
Cell Models and Omics Techniques for the Study of Nonalcoholic Fatty Liver Disease: Focusing on Stem Cell-Derived Cell Models

 ,
,
Abstract
:1. Introduction
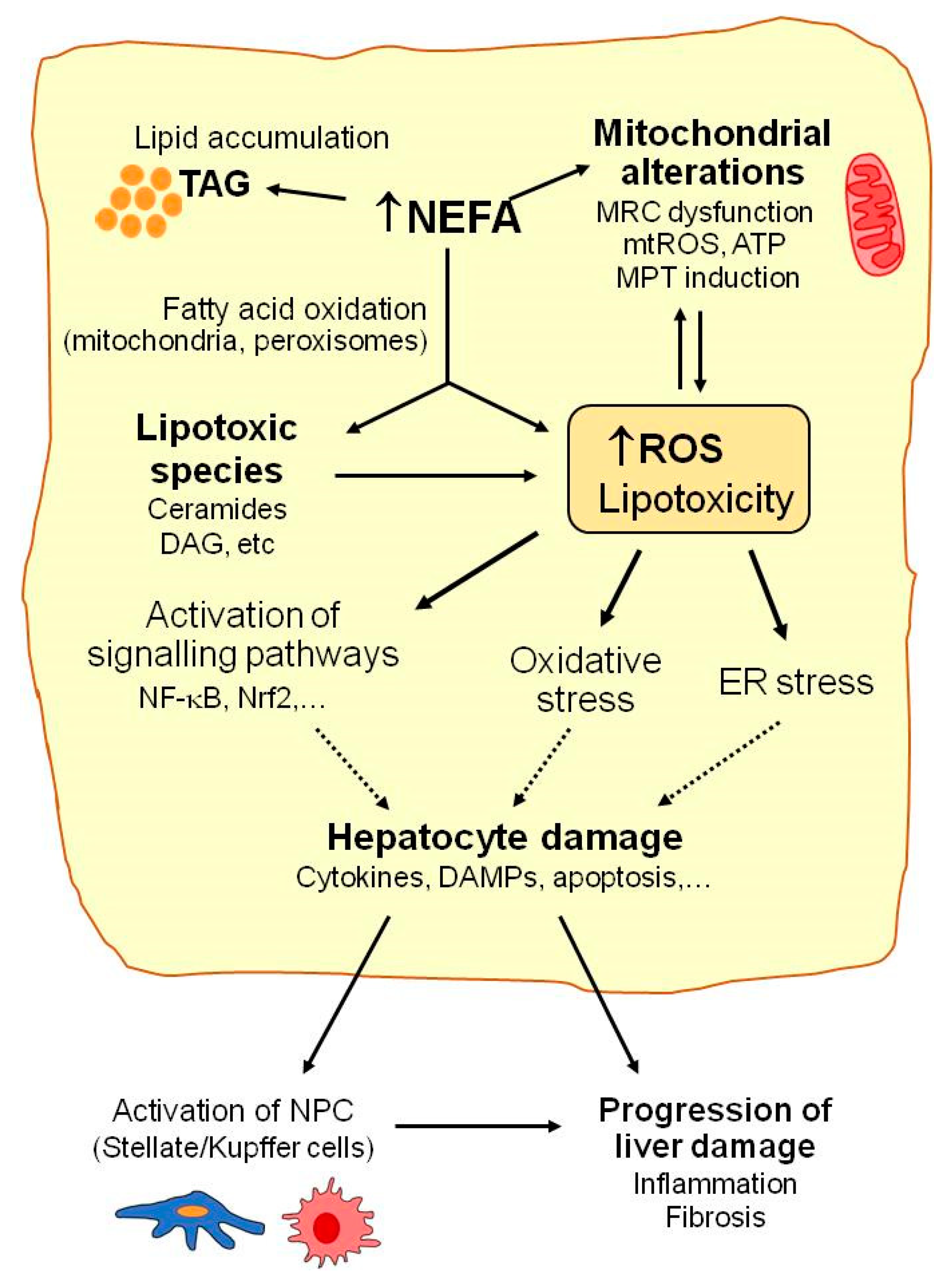
2. Mechanisms of Liver Injury in NAFLD: Mitochondrial Dysfunction, Oxidative Stress, and Lipotoxicity
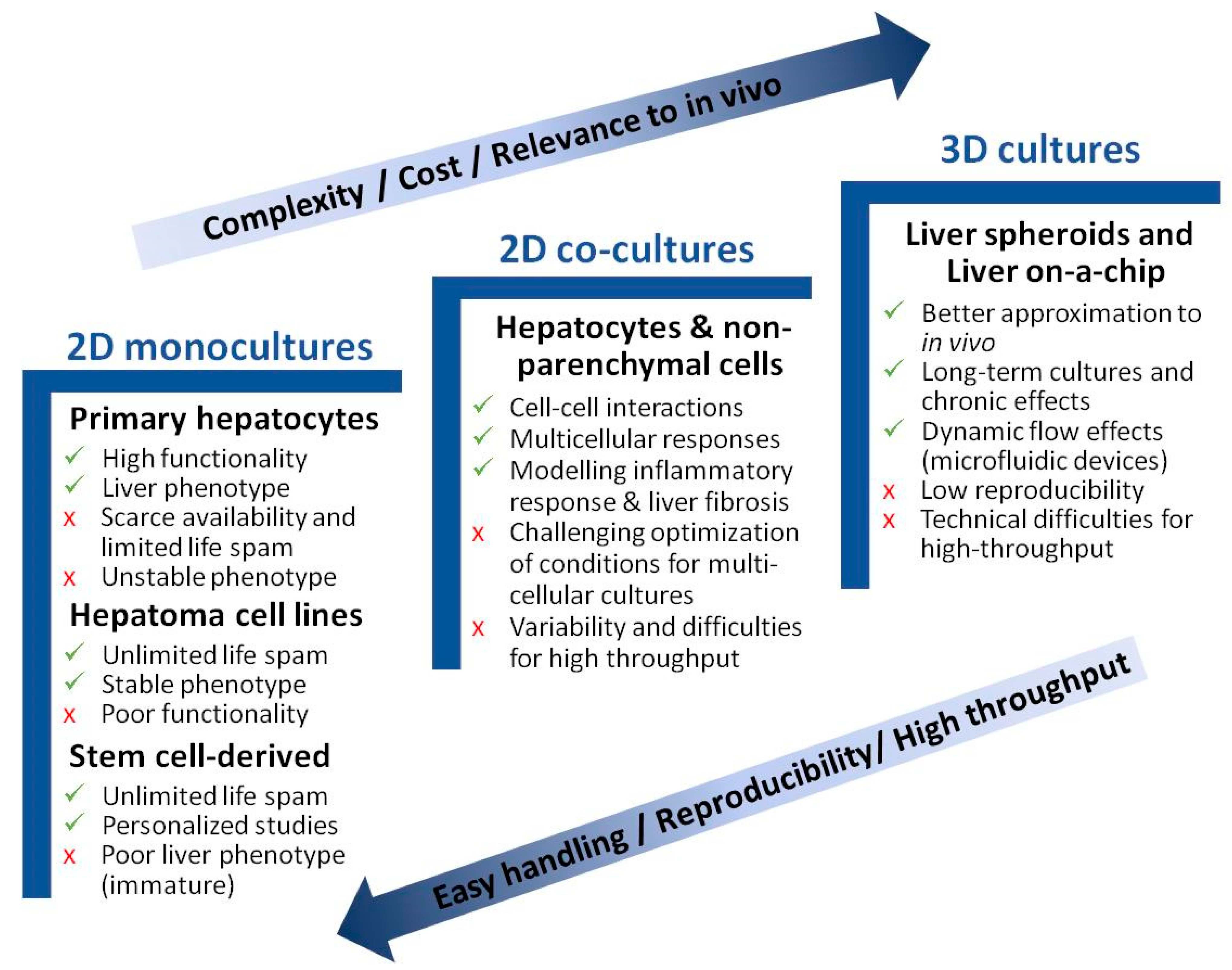
3. Liver Cell Models to Study NAFLD
3.1. Monoculture Models
3.2. Coculture Models
3.3. 3D Models of NAFLD
3.4. Liver-on-a-Chip
4. Human Hepatocyte-like Cells Deriving from Pluripotent Stem Cells for NAFLD Modeling
5. New Approaches and Tools for the In Vitro Assessment of NAFLD
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef]
- Middleton, M.S.; Heba, E.R.; Hooker, C.A.; Bashir, M.R.; Fowler, K.J.; Sandrasegaran, K.; Brunt, E.M.; Kleiner, D.E.; Doo, E.; Van Natta, M.L.; et al. Agreement between Magnetic Resonance Imaging Proton Density Fat Fraction Measurements and Pathologist-Assigned Steatosis Grades of Liver Biopsies From Adults With Nonalcoholic Steatohepatitis. Gastroenterology 2017, 153, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.; Tan, J.; Sun, M.; Hamilton, G.; Bydder, M.; Wolfson, T.; Gamst, A.C.; Middleton, M.; Brunt, E.M.; Loomba, R.; et al. Nonalcoholic fatty liver disease: MR imaging of liver proton density fat fraction to assess hepatic steatosis. Radiology 2013, 267, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Sanchez, G.N.; Dominguez-Perez, M.; Uribe, M.; Chavez-Tapia, N.C.; Nuno-Lambarri, N. Non-alcoholic fatty liver disease and microRNAs expression, how it affects the development and progression of the disease. Ann. Hepatol. 2021, 21, 100212. [Google Scholar] [CrossRef]
- Thiagarajan, P.; Aithal, G.P. Drug Development for Nonalcoholic Fatty Liver Disease: Landscape and Challenges. J. Clin. Exp. Hepatol. 2019, 9, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Suzuki, A. Sexual Dimorphism of NAFLD in Adults. Focus on Clinical Aspects and Implications for Practice and Translational Research. J. Clin. Med. 2020, 9, 1278. [Google Scholar] [CrossRef]
- Lonardo, A.; Arab, J.P.; Arrese, M. Perspectives on Precision Medicine Approaches to NAFLD Diagnosis and Management. Adv. Ther. 2021, 38, 2130–2158. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Patel, P.; Dunn-Valadez, S.; Dao, C.; Khan, V.; Ali, H.; El-Serag, L.; Hernaez, R.; Sisson, A.; Thrift, A.P.; et al. Women Have a Lower Risk of Nonalcoholic Fatty Liver Disease but a Higher Risk of Progression vs Men: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2021, 19, 61–71.e15. [Google Scholar] [CrossRef]
- Liebe, R.; Esposito, I.; Bock, H.H.; Vom Dahl, S.; Stindt, J.; Baumann, U.; Luedde, T.; Keitel, V. Diagnosis and management of secondary causes of steatohepatitis. J. Hepatol. 2021, 74, 1455–1471. [Google Scholar] [CrossRef]
- Miele, L.; Liguori, A.; Marrone, G.; Biolato, M.; Araneo, C.; Vaccaro, F.G.; Gasbarrini, A.; Grieco, A. Fatty liver and drugs: The two sides of the same coin. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 86–94. [Google Scholar] [PubMed]
- Zelber-Sagi, S.; Ratziu, V.; Oren, R. Nutrition and physical activity in NAFLD: An overview of the epidemiological evidence. World J. Gastroenterol. 2011, 17, 3377–3389. [Google Scholar] [CrossRef] [PubMed]
- Drew, L. Drug development: Sprint finish. Nature 2017, 551, S86–S89. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Oseini, A.M.; Cole, B.K.; Issa, D.; Feaver, R.E.; Sanyal, A.J. Translating scientific discovery: The need for preclinical models of nonalcoholic steatohepatitis. Hepatol. Int. 2018, 12, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Arrese, M.; Arab, J.P.; Barrera, F.; Kaufmann, B.; Valenti, L.; Feldstein, A.E. Insights into Nonalcoholic Fatty-Liver Disease Heterogeneity. Semin. Liver Dis. 2021, 41, 421–434. [Google Scholar] [CrossRef]
- Graffmann, N.; Ring, S.; Kawala, M.A.; Wruck, W.; Ncube, A.; Trompeter, H.I.; Adjaye, J. Modeling Nonalcoholic Fatty Liver Disease with Human Pluripotent Stem Cell-Derived Immature Hepatocyte-Like Cells Reveals Activation of PLIN2 and Confirms Regulatory Functions of Peroxisome Proliferator-Activated Receptor Alpha. Stem Cells Dev. 2016, 25, 1119–1133. [Google Scholar] [CrossRef] [Green Version]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef] [PubMed]
- Vatner, D.F.; Majumdar, S.K.; Kumashiro, N.; Petersen, M.C.; Rahimi, Y.; Gattu, A.K.; Bears, M.; Camporez, J.P.; Cline, G.W.; Jurczak, M.J.; et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc. Natl. Acad. Sci. USA 2015, 112, 1143–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, J.; Begriche, K.; Moreau, C.; Fromenty, B. Role of nonalcoholic fatty liver disease as risk factor for drug-induced hepatotoxicity. J. Clin. Transl. Res. 2017, 3, 212–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, C.L.; Steinmetz, F.P.; Cronin, M.T. The identification of nuclear receptors associated with hepatic steatosis to develop and extend adverse outcome pathways. Crit. Rev. Toxicol. 2016, 46, 138–152. [Google Scholar] [CrossRef]
