; Dearborn, J.T.; Culver, J.P.; et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Investig. 2013, 123, 5389–5400. [Google Scholar] [CrossRef] [Green Version]
- Welz, P.S.; Benitah, S.A. Molecular Connections Between Circadian Clocks and Aging. J. Mol. Biol. 2020, 432, 3661–3679. [Google Scholar] [CrossRef]
- Dubrovsky, Y.V.; Samsa, W.E.; Kondratov, R.V. Deficiency of circadian protein CLOCK reduces lifespan and increases age-related cataract development in mice. Aging 2010, 2, 936–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcello, M.; White, M.R. Spatial and temporal information coding and noise in the NF-κB system. Biochem. Soc. Trans. 2010, 38, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Narasimamurthy, R.; Hatori, M.; Nayak, S.K.; Liu, F.; Panda, S.; Verma, I.M. Circadian clock protein cryptochrome regulates the expression of proinflammatory cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 12662–12667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Malkani, G.; Shi, X.; Meyer, M.; Cunningham-Runddles, S.; Ma, X.; Sun, Z.S. The circadian clock Period 2 gene regulates gamma interferon production of NK cells in host response to lipopolysaccharide-induced endotoxic shock. Infect. Immun. 2006, 74, 4750–4756. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.E.; Blaikley, J.; Beesley, S.; Matthews, L.; Simpson, K.D.; Boyce, S.H.; Farrow, S.N.; Else, K.J.; Singh, D.; Ray, D.W.; et al. The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Cavadini, G.; Petrzilka, S.; Kohler, P.; Jud, C.; Tobler, I.; Birchler, T.; Fontana, A. TNF-alpha suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 12843–12848. [Google Scholar] [CrossRef] [Green Version]
- Petrzilka, S.; Taraborrelli, C.; Cavadini, G.; Fontana, A.; Birchler, T. Clock gene modulation by TNF-alpha depends on calcium and p38 MAP kinase signaling. J. Biol. Rhythm. 2009, 24, 283–294. [Google Scholar] [CrossRef]
- Venteclef, N.; Jakobsson, T.; Steffensen, K.R.; Treuter, E. Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends Endocrinol. Metab. 2011, 22, 333–343. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 2010, 10, 365–376. [Google Scholar] [CrossRef]
- Charoensuksai, P.; Xu, W. PPARs in Rhythmic Metabolic Regulation and Implications in Health and Disease. PPAR Res. 2010, 2010, pii243643. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A. Control of macrophage activation and function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Migita, H.; Morser, J.; Kawai, K. Rev-erb alpha upregulates NF-kappaB-responsive genes in vascular smooth muscle cells. FEBS Lett. 2004, 561, 69–74. [Google Scholar] [CrossRef]
- Fontaine, C.; Rigamonti, E.; Pourcet, B.; Duez, H.; Duhem, C.; Fruchart, J.C.; Chinetti-Gbaguidi, G.; Staels, B. The nuclear receptor Rev-erbalpha is a liver X receptor (LXR) target gene driving a negative feedback loop on select LXR-induced pathways in human macrophages. Mol. Endocrinol. 2008, 22, 1797–1811. [Google Scholar] [CrossRef] [Green Version]
- Silver, A.C.; Arjona, A.; Walker, W.E.; Fikrig, E. The circadian clock controls toll-like receptor 9-mediated innate and adaptive immunity. Immunity 2012, 36, 251–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, K.J.; Gibbs, J.E. Adaptive immunity, chronic inflammation and the clock. Semin. Immunopathol. 2022, 44, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Spengler, M.L.; Kuropatwinski, K.K.; Comas, M.; Gasparian, A.V.; Fedtsova, N.; Gleiberman, A.S.; Gitlin, I.I.; Artemicheva, N.M.; Deluca, K.A.; Gudkov, A.V.; et al. Core circadian protein CLOCK is a positive regulator of NF-κB-mediated transcription. Proc. Natl. Acad. Sci. USA 2012, 109, E2457–E2465. [Google Scholar] [CrossRef] [Green Version]
- Cermakian, N.; Lange, T.; Golombek, D.; Sarkar, D.; Nakao, A.; Shibata, S.; Mazzoccoli, G. Crosstalk between the circadian clock circuitry and the immune system. Chronobiol. Int. 2013, 30, 870–888. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, Z.; Mao, K.; Zou, J.; Wang, Y.; Tao, Z.; Lin, G.; Tian, L.; Ji, Y.; Wu, X.; et al. Dec2 promotes Th2 cell differentiation by enhancing IL-2R signaling. J. Immunol. 2009, 183, 6320–6329. [Google Scholar] [CrossRef] [Green Version]
