
Glutathione in the Nervous System as a Potential Therapeutic Target to Control the Development and Progression of Amyotrophic Lateral Sclerosis


Abstract
:1. Introduction
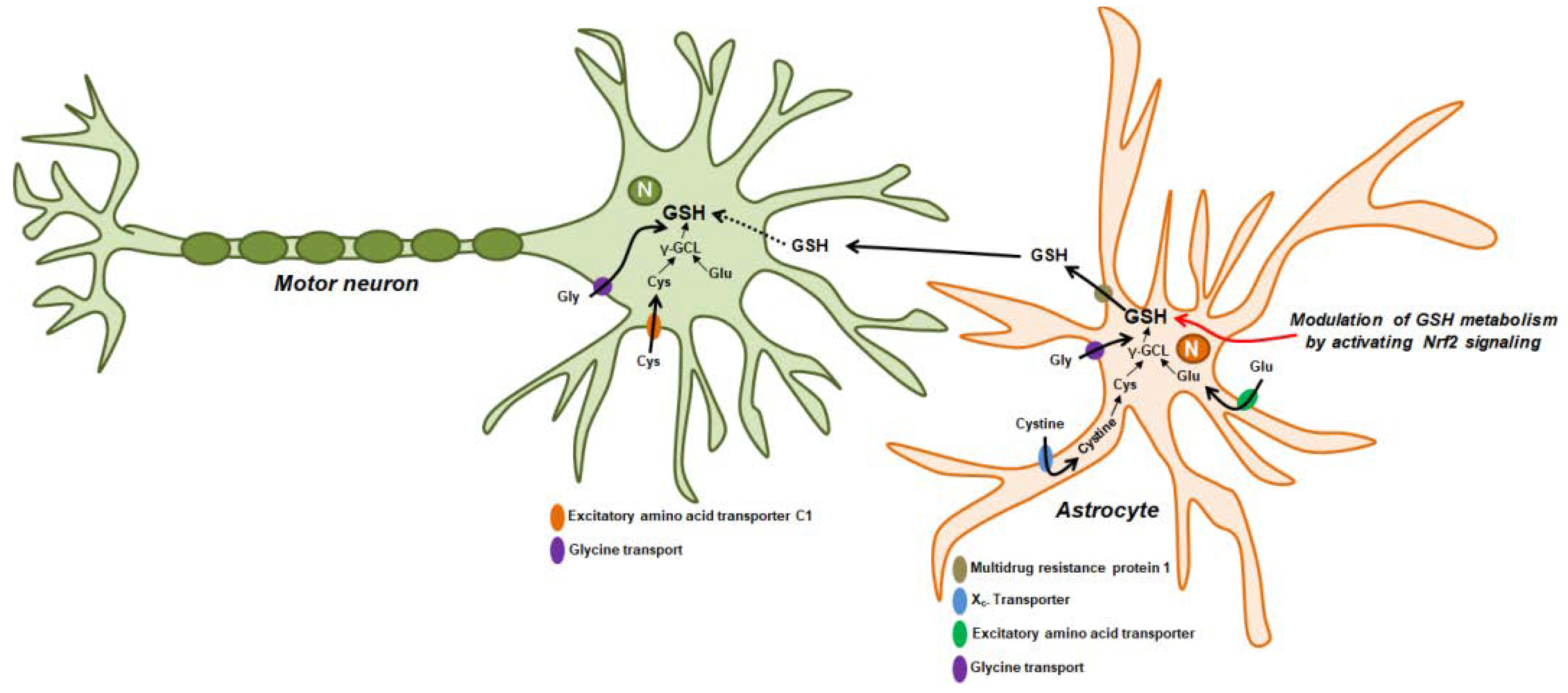
2. The Roles of GSH as an Antioxidant in the Nervous System
GSH Synthesis and Cellular Distribution in Neurons
3. Evidence for the Dysfunction of GSH Metabolism in ALS
3.1. GSH Redox Imbalance in Cellular Models of ALS: In Vitro Studies
3.2. GSH Redox Imbalance in Animal Models of ALS: In Vivo Studies

{kind=link}
| Cell Lines and Animals | Experimental ALS Models | Phenotypes Associated with Oxidative Stress | GSH Status | Reference |
|---|---|---|---|---|
| NT-2 SK-N-MC | Human SOD1WT Human SOD1G37R Human SOD1G85R | Increased protein carbonyl Increased 8-OHG Increased lipid peroxidation | Decrease GSH Increased GSSG | [69] |
| NSC-34 | Ethacrynic acid treatment | Decreased mitochondrial membrane potential Increased ROS generation Increased apoptosis | Decreased GSH | [70] |
| NSC-34 | Human SOD1WT Human SOD1G93A | Decreased cell viability Increased ROS generation Decreased mitochondrial membrane potential Increased mitochondrial toxicity | - | [71] |
| NSC-34 | Ethacrynic acid treatment | Increased ROS generation Increased oxidative response gene expression Increased apoptosis | Decreased GSH | [72] |
| NSC-34 | Human SOD1G93A | Decreased cell proliferation Decreased cell viability Increased apoptosis | Decreased mitochondrial GSH | [73] |
| NSC-34 | Human SOD1WT Human SOD1G93A | Increased mitochondrial dysfunction | Decreased GSH | [76] |
| NSC-34 | Human TDP-43M337V | Increased gene expression of Nrf2 signaling pathway | Decreased GSH | [78] |
| NSC-34 | Human TDP-43WT Human TDP-43A315T | Increased ROS generation Decreased cell viability Increased cell death | Decreased GSH | [79] |
| NSC-34 | Human SOD1G93A | Restored cell viability by treating urate | Increased GSH by treating urate | [98] |
| Rat astrocytes | Human SOD1G93A | Restored cell survival by activating Nrf2 | Increased GSH by activating Nrf2 | [85] |
| Mouse motor neurons | Human SOD1G93A | Increased apoptosis | Decrease GSH Increased GSSG | [72] |
| Mouse astrocytes | Human SOD1G93A | Extended survival by expressing Nrf2 Delayed muscle denervation by expressing Nrf2 | Increased GSH secretion by expressing Nrf2 | [86] |
| Mouse astrocytes | Human SOD1G93A Human SOD1H46R/H48Q | Decreased cell survival by GCLM knockout Increased motor neuron loss by GCLM knockout Increased oxidative stress by GCLM knockout Decreased complex IV activity by GCLM knockout | Decrease GSH by GCLM knockout | [88] |
| Mouse | Human SOD1G93A | Increased cystine uptake by cystine/glutamate antiporter | - | [94] |
| Fly | Human SOD1WT Human SOD1G85R | Extended survival by treatment with urate Improved motor defect by treatment with urate Enhanced antioxidant enzyme activity by treatment with urate Decreased ROS level by treatment with urate | - | [98] |
| Mouse | Human TDP-25 | Increased memory deficit by treatment with dexamethasone | Decreased GSH/GSSG ratio by treatment with dexamethasone | [103] |
| Mouse | Human SOD1G93A | Decreased survival | Decreased GSH in whole blood and spinal cord | [87] |
| Mouse | Human SOD1WT | Decreased cell survival by GCLM knockout Increased motor neuron loss by GCLM knockout | Decrease GSH by GCLM knockout | [89] |

3.3. GSH Redox Imbalance in ALS Patients
4. Clinical Trials in ALS