- Hodson, L.; McQuaid, S.E.; Humphreys, S.M.; Milne, R.; Fielding, B.A.; Frayn, K.N.; Karpe, F. Greater dietary fat oxidation in obese compared with lean men: An adaptive mechanism to prevent liver fat accumulation? Am. J. Physiol. Endocrinol. Metab. 2010, 299, E584–E592. [Google Scholar] [CrossRef] [Green Version]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Romano, A.D.; Giudetti, A.M.; Capitanio, N.; Petrella, A.; Vendemiale, G.; Altomare, E. Alterations of hepatic ATP homeostasis and respiratory chain during development of non-alcoholic steatohepatitis in a rodent model. Eur. J. Clin. Investig. 2008, 38, 245–252. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Serviddio, G.; Sastre, J.; Bellanti, F.; Vina, J.; Vendemiale, G.; Altomare, E. Mitochondrial involvement in non-alcoholic steatohepatitis. Mol. Aspects Med. 2008, 29, 22–35. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Wasilewska, N.; Lebensztejn, D.M. Non-alcoholic fatty liver disease and lipotoxicity. Clin. Exp. Hepatol. 2021, 7, 1–6. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Lake, A.D.; Novak, P.; Hardwick, R.N.; Flores-Keown, B.; Zhao, F.; Klimecki, W.T.; Cherrington, N.J. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol. Sci. 2014, 137, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Deng, X.; Liu, Y.; Tan, Q.; Huang, G.; Che, Q.; Guo, J.; Su, Z. Kupffer Cells in Non-alcoholic Fatty Liver Disease: Friend or Foe? Int. J. Biol. Sci. 2020, 16, 2367–2378. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [Green Version]
- Jimenez Ramos, M.; Bandiera, L.; Menolascina, F.; Fallofield, J.A. In vitro models for non-alcoholic fatty liver disease: Emerging platforms and their applications. iScience 2022, 25, 103549. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Bottger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef] [Green Version]
- Heslop, J.A.; Rowe, C.; Walsh, J.; Sison-Young, R.; Jenkins, R.; Kamalian, L.; Kia, R.; Hay, D.; Jones, R.P.; Malik, H.Z.; et al. Mechanistic evaluation of primary human hepatocyte culture using global proteomic analysis reveals a selective dedifferentiation profile. Arch. Toxicol. 2017, 91, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Lechon, M.J.; Donato, M.T.; Castell, J.V.; Jover, R. Human hepatocytes as a tool for studying toxicity and drug metabolism. Curr. Drug Metab. 2003, 4, 292–312. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Rennert, C.; Heil, T.; Schicht, G.; Stilkerich, A.; Seidemann, L.; Kegel-Hubner, V.; Seehofer, D.; Damm, G. Prolonged Lipid Accumulation in Cultured Primary Human Hepatocytes Rather Leads to ER Stress than Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 7097. [Google Scholar] [CrossRef]
- Brown, M.V.; Compton, S.A.; Milburn, M.V.; Lawton, K.A.; Cheatham, B. Metabolomic signatures in lipid-loaded HepaRGs reveal pathways involved in steatotic progression. Obesity 2013, 21, E561–E570. [Google Scholar] [CrossRef]
- Chavez-Tapia, N.C.; Rosso, N.; Tiribelli, C. Effect of intracellular lipid accumulation in a new model of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Jiang, Y.; Wang, Y.; An, W. Suppression of ABCA1 by unsaturated fatty acids leads to lipid accumulation in HepG2 cells. Biochimie 2010, 92, 958–963. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, L.; Hu, W.; Zheng, Q.; Xiang, W. Mitochondrial dysfunction during in vitro hepatocyte steatosis is reversed by omega-3 fatty acid-induced up-regulation of mitofusin 2. Metabolism 2011, 60, 767–775. [Google Scholar] [CrossRef]
- Antherieu, S.; Rogue, A.; Fromenty, B.; Guillouzo, A.; Robin, M.A. Induction of vesicular steatosis by amiodarone and tetracycline is associated with up-regulation of lipogenic genes in HepaRG cells. Hepatology 2011, 53, 1895–1905. [Google Scholar] [CrossRef]
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of bisphenol A as environmental factor in the promotion of non-alcoholic fatty liver disease: In vitro and clinical study. Aliment Pharmacol. Ther. 2018, 47, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yu, P.; Xie, X.; Wu, L.; Zhou, M.; Huan, F.; Jiang, L.; Gao, R. Bisphenol F induces nonalcoholic fatty liver disease-like changes: Involvement of lysosome disorder in lipid droplet deposition. Environ. Pollut. 2021, 271, 116304. [Google Scholar] [CrossRef]
- Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Negro, A.; Herrera, G.; Castell, J.V.; O’Connor, J.E.; Gomez-Lechon, M.J. Cytometric analysis for drug-induced steatosis in HepG2 cells. Chem. Biol. Interact 2009, 181, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Tolosa, L.; Jimenez, N.; Castell, J.V.; Gomez-Lechon, M.J. High-content imaging technology for the evaluation of drug-induced steatosis using a multiparametric cell-based assay. J. Biomol. Screen 2012, 17, 394–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wandrer, F.; Frangez, Z.; Liebig, S.; John, K.; Vondran, F.; Wedemeyer, H.; Veltmann, C.; Pfeffer, T.J.; Shibolet, O.; Schulze-Osthoff, K.; et al. Autophagy alleviates amiodarone-induced hepatotoxicity. Arch. Toxicol. 2020, 94, 3527–3539. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, Y.; Chen, X.; Zhao, L.; Guo, Y. Involvement of CYP2E1-ROS-CD36/DGAT2 axis in the pathogenesis of VPA-induced hepatic steatosis in vivo and in vitro. Toxicology 2020, 445, 152585. [Google Scholar] [CrossRef]