- Kashiwada, M.; Cassel, S.L.; Colgan, J.D.; Rothman, P.B. NFIL3/E4BP4 controls type 2 T helper cell cytokine expression. EMBO J. 2011, 30, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Kamizono, S.; Duncan, G.S.; Seidel, M.G.; Morimoto, A.; Hamada, K.; Grosveld, G.; Akashi, K.; Lind, E.F.; Haight, J.P.; Ohashi, P.S.; et al. Nfil3/E4bp4 is required for the development and maturation of NK cells in vivo. J. Exp. Med. 2009, 206, 2977–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascoyne, D.M.; Long, E.; Veiga-Fernandes, H.; de Boer, J.; Williams, O.; Seddon, B.; Coles, M.; Kioussis, D.; Brady, H.J. The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat. Immunol. 2009, 10, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Burgdorf, S.; Dani, I.; Saijo, K.; Flossdorf, J.; Hucke, S.; Alferink, J.; Nowak, N.; Beyer, M.; Mayer, G.; et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 2009, 206, 2079–2089. [Google Scholar] [CrossRef]
- Jones, D.C.; Ding, X.; Zhang, T.Y.; Daynes, R.A. Peroxisome proliferator-activated receptor alpha negatively regulates T-bet transcription through suppression of p38 mitogen-activated protein kinase activation. J. Immunol. 2003, 171, 196–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jetten, A.M. Retinoid-related orphan receptors (RORs): Critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal. 2009, 7, e003. [Google Scholar] [CrossRef] [Green Version]
- Solt, L.A.; Kumar, N.; Nuhant, P.; Wang, Y.; Lauer, J.L.; Liu, J.; Istrate, M.A.; Kamenecka, T.M.; Roush, W.R.; Vidović, D.; et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 2011, 472, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Rauen, T.; Juang, Y.T.; Hedrich, C.M.; Kis-Toth, K.; Tsokos, G.C. A novel isoform of the orphan receptor RORγt suppresses IL-17 production in human T cells. Genes Immun. 2012, 13, 346–350. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Rollins, D.; Ruhn, K.A.; Stubblefeld, J.J.; Green, C.B.; Kashiwada, M.; Rothman, P.B.; Takahashi, J.S.; Hooper, L.V. TH17 cell diferentiation is regulated by the circadian clock. Science 2013, 342, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Amir, M.; Chaudhari, S.; Wang, R.; Campbell, S.; Mosure, S.A.; Chopp, L.B.; Lu, Q.; Shang, J.; Pelletier, O.B.; He, Y.; et al. REV-ERBalpha Regulates TH17 Cell Development and Autoimmunity. Cell Rep. 2018, 25, 3733–3749.e8. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.; Walker, J.A.; Jolin, H.E.; Drynan, L.F.; Hams, E.; Camelo, A.; Barlow, J.L.; Neill, D.R.; Panova, V.; Koch, U.; et al. Transcription factor RORα is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.S.; McHeyzer-Williams, L.J.; Okitsu, S.L.; Burris, T.P.; Reiner, S.L.; McHeyzer-Williams, M.G. Divergent transcriptional programming of class-specific B cell memory by T-bet and RORα. Nat. Immunol. 2012, 13, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, C.; Tone, Y.; Tsuda, M.; Peter, C.; Waldmann, H.; Tone, M. TGF-β-mediated Foxp3 gene expression is cooperatively regulated by Stat5, Creb, and AP-1 through CNS2. J. Immunol. 2014, 192, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; Singh, U.P.; Singh, B.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE 2011, 6, e23522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, G.; Beischlag, T.V.; Vinciguerra, M.; Mazzoccoli, G. The circadian clock circuitry and the AHR signaling pathway in physiology and pathology. Biochem. Pharmacol. 2013, 85, 1405–1416. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Uehata, T.; Takeuchi, O. RNA Recognition and Immunity-Innate Immune Sensing and Its Posttranscriptional Regulation Mechanisms. Cells 2020, 9, 1701. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vande Walle, L.; Lamkanfi, M. Inflammasomes: Caspase-1-activating platforms with critical roles in host defense. Front. Microbiol. 2011, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 5, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Li, S.; Molusky, M.M.; Lin, J.D. Circadian autophagy rhythm: A link between clock and metabolism? Trends Endocrinol. Metab. 2012, 23, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Pastore, N.; Ballabio, A. Keeping the autophagy tempo. Autophagy 2019, 15, 1854–1856. [Google Scholar] [CrossRef]