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cleveland, D.W.; Rothstein, J.D. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Fecto, F.; Siddique, T. Making connections: Pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementia. J. Mol. Neurosci. 2011, 45, 663–675. [Google Scholar] [CrossRef]
- Ince, P.G.; Highley, J.R.; Kirby, J.; Wharton, S.B.; Takahashi, H.; Strong, M.J.; Shaw, P.J. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: An emerging molecular pathway and the significance of glial pathology. Acta Neuropathol. 2011, 122, 657–671. [Google Scholar] [CrossRef]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum. Genet. 2017. [Google Scholar] [CrossRef] [Green Version]
- Costa, J.; Gomes, C.; de Carvalho, M. Diagnosis, pathogenesis and therapeutic targets in amyotrophic lateral sclerosis. CNS Neurol. Disord. Drug Targets 2010, 9, 764–778. [Google Scholar] [CrossRef]
- Bhandari, R.; Kuhad, A.; Kuhad, A. Edaravone: A new hope for deadly amyotrophic lateral sclerosis. Drugs Today 2018, 54, 349–360. [Google Scholar] [CrossRef]
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Shorter, J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 2011, 5, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Lagier-Tourenne, C.; Cleveland, D.W. Rethinking ALS: The FUS about TDP-43. Cell 2009, 136, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Ticozzi, N.; Vance, C.; Leclerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156B, 285–290. [Google Scholar] [CrossRef]
- Couthouis, J.; Hart, M.P.; Erion, R.; King, O.D.; Diaz, Z.; Nakaya, T.; Ibrahim, F.; Kim, H.J.; Mojsilovic-Petrovic, J.; Panossian, S.; et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 2899–2911. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Dormann, D.; Haass, C. TDP-43 and FUS: A nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Nguyen, P.T.; Shim, H.S.; Hyeon, S.J.; Im, H.; Choi, M.H.; Chung, S.; Kowall, N.W.; Lee, S.B.; Ryu, H. EWSR1, a multifunctional protein, regulates cellular function and aging via genetic and epigenetic pathways. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1938–1945. [Google Scholar] [CrossRef]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 2011, 134, 2595–2609. [Google Scholar] [CrossRef] [Green Version]
- Couthouis, J.; Hart, M.P.; Shorter, J.; DeJesus-Hernandez, M.; Erion, R.; Oristano, R.; Liu, A.X.; Ramos, D.; Jethava, N.; Hosangadi, D.; et al. A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. USA 2011, 108, 20881–20890. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Singh, R.L.; Kalita, J.; Misra, U.K. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef]
- Cova, E.; Bongioanni, P.; Cereda, C.; Metelli, M.R.; Salvaneschi, L.; Bernuzzi, S.; Guareschi, S.; Rossi, B.; Ceroni, M. Time course of oxidant markers and antioxidant defenses in subgroups of amyotrophic lateral sclerosis patients. Neurochem. Int. 2010, 56, 687–693. [Google Scholar] [CrossRef]
- Kuzma, M.; Jamrozik, Z.; Baranczyk-Kuzma, A. Activity and expression of glutathione S-transferase pi in patients with amyotrophic lateral sclerosis. Clin. Chim. Acta 2006, 364, 217–221. [Google Scholar] [CrossRef]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Increase in oxidized NO products and reduction in oxidized glutathione in cerebrospinal fluid from patients with sporadic form of amyotrophic lateral sclerosis. Neurosci. Lett. 1999, 260, 204–206. [Google Scholar] [CrossRef]
