
Altered Cellular Protein Quality Control System Modulates Cardiomyocyte Function in Volume Overload-Induced Hypertrophy
 , ,
, ,  , and
, and
Abstract
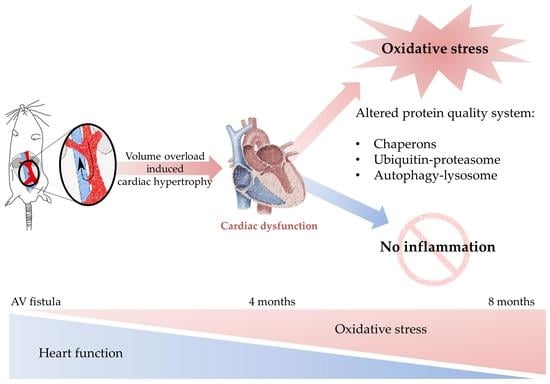
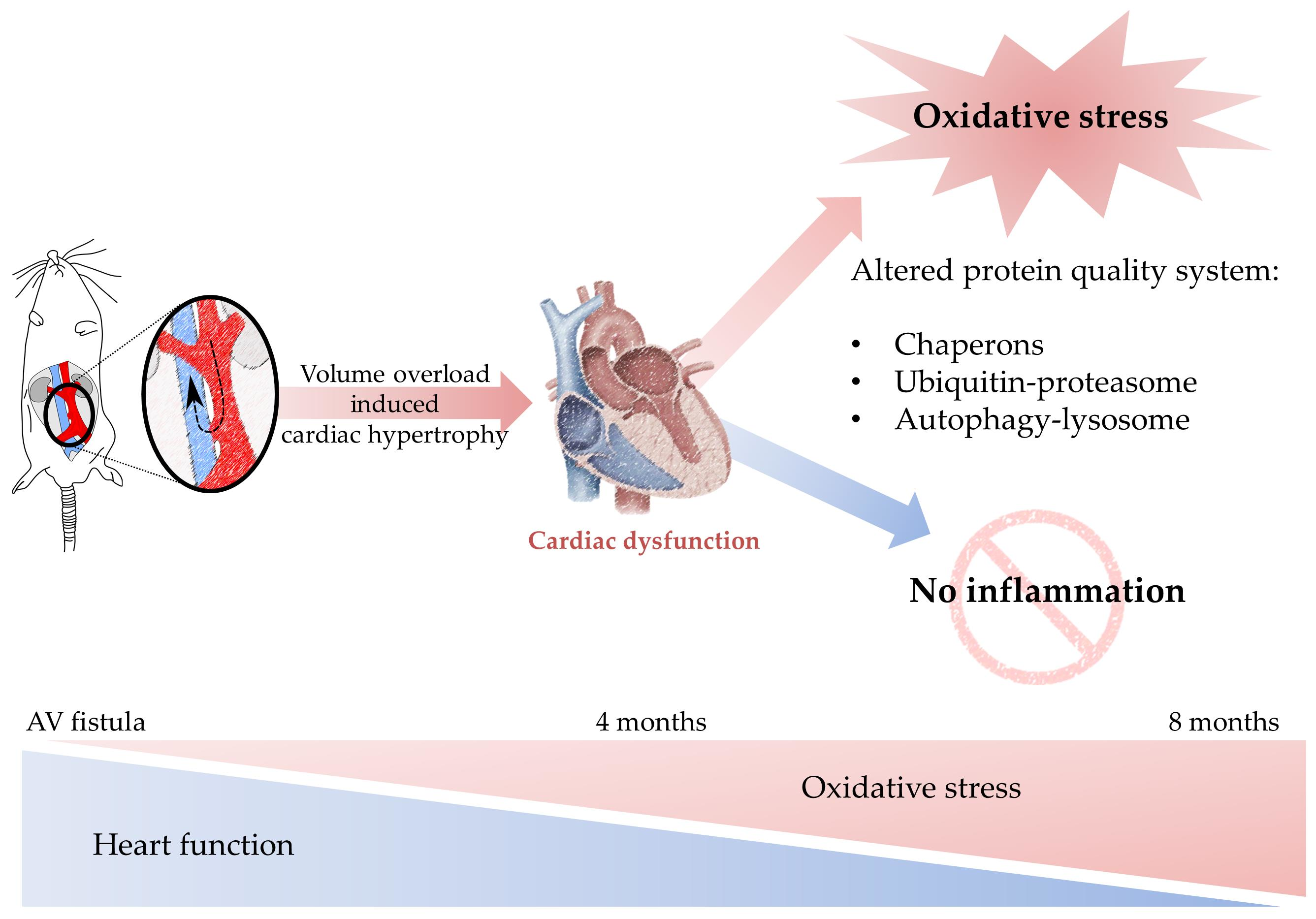
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Volume Overload-Induced Hypertrophy by Aortocaval Fistula
2.3. Transthoracic Echocardiography
2.4. Termination of Animals
2.5. Passive Stiffness Measurement of Cardiac Sarcomere
2.6. Western Blot
2.7. Titin Gel Electrophoresis and Immunoblotting
2.8. Statistical Analysis
3. Results
3.1. Development of Cardiac Hypertrophy in VO Rats with Preserved Ejection Fraction
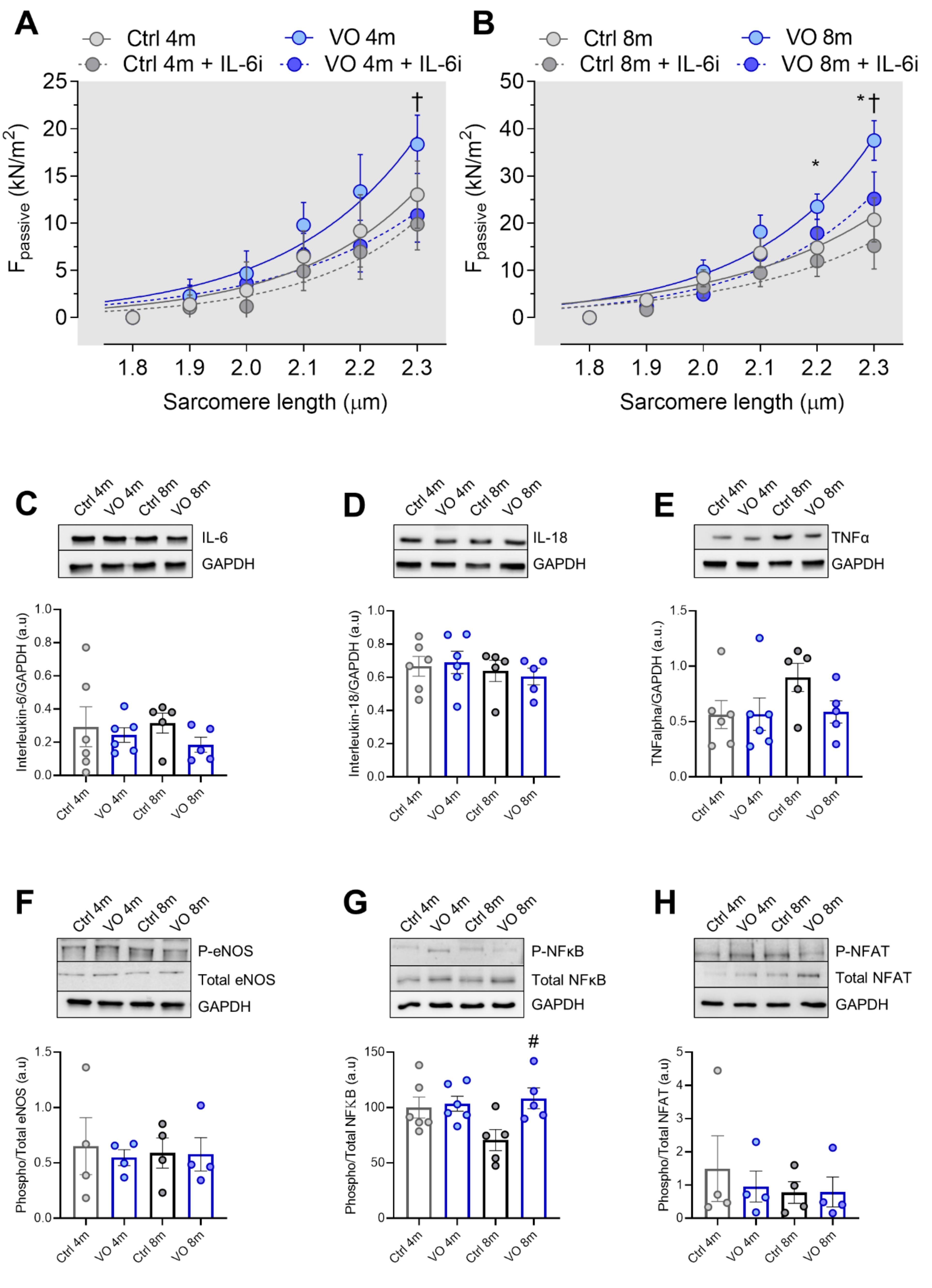
3.2. Increased Titin-Based Myocardial Stiffness and Oxidation of the Giant Sarcomere Protein Titin
3.3. Increased Oxidative Stress and Unaltered Inflammatory Markers in VO Animals
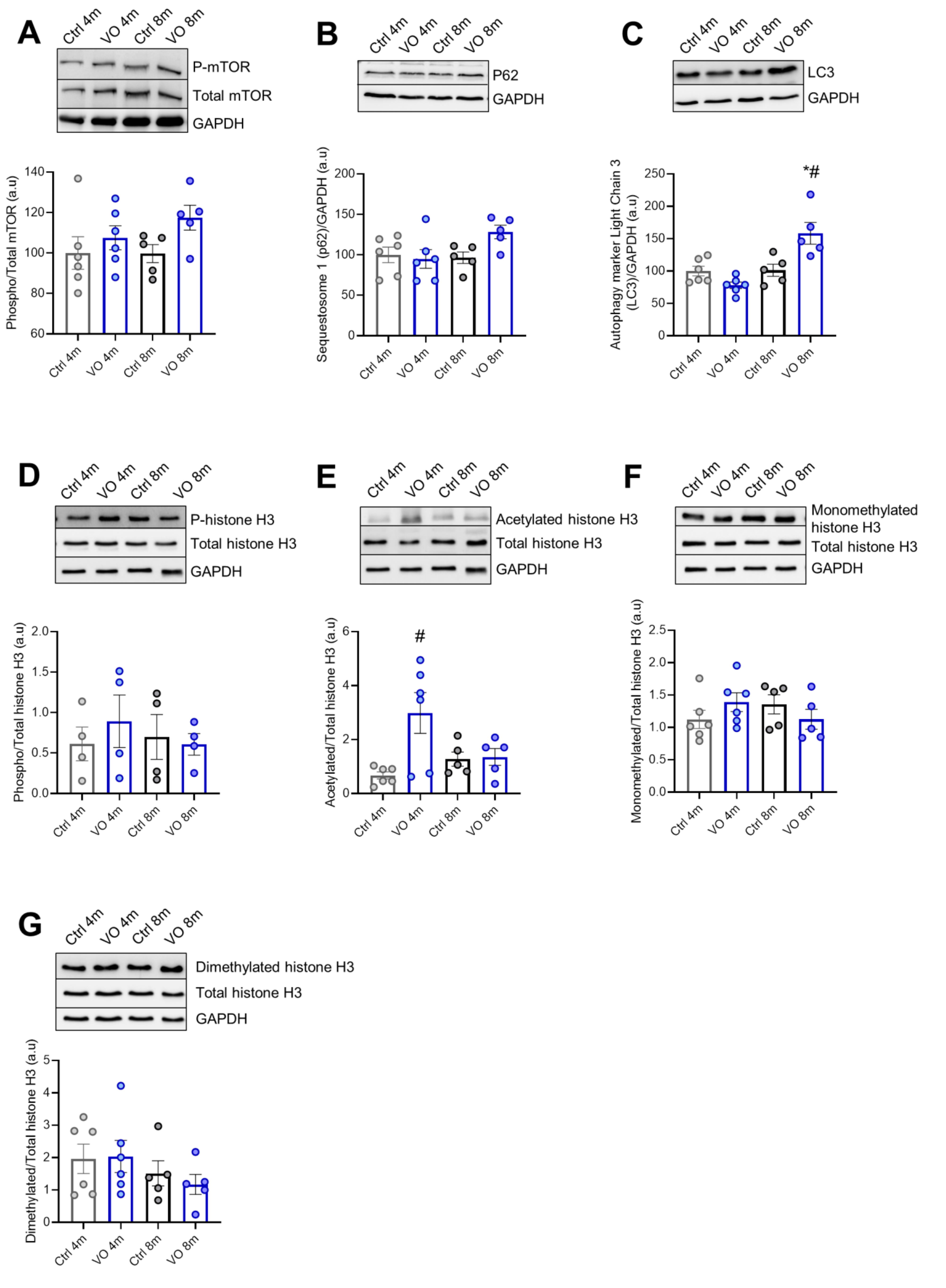
3.4. Alteration of Autophagy Markers in VO Animals
3.5. Histone’s Modifications due to VO
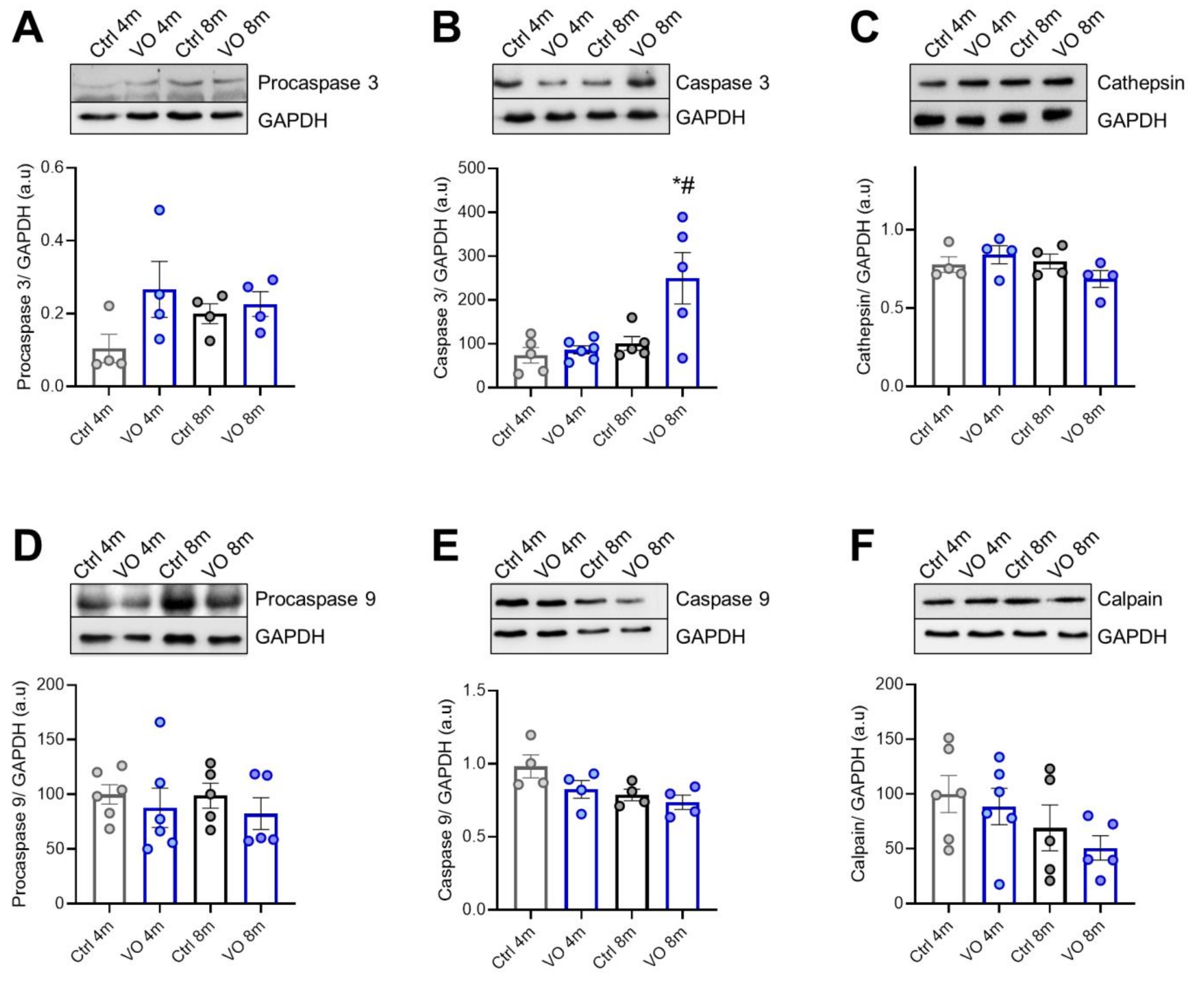
3.6. Apoptotic Markers and Caspases in VO Animals
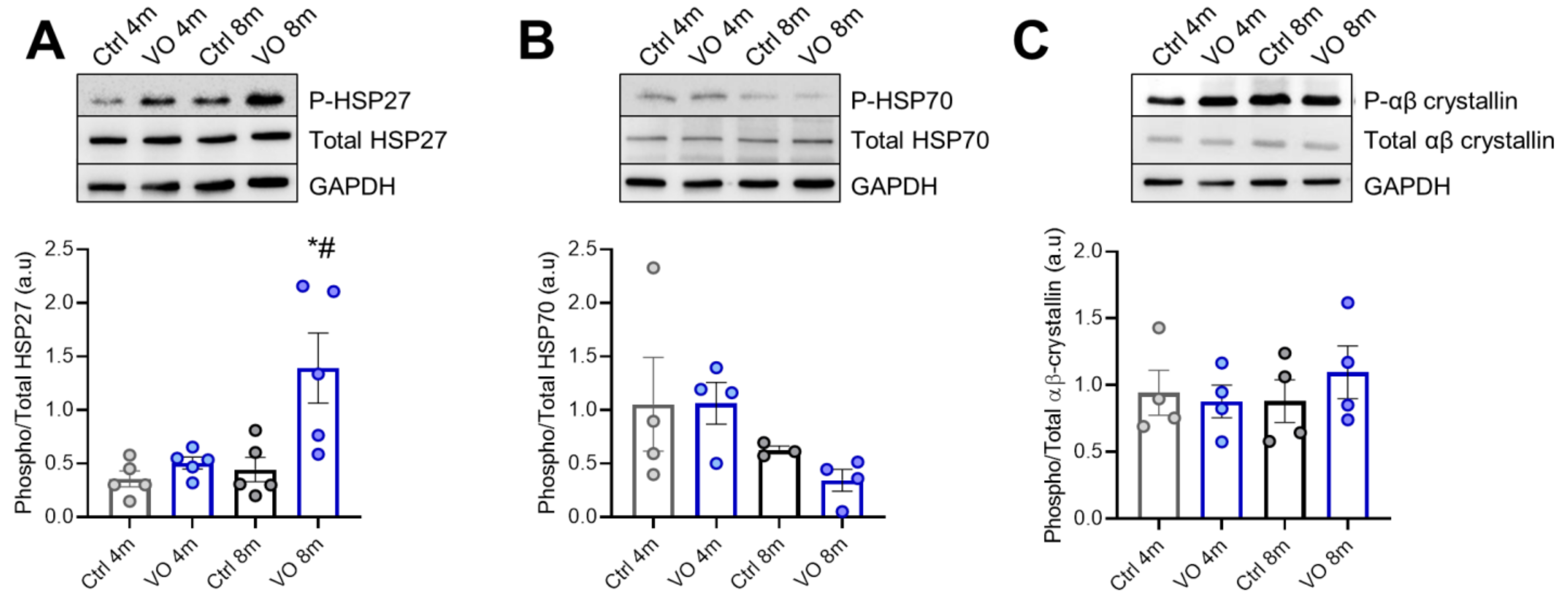
3.7. Alterations in Heat Shock Proteins in VO Animals
4. Discussion
5. Conclusions
6. Clinical Relevance and Future Therapeutic Approaches
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kikuchi, K.; Poss, K.D. Cardiac regenerative capacity and mechanisms. Annu. Rev. Cell Dev. Biol. 2012, 28, 719–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Robbins, J. Heart failure and protein quality control. Circ. Res. 2006, 99, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassoun, R.; Budde, H.; Zhazykbayeva, S.; Herwig, M.; Sieme, M.; Delalat, S.; Mostafi, N.; Gomori, K.; Tangos, M.; Jarkas, M.; et al. Stress activated signalling impaired protein quality control pathways in human hypertrophic cardiomyopathy. Int. J. Cardiol. 2021, 344, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Dorsch, L.M.; Schuldt, M.; dos Remedios, C.G.; Schinkel, A.F.L.; de Jong, P.L.; Michels, M.; Kuster, D.W.D.; Brundel, B.; van der Velden, J. Protein Quality Control Activation and Microtubule Remodeling in Hypertrophic Cardiomyopathy. Cells 2019, 8, 741. [Google Scholar] [CrossRef] [Green Version]
- You, J.; Wu, J.; Zhang, Q.; Ye, Y.; Wang, S.; Huang, J.; Liu, H.; Wang, X.; Zhang, W.; Bu, L.; et al. Differential cardiac hypertrophy and signaling pathways in pressure versus volume overload. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H552–H562. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Akazawa, H.; Naito, A.T.; Komuro, I. Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative roles in heart failure. Circ. Res. 2014, 114, 565–571. [Google Scholar] [CrossRef]
- Leite-Moreira, A.M.; Almeida-Coelho, J.; Neves, J.S.; Pires, A.L.; Ferreira-Martins, J.; Castro-Ferreira, R.; Ladeiras-Lopes, R.; Conceicao, G.; Miranda-Silva, D.; Rodrigues, P.; et al. Stretch-induced compliance: A novel adaptive biological mechanism following acute cardiac load. Cardiovasc. Res. 2018, 114, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Pitoulis, F.G.; Terracciano, C.M. Heart Plasticity in Response to Pressure- and Volume-Overload: A Review of Findings in Compensated and Decompensated Phenotypes. Front. Physiol. 