- Maffei, V.J.; Kim, S.; Blanchard, E.; Luo, M.; Jazwinski, S.M.; Taylor, C.M.; Welsh, D.A. Biological Aging and the Human Gut Microbiota. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1474–1482. [Google Scholar] [CrossRef] [Green Version]
- Ratiner, K.; Abdeen, S.K.; Goldenberg, K.; Elinav, E. Utilization of Host and Microbiome Features in Determination of Biological Aging. Microorganisms 2022, 10, 668. [Google Scholar] [CrossRef]
- Bishehsari, F.; Voigt, R.M.; Keshavarzian, A. Circadian rhythms and the gut microbiota: From the metabolic syndrome to cancer. Nat. Rev. Endocrinol. 2020, 16, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Bushman, F.D.; FitzGerald, G.A. Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc. Natl. Acad. Sci. USA 2015, 112, 10479–10484. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Rao, M.C.; Chang, E.B. Gut microbiota as a transducer of dietary cues to regulate host circadian rhythms and metabolism. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.R.; Nomellini, V.; Faunce, D.E.; Kovacs, E.J. Innate immunity and aging. Exp. Gerontol. 2008, 43, 718–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahbub, S.; Brubaker, A.L.; Kovacs, E.J. Aging of the Innate Immune System: An Update. Curr. Immunol. Rev. 2011, 7, 104–115. [Google Scholar] [CrossRef]
- Ma, S.; Wang, C.; Mao, X.; Hao, Y. B Cell Dysfunction Associated With Aging and Autoimmune Diseases. Front. Immunol. 2019, 10, 318. [Google Scholar] [CrossRef] [Green Version]
- Weng, N.P. Aging of the immune system: How much can the adaptive immune system adapt? Immunity 2006, 24, 495–499. [Google Scholar] [CrossRef] [Green Version]
- Sprent, J.; Surh, C.D. Normal T cell homeostasis: The conversion of naive cells into memory-phenotype cells. Nat. Immunol. 2011, 12, 478–484. [Google Scholar] [CrossRef]
- Lazuardi, L.; Jenewein, B.; Wolf, A.M.; Pfister, G.; Tzankov, A.; Grubeck-Loebenstein, B. Age-related loss of naïve T cells and dysregulation of T-cell/B-cell interactions in human lymph nodes. Immunology 2005, 114, 37–43. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb. Exp. Pharmacol. 2006, 172, 69–91. [Google Scholar]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gökçen, A.; Kracht, M. The Crosstalk of Endoplasmic Reticulum (ER) Stress Pathways with NF-κB: Complex Mechanisms Relevant for Cancer, Inflammation and Infection. Biomedicines 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, L.; Yuan, C.; Wu, X. Endoplasmic Reticulum Stress in Diabetic Nephrology: Regulation, Pathological Role, and Therapeutic Potential. Oxidative Med. Cell. Longev. 2021, 2021, 7277966. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Sterol regulatory element-binding protein (SREBP) cleavage regulates Golgi-to-endoplasmic reticulum recycling of SREBP cleavage-activating protein (SCAP). J. Biol. Chem. 2014, 289, 7547–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Shuangquan, L. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 6129–6140. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Coelho, D.S.; Domingos, P.M. Physiological roles of regulated Ire1 dependent decay. Front. Genet. 2014, 5, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, G.; Staiano, L.; Moretti, M.N.; De Cegli, R.; Fagnocchi, L.; Di Tullio, G.; Polletti, S.; Braccia, C.; Armirotti, A.; Zippo, A.; et al. GADD34 is a modulator of autophagy during starvation. Sci. Adv. 2020, 6, eabb0205. [Google Scholar] [CrossRef] [PubMed]