- Tolosa, L.; Gomez-Lechon, M.J.; Jimenez, N.; Hervas, D.; Jover, R.; Donato, M.T. Advantageous use of HepaRG cells for the screening and mechanistic study of drug-induced steatosis. Toxicol. Appl. Pharmacol. 2016, 302, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kanebratt, K.P.; Andersson, T.B. HepaRG cells as an in vitro model for evaluation of cytochrome P450 induction in humans. Drug Metab. Dispos. 2008, 36, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Allard, J.; Bucher, S.; Massart, J.; Ferron, P.J.; Le Guillou, D.; Loyant, R.; Daniel, Y.; Launay, Y.; Buron, N.; Begriche, K.; et al. Drug-induced hepatic steatosis in absence of severe mitochondrial dysfunction in HepaRG cells: Proof of multiple mechanism-based toxicity. Cell Biol. Toxicol. 2021, 37, 151–175. [Google Scholar] [CrossRef]
- Saito, J.; Okamura, A.; Takeuchi, K.; Hanioka, K.; Okada, A.; Ohata, T. High content analysis assay for prediction of human hepatotoxicity in HepaRG and HepG2 cells. Toxicol. Vitro 2016, 33, 63–70. [Google Scholar] [CrossRef]
- Barbero-Becerra, V.J.; Giraudi, P.J.; Chavez-Tapia, N.C.; Uribe, M.; Tiribelli, C.; Rosso, N. The interplay between hepatic stellate cells and hepatocytes in an in vitro model of NASH. Toxicol. In Vitro 2015, 29, 1753–1758. [Google Scholar] [CrossRef]
- Yu, H.; Jiang, X.; Dong, F.; Zhang, F.; Ji, X.; Xue, M.; Yang, F.; Chen, J.; Hu, X.; Bao, Z. Lipid accumulation-induced hepatocyte senescence regulates the activation of hepatic stellate cells through the Nrf2-antioxidant response element pathway. Exp. Cell Res. 2021, 405, 112689. [Google Scholar] [CrossRef]
- Cole, B.K.; Feaver, R.E.; Wamhoff, B.R.; Dash, A. Non-alcoholic fatty liver disease (NAFLD) models in drug discovery. Expert Opin. Drug Discov. 2018, 13, 193–205. [Google Scholar] [CrossRef]
- Zhou, Z.; Qi, J.; Lim, C.W.; Kim, J.W.; Kim, B. Dual TBK1/IKKepsilon inhibitor amlexanox mitigates palmitic acid-induced hepatotoxicity and lipoapoptosis in vitro. Toxicology 2020, 444, 152579. [Google Scholar] [CrossRef]
- Orbach, S.M.; Ehrich, M.F.; Rajagopalan, P. High-throughput toxicity testing of chemicals and mixtures in organotypic multi-cellular cultures of primary human hepatic cells. Toxicol. In Vitro 2018, 51, 83–94. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Hendriks, D.F.; Bell, C.C.; Andersson, T.B.; Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chem. Res. Toxicol. 2016, 29, 1936–1955. [Google Scholar] [CrossRef]
- Liu, D.; Chen, S.; Win Naing, M. A review of manufacturing capabilities of cell spheroid generation technologies and future development. Biotechnol. Bioeng. 2021, 118, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Johansson, I.; Nordling, A.; Ullah, S.; Lauschke, V.M.; Ingelman-Sundberg, M. Human hepatic 3D spheroids as a model for steatosis and insulin resistance. Sci. Rep. 2018, 8, 14297. [Google Scholar] [CrossRef] [Green Version]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Linden, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Zhelnin, L.; Sanfiz, A.; Pan, J.; Li, Z.; Yarde, M.; McCarty, J.; Jarai, G. Development and validation of an in vitro 3D model of NASH with severe fibrotic phenotype. Am. J. Transl. Res. 2019, 11, 1531–1540. [Google Scholar] [PubMed]
- Jiang, J.; Messner, S.; Kelm, J.M.; van Herwijnen, M.; Jennen, D.G.J.; Kleinjans, J.C.; de Kok, T.M. Human 3D multicellular microtissues: An upgraded model for the in vitro mechanistic investigation of inflammation-associated drug toxicity. Toxicol. Lett. 2019, 312, 34–44. [Google Scholar] [CrossRef]
- Rowe, C.; Gerrard, D.T.; Jenkins, R.; Berry, A.; Durkin, K.; Sundstrom, L.; Goldring, C.E.; Park, B.K.; Kitteringham, N.R.; Hanley, K.P.; et al. Proteome-wide analyses of human hepatocytes during differentiation and dedifferentiation. Hepatology 2013, 58, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Feaver, R.E.; Cole, B.K.; Lawson, M.J.; Hoang, S.A.; Marukian, S.; Blackman, B.R.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R.; Dash, A. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 2016, 1, e90954. [Google Scholar] [CrossRef]
- Duriez, M.; Jacquet, A.; Hoet, L.; Roche, S.; Bock, M.D.; Rocher, C.; Haussy, G.; Vige, X.; Bocskei, Z.; Slavnic, T.; et al. A 3D Human Liver Model of Nonalcoholic Steatohepatitis. J. Clin. Transl. Hepatol. 2020, 8, 359–370. [Google Scholar] [CrossRef]
- Banaeiyan, A.A.; Theobald, J.; Paukstyte, J.; Wolfl, S.; Adiels, C.B.; Goksor, M. Design and fabrication of a scalable liver-lobule-on-a-chip microphysiological platform. Biofabrication 2017, 9, 015014. [Google Scholar] [CrossRef]
- Knowlton, S.; Tasoglu, S. A Bioprinted Liver-on-a-Chip for Drug Screening Applications. Trends Biotechnol. 2016, 34, 681–682. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Deng, P.; Tao, T.; Liu, H.; Wu, S.; Chen, W.; Qin, J. Modeling Human Nonalcoholic Fatty Liver Disease (NAFLD) with an Organoids-on-a-Chip System. ACS Biomater. Sci. Eng. 2020, 6, 5734–5743. [Google Scholar] [CrossRef]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating Nonalcoholic Fatty Liver Disease in a Liver-on-a-Chip Microfluidic Device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef]