2020, 11, 92. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Schulz, E.; Jansen, T.; Wenzel, P.; Daiber, A.; Munzel, T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid. Redox Signal. 2008, 10, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Gomori, K.; Herwig, M.; Budde, H.; Hassoun, R.; Mostafi, N.; Zhazykbayeva, S.; Sieme, M.; Modi, S.; Szabados, T.; Pipis, J.; et al. Ca2+/calmodulin-dependent protein kinase II and protein kinase G oxidation contributes to impaired sarcomeric proteins in hypertrophy model. ESC Heart Fail. 2022, 9, 2585–2600. [Google Scholar] [CrossRef] [PubMed]
- Budde, H.; Hassoun, R.; Tangos, M.; Zhazykbayeva, S.; Herwig, M.; Varatnitskaya, M.; Sieme, M.; Delalat, S.; Sultana, I.; Kolijn, D.; et al. The Interplay between S-Glutathionylation and Phosphorylation of Cardiac Troponin I and Myosin Binding Protein C in End-Stage Human Failing Hearts. Antioxidants 2021, 10, 1134. [Google Scholar] [CrossRef]
- Breitkreuz, M.; Hamdani, N. A change of heart: Oxidative stress in governing muscle function? Biophys. Rev. 2015, 7, 321–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P. Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid. Redox Signal. 2006, 8, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Ranek, M.J.; Stachowski, M.J.; Kirk, J.A.; Willis, M.S. The role of heat shock proteins and co-chaperones in heart failure. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160530. [Google Scholar] [CrossRef] [Green Version]