- Cretenet, G.; Le Clech, M.; Gachon, F. Circadian clock-coordinated 12 Hr period rhythmic activation of the IRE1alpha pathway controls lipid metabolism in mouse liver. Cell Metab. 2010, 11, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Bu, Y.; Yoshida, A.; Chitnis, N.; Altman, B.J.; Tameire, F.; Oran, A.; Gennaro, V.; Armeson, K.E.; McMahon, S.B.; Wertheim, G.B.; et al. A PERK-miR-211 axis suppresses circadian regulators and protein synthesis to promote cancer cell survival. Nat. Cell Biol. 2018, 20, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Milev, N.B.; Gatfield, D. Circadian Clocks and UPR: New Twists as the Story Unfolds. Dev. Cell 2018, 44, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Chen, H.; Li, C.; Xiao, Y.; Yang, D.; Zhang, M.; Zhou, D.; Liu, W.; Wang, A.; Jin, Y. ER stress activation impairs the expression of circadian clock and clock-controlled genes in NIH3T3 cells via an ATF4-dependent mechanism. Cell Signal. 2019, 57, 89–101. [Google Scholar] [CrossRef]
- So, J.S. Roles of Endoplasmic Reticulum Stress in Immune Responses. Mol. Cells 2018, 41, 705–716. [Google Scholar] [CrossRef]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. ER stress-induced inflammation: Does it aid or impede disease progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef]
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Lencer, W.I.; DeLuca, H.; Grey, M.J.; Cho, J.A. Innate immunity at mucosal surfaces: The IRE1-RIDD-RIG-I pathway. Trends Immunol. 2015, 36, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junjappa, R.P.; Patil, P.; Bhattarai, K.R.; Kim, H.R.; Chae, H.J. IRE1α Implications in Endoplasmic Reticulum Stress-Mediated Development and Pathogenesis of Autoimmune Diseases. Front. Immunol. 2018, 9, 1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Joe, Y.; Kim, H.J.; Kim, Y.S.; Jeong, S.O.; Pae, H.O.; Ryter, S.W.; Surh, Y.J.; Chung, H.T. Endoplasmic reticulum stress-induced IRE1α activation mediates cross-talk of GSK-3β and XBP-1 to regulate inflammatory cytokine production. J. Immunol. 2015, 194, 4498–4506. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Hegde, R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010, 189, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol. Cell Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Zhai, Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am. J. Transplant. 2014, 14, 1552–1561. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Kim, S.M.; Kim, K.P.; Lee, S.H.; Moon, J.Y. The Role of Inflammasome-Dependent and Inflammasome-Independent NLRP3 in the Kidney. Cells 2019, 8, 1389. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nuñez, G.; He, Y.; Yin, X.M.; O’Riordan, M.X. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Murugan, D.D.; Khan, H.; Huang, Y.; Cheang, W.S. Roles and Therapeutic Implications of Endoplasmic Reticulum Stress and Oxidative Stress in Cardiovascular Diseases. Antioxidants 2021, 10, 1167. [Google Scholar] [CrossRef] [PubMed]
- Thoudam, T.; Jeon, J.H.; Ha, C.M.; Lee, I.K. Role of Mitochondria-Associated Endoplasmic Reticulum Membrane in Inflammation-Mediated Metabolic Diseases. Mediat. Inflamm. 2016, 2016, 1851420. [Google Scholar] [CrossRef] [PubMed]
- Pourcet, B.; Zecchin, M.; Ferri, L.; Beauchamp, J.; Sitaula, S.; Billon, C.; Delhaye, S.; Vanhoutte, J.; Mayeuf-Louchart, A.; Thorel, Q.; et al. Nuclear Receptor Subfamily 1 Group D Member 1 Regulates Circadian Activity of NLRP3 Inflammasome to Reduce the Severity of Fulminant Hepatitis in Mice. Gastroenterology 2018, 154, 1449–1464.e20. [Google Scholar] [CrossRef]
- Billon, C.; Murray, M.H.; Avdagic, A.; Burris, T.P. RORγ regulates the NLRP3 inflammasome. J. Biol. Chem. 2019, 294, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, S.; Sassone-Corsi, P. Plasticity and specificity of the circadian epigenome. Nat. Neurosci. 2010, 13, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Masri, S.; Orozco-Solis, R.; Aguilar-Arnal, L.; Cervantes, M.; Sassone-Corsi, P. Coupling circadian rhythms of metabolism and chromatin remodelling. Diabetes Obes. Metab. 2015, 17 (Suppl. S1), 17–22. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+ −dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Cela, O.; Scrima, R.; Pazienza, V.; Merla, G.; Benegiamo, G.; Augello, B.; Fugetto, S.; Menga, M.; Rubino, R.; Fuhr, L.; et al. Clock genes-dependent acetylation of complex I sets rhythmic activity of mitochondrial OxPhos. Biochim. Biophys. Acta 2016, 1863, 596–606. [Google Scholar] [CrossRef]