- Gori, M.; Giannitelli, S.M.; Zancla, A.; Mozetic, P.; Trombetta, M.; Merendino, N.; Rainer, A. Quercetin and hydroxytyrosol as modulators of hepatic steatosis: A NAFLD-on-a-chip study. Biotechnol. Bioeng. 2021, 118, 142–152. [Google Scholar] [CrossRef]
- Freag, M.S.; Namgung, B.; Reyna Fernandez, M.E.; Gherardi, E.; Sengupta, S.; Jang, H.L. Human Nonalcoholic Steatohepatitis on a Chip. Hepatol. Commun. 2021, 5, 217–233. [Google Scholar] [CrossRef]
- Lee, S.Y.; Sung, J.H. Gut-liver on a chip toward an in vitro model of hepatic steatosis. Biotechnol. Bioeng. 2018, 115, 2817–2827. [Google Scholar] [CrossRef]
- Slaughter, V.L.; Rumsey, J.W.; Boone, R.; Malik, D.; Cai, Y.; Sriram, N.N.; Long, C.J.; McAleer, C.W.; Lambert, S.; Shuler, M.L.; et al. Validation of an adipose-liver human-on-a-chip model of NAFLD for preclinical therapeutic efficacy evaluation. Sci. Rep. 2021, 11, 13159. [Google Scholar] [CrossRef]
- Wang, X.H.; Tian, Y.; Guo, Z.J.; Fan, Z.P.; Qiu, D.K.; Zeng, M.D. Cholesterol metabolism and expression of its relevant genes in cultured steatotic hepatocytes. J. Dig. Dis. 2009, 10, 310–314. [Google Scholar] [CrossRef]
- Tolosa, L.; Gomez-Lechon, M.J.; Lopez, S.; Guzman, C.; Castell, J.V.; Donato, M.T.; Jover, R. Human Upcyte Hepatocytes: Characterization of the Hepatic Phenotype and Evaluation for Acute and Long-Term Hepatotoxicity Routine Testing. Toxicol. Sci. 2016, 152, 214–229. [Google Scholar] [CrossRef] [Green Version]
- Chagastelles, P.C.; Nardi, N.B. Biology of stem cells: An overview. Kidney Int. Suppl. 2011, 1, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Larsen, L.E.; Smith, M.A.; Abbey, D.; Korn, A.; Reeskamp, L.F.; Hand, N.J.; Holleboom, A.G. Hepatocyte-like cells derived from induced pluripotent stem cells: A versatile tool to understand lipid disorders. Atherosclerosis 2020, 303, 8–14. [Google Scholar] [CrossRef]
- Mannino, G.; Russo, C.; Maugeri, G.; Musumeci, G.; Vicario, N.; Tibullo, D.; Giuffrida, R.; Parenti, R.; Lo Furno, D. Adult stem cell niches for tissue homeostasis. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef]
- Lee, K.D.; Kuo, T.K.; Whang-Peng, J.; Chung, Y.F.; Lin, C.T.; Chou, S.H.; Chen, J.R.; Chen, Y.P.; Lee, O.K. In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology 2004, 40, 1275–1284. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; De Kock, J.; Branson, S.; Vinken, M.; Meganathan, K.; Chaudhari, U.; Sachinidis, A.; Govaere, O.; Roskams, T.; De Boe, V.; et al. Human skin-derived stem cells as a novel cell source for in vitro hepatotoxicity screening of pharmaceuticals. Stem Cells Dev. 2014, 23, 44–55. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; Branson, S.; De Boe, V.; Sachinidis, A.; Rogiers, V.; De Kock, J.; Vanhaecke, T. In vitro assessment of drug-induced liver steatosis based on human dermal stem cell-derived hepatic cells. Arch. Toxicol. 2016, 90, 677–689. [Google Scholar] [CrossRef]
- Boeckmans, J.; Buyl, K.; Natale, A.; Vandenbempt, V.; Branson, S.; De Boe, V.; Rogiers, V.; De Kock, J.; Rodrigues, R.M.; Vanhaecke, T. Elafibranor restricts lipogenic and inflammatory responses in a human skin stem cell-derived model of NASH. Pharmacol. Res. 2019, 144, 377–389. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Vanhaecke, T.; Rogiers, V.; Rodrigues, R.M.; De Kock, J. Flow cytometric quantification of neutral lipids in a human skin stem cell-derived model of NASH. MethodsX 2020, 7, 101068. [Google Scholar] [CrossRef]
- Roh, J.K.; Jung, K.H.; Chu, K. Adult stem cell transplantation in stroke: Its limitations and prospects. Curr. Stem Cell Res. Ther. 2008, 3, 185–196. [Google Scholar] [CrossRef]
- Saito, Y.; Ikemoto, T.; Morine, Y.; Shimada, M. Current status of hepatocyte-like cell therapy from stem cells. Surg. Today 2021, 51, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Collin de l’Hortet, A.; Takeishi, K.; Guzman-Lepe, J.; Morita, K.; Achreja, A.; Popovic, B.; Wang, Y.; Handa, K.; Mittal, A.; Meurs, N.; et al. Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab. 2019, 30, 385–401.e9. [Google Scholar] [CrossRef]
- Gurevich, I.; Burton, S.A.; Munn, C.; Ohshima, M.; Goedland, M.E.; Czysz, K.; Rajesh, D. iPSC-derived hepatocytes generated from NASH donors provide a valuable platform for disease modeling and drug discovery. Biol. Open 2020, 9, bio055087. [Google Scholar] [CrossRef] [PubMed]
- Parafati, M.; Kirby, R.J.; Khorasanizadeh, S.; Rastinejad, F.; Malany, S. A nonalcoholic fatty liver disease model in human induced pluripotent stem cell-derived hepatocytes, created by endoplasmic reticulum stress-induced steatosis. Dis. Model Mech. 2018, 11, dmm033530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486.e12. [Google Scholar] [CrossRef] [PubMed]
- Tilson, S.G.; Morell, C.M.; Lenaerts, A.S.; Park, S.B.; Hu, Z.; Jenkins, B.; Koulman, A.; Liang, T.J.; Vallier, L. Modeling PNPLA3-Associated NAFLD Using Human-Induced Pluripotent Stem Cells. Hepatology 2021, 74, 2998–3017. [Google Scholar] [CrossRef]