- Kalmar, B.; Greensmith, L. Induction of heat shock proteins for protection against oxidative stress. Adv. Drug Deliv. Rev. 2009, 61, 310–318. [Google Scholar] [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschope, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Kotter, S.; Unger, A.; Hamdani, N.; Lang, P.; Vorgerd, M.; Nagel-Steger, L.; Linke, W.A. Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J. Cell Biol. 2014, 204, 187–202. [Google Scholar] [CrossRef]
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1–26. [Google Scholar]
- Parry, T.L.; Willis, M.S. Cardiac ubiquitin ligases: Their role in cardiac metabolism, autophagy, cardioprotection and therapeutic potential. Biochim. Biophys. Acta 2016, 1862, 2259–2269. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, J.; He, L.; Peng, L.; Zhong, Q.; Chen, L.; Jiang, Z. The role of autophagy in cardiac hypertrophy. Acta Biochim. Biophys. Sin. 2016, 48, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Wang, J.; Yang, X. Functions of autophagy in pathological cardiac hypertrophy. Int. J. Biol. Sci. 2015, 11, 672–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwig, M.; Kolijn, D.; Lodi, M.; Holper, S.; Kovacs, A.; Papp, Z.; Jaquet, K.; Haldenwang, P.; Dos Remedios, C.; Reusch, P.H.; et al. Modulation of Titin-Based Stiffness in Hypertrophic Cardiomyopathy via Protein Kinase D. Front. Physiol. 2020, 11, 240. [Google Scholar] [CrossRef]
- Henning, R.H.; Brundel, B. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Loescher, C.M.; Breitkreuz, M.; Li, Y.; Nickel, A.; Unger, A.; Dietl, A.; Schmidt, A.; Mohamed, B.A.; Kotter, S.; Schmitt, J.P.; et al. Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx). Proc. Natl. Acad. Sci. USA 2020, 117, 24545–24556. [Google Scholar] [CrossRef]