- de Goede, P.; Wefers, J.; Brombacher, E.C.; Schrauwen, P.; Kalsbeek, A. Circadian rhythms in mitochondrial respiration. J. Mol. Endocrinol. 2018, 60, R115–R130. [Google Scholar] [CrossRef] [Green Version]
- Katada, S.; Sassone-Corsi, P. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat. Struct. Mol. Biol. 2010, 17, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Valekunja, U.K.; Edgar, R.S.; Oklejewicz, M.; van der Horst, G.T.; O’Neill, J.S.; Tamanini, F.; Turner, D.J.; Reddy, A.B. Histone methyltransferase MLL3 contributes to genome-scale circadian transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 1554–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etchegaray, J.P.; Yang, X.; DeBruyne, J.P.; Peters, A.H.F.M.; Weaver, D.R.; Jenuwein, T.; Reppert, S.M. The polycomb group protein EZH2 is required for mammalian circadian clock function. J. Biol. Chem. 2006, 281, 21209–21215. [Google Scholar] [CrossRef] [PubMed]
- Di Tacchio, L.; Le, H.D.; Vollmers, C.; Hatori, M.; Witcher, M.; Secombe, J.; Panda, S. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science 2011, 333, 1881–1885. [Google Scholar] [CrossRef] [Green Version]
- Nam, H.J.; Boo, K.; Kim, D.; Han, D.; Choe, H.K.; Kim, C.R.; Sun, W.; Kim, H.; Kim, K.; Lee, H.; et al. Phosphorylation of LSD1 by PKCalpha is crucial for circadian rhythmicity and phase resetting. Mol. Cell 2014, 53, 791–805. [Google Scholar] [CrossRef] [Green Version]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Maekawa, F.; Shimba, S.; Takumi, S.; Sano, T.; Suzuki, T.; Bao, J.; Ohwada, M.; Ehara, T.; Ogawa, Y.; Nohara, K. Diurnal expression of Dnmt3b mRNA in mouse liver is regulated by feeding and hepatic clockwork. Epigenetics 2012, 7, 1046–1056. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Ma, S.; Zhang, Y.; Wang, T.; Zhou, M.; Wang, Z.; Zhang, J. Daily variation in global and local DNA methylation in mouse livers. PLoS ONE 2015, 10, e0118101. [Google Scholar] [CrossRef] [Green Version]
- Pazienza, V.; Tavano, F.; Francavilla, M.; Fontana, A.; Pellegrini, F.; Benegiamo, G.; Corbo, V.; di Mola, F.F.; Di Sebastiano, P.; Andriulli, A.; et al. Time-Qualified Patterns of Variation of PPARγ, DNMT1, and DNMT3B Expression in Pancreatic Cancer Cell Lines. PPAR Res. 2012, 2012, 890875. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Poole, J.; Ray, D. The role of circadian clock genes in critical illness: The potential role of translational clock gene therapies to target inflammation, mitochondrial function and muscle mass in ICU. J. Biol. Rhythm. 2022, 37, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Solis, R.; Sassone-Corsi, P. Circadian clock: Linking epigenetics to aging. Curr. Opin. Genet. Dev. 2014, 26, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, R.; Fu, Q.; Sun, Y.; Li, Q. Epigenetic clock: A promising biomarker and practical tool in aging. Res. Aging Rev. 2022, 81, 101743. [Google Scholar] [CrossRef] [PubMed]
- Galow, A.M.; Peleg, S. How to slow down the tick of time: Age-associated epigenetic alterations and related interventions to prolong lifespan. Cells 2022, 11, 468. [Google Scholar] [CrossRef]
- Sillanpää, E.; Laakkonen, E.K.; Vaara, E.; Rantanen, T.; Kovanen, V.; Sipilä, S.; Kaprio, J.; Ollikainen, M. Biological clocks and physical functioning in monozygotic female twins. BMC Geriatr. 2018, 18, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milicic, L.; Vacher, M.; Porter, T.; Doré, V.; Burnham, S.C.; Bourgeat, P.; Shishegar, R.; Doecke, J.; Armstrong, N.J.; Tankard, R.; et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI); Australian Imaging Biomarkers and Lifestyle (AIBL) Study. Comprehensive analysis of epigenetic clocks reveals associations between disproportionate biological ageing and hippocampal volume. Geroscience 2022, 44, 1807–1823. [Google Scholar] [CrossRef]