- Lyall, M.J.; Cartier, J.; Thomson, J.P.; Cameron, K.; Meseguer-Ripolles, J.; O’Duibhir, E.; Szkolnicka, D.; Villarin, B.L.; Wang, Y.; Blanco, G.R.; et al. Modelling non-alcoholic fatty liver disease in human hepatocyte-like cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170362. [Google Scholar] [CrossRef] [Green Version]
- Pradip, A.; Steel, D.; Jacobsson, S.; Holmgren, G.; Ingelman-Sundberg, M.; Sartipy, P.; Bjorquist, P.; Johansson, I.; Edsbagge, J. High Content Analysis of Human Pluripotent Stem Cell Derived Hepatocytes Reveals Drug Induced Steatosis and Phospholipidosis. Stem Cells Int. 2016, 2016, 2475631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, M.T.; Tolosa, L. Stem-cell derived hepatocyte-like cells for the assessment of drug-induced liver injury. Differentiation 2019, 106, 15–22. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Graffmann, N.; Bohndorf, M.; Ncube, A.; Kawala, M.A.; Wruck, W.; Kashofer, K.; Zatloukal, K.; Adjaye, J. Establishment and characterization of an iPSC line from a 35 years old high grade patient with nonalcoholic fatty liver disease (30–40% steatosis) with homozygous wildtype PNPLA3 genotype. Stem Cell Res. 2018, 31, 113–116. [Google Scholar] [CrossRef]
- Graffmann, N.; Bohndorf, M.; Ncube, A.; Wruck, W.; Kashofer, K.; Zatloukal, K.; Adjaye, J. Establishment and characterization of an iPSC line from a 58 years old high grade patient with nonalcoholic fatty liver disease (70% steatosis) with homozygous wildtype PNPLA3 genotype. Stem Cell Res. 2018, 31, 131–134. [Google Scholar] [CrossRef]
- Graffmann, N.; Ncube, A.; Martins, S.; Fiszl, A.R.; Reuther, P.; Bohndorf, M.; Wruck, W.; Beller, M.; Czekelius, C.; Adjaye, J. A stem cell based in vitro model of NAFLD enables the analysis of patient specific individual metabolic adaptations in response to a high fat diet and AdipoRon interference. Biol. Open 2021, 10, bio054189. [Google Scholar] [CrossRef]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [Green Version]
- Kammerer, S. Three-Dimensional Liver Culture Systems to Maintain Primary Hepatic Properties for Toxicological Analysis In Vitro. Int. J. Mol. Sci. 2021, 22, 214. [Google Scholar] [CrossRef] [PubMed]
- Segovia-Miranda, F.; Morales-Navarrete, H.; Kucken, M.; Moser, V.; Seifert, S.; Repnik, U.; Rost, F.; Brosch, M.; Hendricks, A.; Hinz, S.; et al. Three-dimensional spatially resolved geometrical and functional models of human liver tissue reveal new aspects of NAFLD progression. Nat. Med. 2019, 25, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.; Withey, S.; Harrison, S.; Segeritz, C.P.; Zhang, F.; Atkinson-Dell, R.; Rowe, C.; Gerrard, D.T.; Sison-Young, R.; Jenkins, R.; et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015, 62, 581–589. [Google Scholar] [CrossRef]
- Schwartz, R.E.; Fleming, H.E.; Khetani, S.R.; Bhatia, S.N. Pluripotent stem cell-derived hepatocyte-like cells. Biotechnol. Adv. 2014, 32, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parlati, L.; Regnier, M.; Guillou, H.; Postic, C. New targets for NAFLD. JHEP Rep. 2021, 3, 100346. [Google Scholar] [CrossRef]
- Taliento, A.E.; Dallio, M.; Federico, A.; Prati, D.; Valenti, L. Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 2755. [Google Scholar] [CrossRef] [Green Version]
- Bell, L.N.; Theodorakis, J.L.; Vuppalanchi, R.; Saxena, R.; Bemis, K.G.; Wang, M.; Chalasani, N. Serum proteomics and biomarker discovery across the spectrum of nonalcoholic fatty liver disease. Hepatology 2010, 51, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, J.; Caballeria, J.; Martinez-Arranz, I.; Dominguez-Diez, A.; Alonso, C.; Muntane, J.; Perez-Cormenzana, M.; Garcia-Monzon, C.; Mayo, R.; Martin-Duce, A.; et al. Obesity-dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J. Proteome Res. 2012, 11, 2521–2532. [Google Scholar] [CrossRef]
- Garcia-Canaveras, J.C.; Donato, M.T.; Castell, J.V.; Lahoz, A. A comprehensive untargeted metabonomic analysis of human steatotic liver tissue by RP and HILIC chromatography coupled to mass spectrometry reveals important metabolic alterations. J. Proteome Res. 2011, 10, 4825–4834. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Yamaguchi, K.; Itoh, Y. The genetic backgrounds in nonalcoholic fatty liver disease. Clin. J. Gastroenterol. 2018, 11, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Z.; Zou, W.; Hong, H.; Fang, H.; Tong, W. Molecular regulation of miRNAs and potential biomarkers in the progression of hepatic steatosis to NASH. Biomark. Med. 2015, 9, 1189–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, E.; Helbig, K.J.; Van der Hoek, K.; McCartney, E.M.; Van der Hoek, M.; George, J.; Beard, M.R. Fatty Acids Induce a Pro-Inflammatory Gene Expression Profile in Huh-7 Cells That Attenuates the Anti-HCV Action of Interferon. J. Interferon Cytokine Res. 2015, 35, 392–400. [Google Scholar] [CrossRef]
- Hetherington, A.M.; Sawyez, C.G.; Zilberman, E.; Stoianov, A.M.; Robson, D.L.; Borradaile, N.M. Differential Lipotoxic Effects of Palmitate and Oleate in Activated Human Hepatic Stellate Cells and Epithelial Hepatoma Cells. Cell Physiol. Biochem. 2016, 39, 1648–1662. [Google Scholar] [CrossRef] [PubMed]