- Hamdani, N.; Franssen, C.; Lourenco, A.; Falcao-Pires, I.; Fontoura, D.; Leite, S.; Plettig, L.; Lopez, B.; Ottenheijm, C.A.; Becher, P.M.; et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ. Heart Fail. 2013, 6, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Michel, K.; Herwig, M.; Werner, F.; Spiranec Spes, K.; Abesser, M.; Schuh, K.; Dabral, S.; Mugge, A.; Baba, H.A.; Skryabin, B.V.; et al. C-type natriuretic peptide moderates titin-based cardiomyocyte stiffness. JCI Insight 2020, 5, e139910. [Google Scholar] [CrossRef]
- Qin, J.; Guo, N.; Tong, J.; Wang, Z. Function of histone methylation and acetylation modifiers in cardiac hypertrophy. J. Mol. Cell. Cardiol. 2021, 159, 120–129. [Google Scholar] [CrossRef]
- Fiorillo, C.; Nediani, C.; Ponziani, V.; Giannini, L.; Celli, A.; Nassi, N.; Formigli, L.; Perna, A.M.; Nassi, P. Cardiac volume overload rapidly induces oxidative stress-mediated myocyte apoptosis and hypertrophy. Biochim. Biophys. Acta 2005, 1741, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Zhazykbayeva, S.; Pabel, S.; Mugge, A.; Sossalla, S.; Hamdani, N. The molecular mechanisms associated with the physiological responses to inflammation and oxidative stress in cardiovascular diseases. Biophys. Rev. 2020, 12, 947–968. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, D.B.; Siwik, D.A.; Xiao, L.; Pimentel, D.R.; Singh, K.; Colucci, W.S. Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell. Cardiol. 2002, 34, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Siwik, D.A.; Tzortzis, J.D.; Pimental, D.R.; Chang, D.L.; Pagano, P.J.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Inhibition of copper-zinc superoxide dismutase induces cell growth, hypertrophic phenotype, and apoptosis in neonatal rat cardiac myocytes in vitro. Circ. Res. 1999, 85, 147–153. [Google Scholar] [CrossRef]
- Pimentel, D.R.; Amin, J.K.; Xiao, L.; Miller, T.; Viereck, J.; Oliver-Krasinski, J.; Baliga, R.; Wang, J.; Siwik, D.A.; Singh, K.; et al. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ. Res. 2001, 89, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, Y.; Otsu, K.; Nishida, K.; Hirotani, S.; Nakayama, H.; Yamaguchi, O.; Matsumura, Y.; Ueno, H.; Tada, M.; Hori, M. Involvement of reactive oxygen species-mediated NF-kappa B activation in TNF-alpha-induced cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2002, 34, 233–240. [Google Scholar] [CrossRef]
- Modesti, P.A.; Vanni, S.; Bertolozzi, I.; Cecioni, I.; Lumachi, C.; Perna, A.M.; Boddi, M.; Gensini, G.F. Different growth factor activation in the right and left ventricles in experimental volume overload. Hypertension 2004, 43, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Onodi, Z.; Ruppert, M.; Kucsera, D.; Sayour, A.A.; Toth, V.E.; Koncsos, G.; Novak, J.; Brenner, G.B.; Makkos, A.; Baranyai, T.; et al. AIM2-driven inflammasome activation in heart failure. Cardiovasc. Res. 2021, 117, 2639–2651. [Google Scholar] [CrossRef]