- Chang, H.C.; Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Solanas, G.; Peixoto, F.O.; Bee, L.; Symeonidi, A.; Schmidt, M.S.; Brenner, C.; Masri, S.; Aznar Benitah, S.; Sassone-Corsi, P. Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell 2017, 170, 664–677.e11. [Google Scholar] [CrossRef]
- Solanas, G.; Peixoto, F.O.; Perdiguero, E.; Jardì, M.; Ruiz-Bonilla, V.; Datta, D.; Symeonidi, A.; Castellanos, A.; Welz, P.; Caballero, J.M.; et al. Aged Stem Cells Reprogram Their Daily Rhythmic Functions to Adapt to Stress. Cell 2017, 170, 678–692.e20. [Google Scholar] [CrossRef] [Green Version]
- Marcheva, B.; Ramsey, K.M.; Buhr, E.D.; Kobayashi, Y.; Su, H.; Ko, C.H.; Ivanova, G.; Omura, C.; Mo, S.; Vitaterna, M.H.; et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010, 466, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Morris, C.J.; Purvis, T.E.; Hu, K.; Scheer, F.A. Circadian misalignment increases cardiovascular disease risk factors in humans. Proc. Natl. Acad. Sci. USA 2016, 113, E1402–E1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonten, T.N.; Snoep, J.D.; Assendelft, W.J.; Zwaginga, J.J.; Eikenboom, J.; Huisman, M.V.; Rosendaal, F.R.; Bom, J.G. Time-dependent effects of aspirin on blood pressure and morning platelet reactivity: A randomized cross-over trial. Hypertension 2015, 65, 743–750. [Google Scholar] [CrossRef] [PubMed]
- McNeil, J.J.; Nelson, M.R.; Woods, R.L.; Lockery, J.E.; Wolfe, R.; Reid, C.M.; Kirpach, B.; Shah, R.C.; Ives, D.G.; Storey, E.; et al. Effect of Aspirin on All-Cause Mortality in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Hermida, R.C.; Ayala, D.E.; Mojón, A.; Fernández, J.R. Chronotherapy with nifedipine GITS in hypertensive patients: Improved efficacy and safety with bedtime dosing. Am. J. Hypertens. 2008, 21, 948–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, C.M.; Sassone-Corsi, P. Personalized medicine and circadian rhythms: Opportunities for modern society. J. Exp. Med. 2020, 217, e20200702. [Google Scholar] [CrossRef] [PubMed]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in aging and disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Nohara, K.; Park, N.; Park, Y.; Guillory, B.; Zhao, Z.; Garcia, J.M.; Koike, N.; Lee, C.; Takahashi, J.S.; et al. The Small Molecule Nobiletin Targets the Molecular Oscillator to Enhance Circadian Rhythms and Protect against Metabolic Syndrome. Cell Metab. 2016, 23, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Nohara, K.; Mallampalli, V.; Nemkov, T.; Wirianto, M.; Yang, J.; Ye, Y.; Sun, Y.; Han, L.; Esser, K.A.; Meleykovskaya, E.; et al. Nobiletin fortifies mitochondrial respiration in skeletal muscle to promote healthy aging against metabolic challenge. Nat. Commun. 2019, 10, 3923. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Rodríguez, V.A.; Rijo-Ferreira, F.; Green, C.B.; Takahashi, J.S. Importance of circadian timing for aging and longevity. Nat. Commun. 2021, 12, 2862. [Google Scholar] [CrossRef]
- Lotti, S.; Pagliai, G.; Colombini, B.; Sofi, F.; Dinu, M. Chronotype Differences in Energy Intake, Cardiometabolic Risk Parameters, Cancer, and Depression: A Systematic Review with Meta-Analysis of Observational Studies. Adv. Nutr. 2022, 13, 269–281. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colombini, B.; Dinu, M.; Murgo, E.; Lotti, S.; Tarquini, R.; Sofi, F.; Mazzoccoli, G. Ageing and Low-Level Chronic Inflammation: The Role of the Biological Clock. Antioxidants 2022, 11, 2228. https://doi.org/10.3390/antiox11112228
Colombini B, Dinu M, Murgo E, Lotti S, Tarquini R, Sofi F, Mazzoccoli G. Ageing and Low-Level Chronic Inflammation: The Role of the Biological Clock. Antioxidants. 2022; 11(11):2228. https://doi.org/10.3390/antiox11112228
Chicago/Turabian StyleColombini, Barbara, Monica Dinu, Emanuele Murgo, Sofia Lotti, Roberto Tarquini, Francesco Sofi, and Gianluigi Mazzoccoli. 2022. "Ageing and Low-Level Chronic Inflammation: The Role of the Biological Clock" Antioxidants 11, no. 11: 2228. https://doi.org/10.3390/antiox11112228