- Breher-Esch, S.; Sahini, N.; Trincone, A.; Wallstab, C.; Borlak, J. Genomics of lipid-laden human hepatocyte cultures enables drug target screening for the treatment of non-alcoholic fatty liver disease. BMC Med. Genom. 2018, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Atanasovska, B.; Rensen, S.S.; Marsman, G.; Shiri-Sverdlov, R.; Withoff, S.; Kuipers, F.; Wijmenga, C.; van de Sluis, B.; Fu, J. Long Non-Coding RNAs Involved in Progression of Non-Alcoholic Fatty Liver Disease to Steatohepatitis. Cells 2021, 10, 1883. [Google Scholar] [CrossRef]
- Errafii, K.; Al-Akl, N.S.; Khalifa, O.; Arredouani, A. Comprehensive analysis of LncRNAs expression profiles in an in vitro model of steatosis treated with Exendin-4. J. Transl. Med. 2021, 19, 235. [Google Scholar] [CrossRef]
- Lockman, K.A.; Htun, V.; Sinha, R.; Treskes, P.; Nelson, L.J.; Martin, S.F.; Rogers, S.M.; Le Bihan, T.; Hayes, P.C.; Plevris, J.N. Proteomic profiling of cellular steatosis with concomitant oxidative stress in vitro. Lipids Health Dis. 2016, 15, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chienwichai, P.; Reamtong, O.; Boonyuen, U.; Pisitkun, T.; Somparn, P.; Tharnpoophasiam, P.; Worakhunpiset, S.; Topanurak, S. Hepatic protein Carbonylation profiles induced by lipid accumulation and oxidative stress for investigating cellular response to non-alcoholic fatty liver disease in vitro. Proteome Sci. 2019, 17, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuykx, M.; Claes, L.; Rodrigues, R.M.; Vanhaecke, T.; Covaci, A. Metabolomics profiling of steatosis progression in HepaRG. Toxicol. Lett. 2018, 286, 22–30. [Google Scholar] [CrossRef] [PubMed]
- García-Cañaveras, J.C.; Peris-Díaz, M.D.; Alcoriza-Balaguer, M.I.; Cerdán-Calero, M.; Donato, M.T.; Lahoz, A. A lipidomic cell-based assay for studying drug-induced phospholipidosis and steatosis. Electrophoresis 2017, 38, 2331–2340. [Google Scholar] [CrossRef]
- Yeung, E.N.; Treskes, P.; Martin, S.F.; Manning, J.R.; Dunbar, D.R.; Rogers, S.M.; Le Bihan, T.; Lockman, K.A.; Morley, S.D.; Hayes, P.C.; et al. Fibrinogen production is enhanced in an in-vitro model of non-alcoholic fatty liver disease: An isolated risk factor for cardiovascular events? Lipids Health Dis. 2015, 14, 86. [Google Scholar] [CrossRef] [Green Version]
- Gunn, P.J.; Pramfalk, C.; Millar, V.; Cornfield, T.; Hutchinson, M.; Johnson, E.M.; Nagarajan, S.R.; Troncoso-Rey, P.; Mithen, R.F.; Pinnick, K.E.; et al. Modifying nutritional substrates induces macrovesicular lipid droplet accumulation and metabolic alterations in a cellular model of hepatic steatosis. Physiol. Rep. 2020, 8, e14482. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Freeman, B.L.; Bronk, S.F.; LeBrasseur, N.K.; White, T.A.; Hirsova, P.; Ibrahim, S.H. CXCL10-Mediates Macrophage, but not Other Innate Immune Cells-Associated Inflammation in Murine Nonalcoholic Steatohepatitis. Sci. Rep. 2016, 6, 28786. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shen, J.; Man, K.; Chu, E.S.; Yau, T.O.; Sung, J.C.; Go, M.Y.; Deng, J.; Lu, L.; Wong, V.W.; et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J. Hepatol. 2014, 61, 1365–1375. [Google Scholar] [CrossRef] [Green Version]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Donato, M.T.; Gomez-Lechon, M.J. Drug-induced liver steatosis and phospholipidosis: Cell-based assays for early screening of drug candidates. Curr. Drug Metab. 2012, 13, 1160–1173. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cellular System | NAFLD Induction | NAFLD Outcome | Observations | Ref. |
|---|---|---|---|---|
| 2D Monocultures | ||||
| PHH | NEFA | Steatosis, ER stress | Lipid accumulation, apoptosis, activation of autophagy (IRE1a), and lipid metabolism (ATF6a) | [41,42] |
| HuH7 | NEFA | Steatosis, oxidative stress, inflammation | Lipid accumulation, apoptosis, expression IL-6, IL-8, TNFα, increased ROS, TGFB-1, TGFB -2, VEGF2 | [44] |
| HepG2 | NEFA | Steatosis, oxidative stress | Lipid accumulation, increased ROS, mitochondria changes (ATP levels, mitofusin-2 expression), impaired cholesterol efflux, and ABCA1 expression | [45,46] |
| Endocrine disruptors | Steatosis, oxidative stress, lipoperoxidation, blocking autophagy | Lipid accumulation, TBARS expression, accumulation of autophagosomes, decreased SQSTM1/p62 degradation | [48,49] | |
| Drugs | Steatosis, oxidative stress, blocking autophagy | Lipid accumulation, increased lipogenesis (SREBP1c) and triglyceride formation (DGAT1), ROS generation, decreased SQSTM1/p62 degradation | [50,51,52] | |
| L02 | NEFA | Steatosis | Lipid accumulation, up-regulation of relevant cholesterol synthesis genes | [80] |
| Valproic acid | Steatosis, oxidative stress | Lipid accumulation, decreased GSH level, increased MDA and ROS levels | [53] | |
| HepaRG | Drugs | Steatosis | Decreased β oxidation, expression of enzymes involved in lipogenesis or decreased proteins involved in VLDL secretion | [56] |
| Upcytes | Drugs | Steatosis, oxidative stress | Lipid accumulation, decreased FOXA1 expression, increased ROS | [81] |
| 2D cocultures | ||||
| HuH7 and LX2 | NEFA | Steatosis, HSCs activation | Lipid accumulation, α-SMA expression | [58] |