- Aimo, A.; Castiglione, V.; Borrelli, C.; Saccaro, L.F.; Franzini, M.; Masi, S.; Emdin, M.; Giannoni, A. Oxidative stress and inflammation in the evolution of heart failure: From pathophysiology to therapeutic strategies. Eur. J. Prev. Cardiol. 2020, 27, 494–510. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Button, R.W.; Roberts, S.L.; Willis, T.L.; Hanemann, C.O.; Luo, S. Accumulation of autophagosomes confers cytotoxicity. J. Biol. Chem. 2017, 292, 13599–13614. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, N.; Zhai, P.; Sadoshima, J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 2011, 14, 2179–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullgrabe, J.; Klionsky, D.J.; Joseph, B. Histone post-translational modifications regulate autophagy flux and outcome. Autophagy 2013, 9, 1621–1623. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef]
- Higuchi, M.; Proske, R.J.; Yeh, E.T. Inhibition of mitochondrial respiratory chain complex I by TNF results in cytochrome c release, membrane permeability transition, and apoptosis. Oncogene 1998, 17, 2515–2524. [Google Scholar] [CrossRef] [Green Version]
- Kumarapeli, A.R.; Su, H.; Huang, W.; Tang, M.; Zheng, H.; Horak, K.M.; Li, M.; Wang, X. Alpha B-crystallin suppresses pressure overload cardiac hypertrophy. Circ. Res. 2008, 103, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernandez, J.M. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Bullard, B.; Ferguson, C.; Minajeva, A.; Leake, M.C.; Gautel, M.; Labeit, D.; Ding, L.; Labeit, S.; Horwitz, J.; Leonard, K.R.; et al. Association of the chaperone alphaB-crystallin with titin in heart muscle. J. Biol. Chem. 2004, 279, 7917–7924. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Ke, L.; Mackovicova, K.; Van Der Want, J.J.; Sibon, O.C.; Tanguay, R.M.; Morrow, G.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J. Mol. Cell. Cardiol. 2011, 51, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.; Bosch, J.; Yusuf, S.; Sheridan, P.; Pogue, J.; Arnold, J.M.; Ross, C.; Arnold, A.; Sleight, P.; Probstfield, J.; et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: A randomized controlled trial. JAMA 2005, 293, 1338–1347. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gömöri, K.; Herwig, M.; Hassoun, R.; Budde, H.; Mostafi, N.; Delalat, S.; Modi, S.; Begovic, M.; Szabados, T.; Pipis, J.; et al. Altered Cellular Protein Quality Control System Modulates Cardiomyocyte Function in Volume Overload-Induced Hypertrophy. Antioxidants 2022, 11, 2210. https://doi.org/10.3390/antiox11112210
Gömöri K, Herwig M, Hassoun R, Budde H, Mostafi N, Delalat S, Modi S, Begovic M, Szabados T, Pipis J, et al. Altered Cellular Protein Quality Control System Modulates Cardiomyocyte Function in Volume Overload-Induced Hypertrophy. Antioxidants. 2022; 11(11):2210. https://doi.org/10.3390/antiox11112210
Chicago/Turabian StyleGömöri, Kamilla, Melissa Herwig, Roua Hassoun, Heidi Budde, Nusratul Mostafi, Simin Delalat, Suvasini Modi, Merima Begovic, Tamara Szabados, Judit Pipis, and et al. 2022. "Altered Cellular Protein Quality Control System Modulates Cardiomyocyte Function in Volume Overload-Induced Hypertrophy" Antioxidants 11, no. 11: 2210. https://doi.org/10.3390/antiox11112210