| AML12 and HSC | NEFA | Steatosis, oxidative stress, HSCs activation | Lipid accumulation, ROS induction, decreased CAT, SOD, and GPx, expression of profibrotic molecules (α-SMA, Col I, MMP-2, MMP-9, fibronectin) | [59] |
| PHH and KC | NEFA | Steatosis, inflammation | Lipid accumulation, expression of lipogenesis enzymes (FASN, SREBP1c),expression of TNFα, IL-1β, IL6 | [61] |
| 3D models | ||||
| PHH | NEFA and insulin | Steatosis, insulin resistance | Lipid accumulation, increased expression of PCK1 and PDK4, and reduced GSK3β phosphorylation | [65] |
| HepG2 and LX2 | NEFA | Steatosis, fibrosis | Lipid accumulation, Col1A1 expression | [66] |
| 3D InSightTM | NEFA | Fibrosis, inflammation | Expression of collagen genes, fibronectin, α-SMA, IL-8 expression | [67] |
| PHH, HSC and macrophages | NEFA, insulin and glucose | Steatosis, insulin resistance, inflammation, fibrosis | Lipid accumulation, increased TAG, DAG and CE and PCK1 expression, reduced Akt phosphorylation, expression of IL-8, IL-6, and CXCL10, expression of TGF-β, OPN and α-SMA | [70] |
| PHH, HSC, LEC and KC | NEFA, TNFα and glucose | Steatosis, inflammation, fibrosis | Lipid accumulation, expression of IL-6, CXCL8, CXCL10, expression of MMP2 and MMP9 | [71] |
| Liver-on-a-chip | ||||
| HepG2 | NEFA | Steatosis | Lipid accumulation, increased TAG | [75] |
| PHH, HSC, KC, and LSEC | NEFA and LPS | Steatosis, liver injury, fibrosis, inflammation | Lipid accumulation, ballooned hepatocytes, increased Caspase 3, expression of α-sma, col1a, timp-1, tgf-β and opn, increases in tnf-α, mip1a, and mcp1 | [77] |
| HepG2 and gut cells | NEFA | Steatosis | Lipid accumulation | [78] |
| Cell Model | HLCs Characterization | NAFLD Induction and Model Observations | Ref. |
|---|---|---|---|
| ESC line H1 and iPSCs from healthy donors | Polygonal shape, alb+, ecad+, hnf-4α+, urea synthesis, CYP3A4/3A5/3A7 activities, release of indocyanine dye | NEFA for 48 h. Lipid accumulation. Up-regulation of lipid metabolism regulators PPARα and PLIN2. Down-regulation of certain microRNAs (microRNA hsa-miR-122 and hsa-miR-106b). | [19] |
| HLCs from NAFLD donors (distinct grades of steatosis) | Polygonal shape, alb+, afp+, ecad+, hnf-4α+, a1at+, and ttr; low CYP3A4 expression | NEFA for several days. Lipid accumulation with a donor-specific pattern. Increased PLIN2 expression with differences between donors. Low expression of the genes associated with FGF21 signaling, lipid and cholesterol biosynthesis, and gluconeogenesis, with a low expression of CPT1A in high steatosis lines. AdipoRon effect on metabolism, transport, and signaling pathways. | [103] |
| iPSCs-Hep from female healthy donors | Polygonal shape, alb+, afp+, pou5f1+, hnf-4α+, expression of phase I, II, and III enzymes | NEFA and TAG for 18 h. PA dose-dependent lipid accumulation. Lipid accumulation exacerbated by TAG treatment, which induces ER stress by UPR dysregulation. Up-regulation of the genes related in fat storage in lipid droplets. Down-regulation of β-oxidation genes (ACADM) with FA-TAG treatment. Reduced TAG accumulation after inhibition of ER stress by therapeutic molecules tauroursodeoxycholic acid or obeticholic acid. | [94] |
| iPSC lines FSPS13B and A1ATDR/R obtained by CRISPR/Cas9 technology | Polygonal shape in HLCs alb+, a1at+, hnf-4α+ Detected CYP3A4 activity | NEFA for 7 days. HLCs-PNPLA3KO and HLC-PNPLA3I148M do not activate UPR markers (BIP, GADD34, CHOP, and PERK) with PA treatment, which indicates lipid-associated ER stress alterations. Lower levels of several β-oxidation gene expressions. Down-regulation of the genes implicated in drug detoxification, glucose metabolism and cell stress, but sensitivity to insulin remaining. HLCs-PNPLA3I148M recapitulate the main PNPLA3-associated NAFLD features. | [96] |
| Female H9 ESCs | Polygonal shape, alb+, hnf-4α+ Detected secreted albumin and CYP1A2 and CYP3A4 activities | Lactate, pyruvate and octanoate for 48 h or 96 h. Increased lipid accumulation. Tricarboxylic acid cycle dysregulation and altered expression of related enzymes. Alteration of β-oxidation and oxidative phosphorylation. Up-regulation of lipid vesicle transport proteins PLIN1, PLIN2 or APOA4, and gluconeogenesis genes. Transcriptional dysregulation in insulin resistance mediators. Induction of oxidative stress. | [97] |
| iPSCs from NASH and healthy patients | Polygonal shape with bile canaliculi formation. Hepatic markers (aat+, alb+), mRNA expressions of CYP3A4, CYP3A7, SERPINA, ASGR1, and ALB | NEFA for 24 h. Increased dose-dependent intracellular lipid accumulation. NAFLD patient derived HLCs organoids spontaneously accumulate lipids. HLCs were able to successfully integrate into 3D liver organoids with macrophages, MSCs and endothelial cells allowed NAFLD to be more accurately modeled. | [93] |
| 3D coculture (HLCs and other cells in decellularized rat liver) | Polygonal shape, alb+, hnf-4α+ mRNA expressions of the enzymes, transporters, hepatic nuclear receptors, and transcription factors involved in liver metabolism | Linoleic acid and OA in the presence of DOX for 2–4 days. Increased lipid accumulation, lipid peroxidation levels, and total cholesterol levels. Increased mRNA expression of the genes implicated in de novo lipogenesis (SREBP1c). Decreased mRNA expression of β-oxidation key modulators (PPARα and PGC1α). Bioengineered tissue exhibits steatosis and expresses pro-inflammatory markers. | [92] |
| 3D organoids (HLCs and CLCs deriving from PSCs) | alb+ (HLCs), ck7+ (CLCs), mRNA expression of proteins related to bile acid synthesis/secretion, cholesterol, fat and carbohydrate metabolism, drug detoxification, and hepatic-specific transcription factors | NEFA exposure. Increased lipid accumulation, ROS levels, and lipid peroxidation. Increased expression of the genes related to lipid and carbohydrate metabolism. Bile canaliculi network disruption. | [95] |
| Omics/ Technique | Cell Model (NAFLD Induction) | Observations | Ref. |
|---|---|---|---|
| Transcriptomics | |||
| Microarray | HuH7 (NEFA) | Increased expression of interferon-stimulated genes and NF-kB-dependent pro-inflammatory genes | [116] |
| HLCs and HepG2 (NEFA) | Increase in the PPAR pathway genes and Perilipin-2 | [19] | |
| HepG2 and HSCs (NEFA) | Up-regulation in the ER-stress pathway genes | [117] | |
| PHH, HepG2, and HuH7 (NEFA and TNFα) | Comparison of different test systems. Changes in the genes linked with lipid droplet formation and metabolism (i.e., HSDL2) | [118] | |
| HLCs treated (NEFA, TNFα, IL1β, glucose, Insulin, and TGF1β) | Testing the anti-NASH compound (elafibranor). Gene expression profile and inflammatory markers of NASH | [88] | |
| RNASeq | 3D cocultures of PHH, HSCs, KC, and LSEC (NEFA, glucose, and TNFα) | Time course effects (3, 8, 10 days). 468 differentially expressed genes related to immune cell adhesion and inflammatory pathways | [71] |
| HepG2 (NEFA & TNFα) | Evaluation of lncRNAs profiling in a model of steatohepatitis | [119] | |
| HepG2 (NEFA) | Differential expression of lncRNAs in untreated and steatotic cells with and without treatment with exendin-4 | [120] | |
| Proteomics | |||
| HPLC-MS | C3A cells (lactate, pyruvate, octanoate, and ammonia) | 104 differentially expressed proteins as indicators of enhanced protein synthesis accompanied by a down-regulation of histones | [121] |
| NLC-MS | HepG2 (NEFA, and menadione) | Identification of the differentially expressed carbonylated proteins (i.e., ATP5A) in NASH | [122] |
| Metabolomics | |||
| GC-MS, UHPLC-MS | HepaRG (NEFA) | Global metabolomic analysis. Increased levels of branched chain amino acids and TCA cycle intermediates. Reduced carnitine and GSH levels | [43] |
| HPLC-MS | HepaRG (valproic acid) | Exposure to different concentrations and exposure times of VPA resulted in the identification of a typical steatotic profile: decreased carnitine, SAMe, and PEs in combination with the up-regulation of neutral heavy chain lipids | [123] |
| HPLC-MS | 3D PHH spheroids (NEFA, insulin, glucose, and fructose) | Identification of the metabolites up-regulated in steatosis after 7 and 21 days of treatment. Study of the response to drug treatments | [65] |
| HPLC-MS | HepG2 (NEFA and drugs) | Identification of phospholipidosis- and steatosis-specific metabolites (NEFA, acylcarnitines, monoacylglycerides, diacylglycerides, and TAG) after incubation with phospholipidogenic and steatogenic compounds | [124] |
| Combined strategy | |||
| Microarray & HPLC-MS | C3A (NEFA, lactate, pyruvate, octanoate, & ammonia) | Proteogenomics analysis revealed three candidate genes (fibrinogen α, β and γ chains) and their relation to cardiovascular risk associated with NAFLD patients | [125] |
| RNASeq & GC-MS (lipidomics) | HuH7 and PHH (NEFA, fructose, & insulin) | Studying the effects of media nutritional substrates on intracellular lipid accumulation by means of lipidomics (altered glucose metabolism, FA oxidation, and lipoprotein secretion) and transcriptomics | [126] |
| Microarray & UHPLC-MS | HLCs (lactate, pyruvate, & octanoate) | HLCs treated with lactate, pyruvate, and octanoate recapitulate the transcriptional and metabolic dysregulation of NAFLD The epigenomic analysis revealed the retained expression of TET enzymes and 5hmC | [97] |
| RNASeq & UHPLC-MS (lipidomics) | HPP, HSCs, and hMP (NEFA, glucose, & insulin) | The model recapitulated lipotoxic stress with a similar therapeutic drug response of NASH patients. High ATP and β-oxidation levels | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelechá, M.; Villanueva-Bádenas, E.; Timor-López, E.; Donato, M.T.; Tolosa, L. Cell Models and Omics Techniques for the Study of Nonalcoholic Fatty Liver Disease: Focusing on Stem Cell-Derived Cell Models. Antioxidants 2022, 11, 86. https://doi.org/10.3390/antiox11010086
Pelechá M, Villanueva-Bádenas E, Timor-López E, Donato MT, Tolosa L. Cell Models and Omics Techniques for the Study of Nonalcoholic Fatty Liver Disease: Focusing on Stem Cell-Derived Cell Models. Antioxidants. 2022; 11(1):86. https://doi.org/10.3390/antiox11010086
Chicago/Turabian StylePelechá, María, Estela Villanueva-Bádenas, Enrique Timor-López, María Teresa Donato, and Laia Tolosa. 2022. "Cell Models and Omics Techniques for the Study of Nonalcoholic Fatty Liver Disease: Focusing on Stem Cell-Derived Cell Models" Antioxidants 11, no. 1: 86. https://doi.org/10.3390/antiox11010086