- Kang, Y.; Viswanath, V.; Jha, N.; Qiao, X.; Mo, J.Q.; Andersen, J.K. Brain gamma-glutamyl cysteine synthetase (GCS) mRNA expression patterns correlate with regional-specific enzyme activities and glutathione levels. J. Neurosci. Res. 1999, 58, 436–441. [Google Scholar] [CrossRef]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Cooper, A.J.; Kristal, B.S. Multiple roles of glutathione in the central nervous system. Biol. Chem. 1997, 378, 793–802. [Google Scholar] [PubMed]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, G.; Peana, M.; Maes, M.; Dadar, M.; Severin, B. The glutathione system in Parkinson’s disease and its progression. Neurosci. Biobehav. Rev. 2021, 120, 470–478. [Google Scholar] [CrossRef]
- Toroser, D.; Yarian, C.S.; Orr, W.C.; Sohal, R.S. Mechanisms of gamma-glutamylcysteine ligase regulation. Biochim. Biophys. Acta 2006, 1760, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Rosca, M.; Fan, Y.; Hu, Y.; Feng, P.; Lee, H.G.; Monnier, V.M.; Fan, X. Gclc deficiency in mouse CNS causes mitochondrial damage and neurodegeneration. Hum. Mol. Genet. 2017, 26, 1376–1390. [Google Scholar] [CrossRef]
- Chen, Y.; Curran, C.P.; Nebert, D.W.; Patel, K.V.; Williams, M.T.; Vorhees, C.V. Effect of chronic glutathione deficiency on the behavioral phenotype of Gclm-/- knockout mice. Neurotoxicol. Teratol. 2012, 34, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Gu, F.; Chauhan, V.; Chauhan, A. Impaired synthesis and antioxidant defense of glutathione in the cerebellum of autistic subjects: Alterations in the activities and protein expression of glutathione-related enzymes. Free Radic. Biol. Med. 2013, 65, 488–496. [Google Scholar] [CrossRef]
- Saharan, S.; Mandal, P.K. The emerging role of glutathione in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 40, 519–529. [Google Scholar] [CrossRef]
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Mason, R.P.; Casu, M.; Butler, N.; Breda, C.; Campesan, S.; Clapp, J.; Green, E.W.; Dhulkhed, D.; Kyriacou, C.P.; Giorgini, F. Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat. Genet. 2013, 45, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Oestreicher, J.; Morgan, B. Glutathione: Subcellular distribution and membrane transport. Biochem. Cell Biol. 2019, 97, 270–289. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Checa, J.C.; Kaplowitz, N.; Garcia-Ruiz, C.; Colell, A.; Miranda, M.; Mari, M.; Ardite, E.; Morales, A. GSH transport in mitochondria: Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. 1997, 273, G7–G17. [Google Scholar] [CrossRef]
- Muyderman, H.; Wadey, A.L.; Nilsson, M.; Sims, N.R. Mitochondrial glutathione protects against cell death induced by oxidative and nitrative stress in astrocytes. J. Neurochem. 2007, 102, 1369–1382. [Google Scholar] [CrossRef]
- Wullner, U.; Seyfried, J.; Groscurth, P.; Beinroth, S.; Winter, S.; Gleichmann, M.; Heneka, M.; Loschmann, P.; Schulz, J.B.; Weller, M.; et al. Glutathione depletion and neuronal cell death: The role of reactive oxygen intermediates and mitochondrial function. Brain Res. 1999, 826, 53–62. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Kirchhof, D.; Manning, E.; Joseph, J.W.; Linseman, D.A. Mitochondrial glutathione transport is a key determinant of neuronal susceptibility to oxidative and nitrosative stress. J. Biol. Chem. 2013, 288, 5091–5101. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Bass, R.; Ruddock, L.W.; Klappa, P.; Freedman, R.B. A major fraction of endoplasmic reticulum-located glutathione is present as mixed disulfides with protein. J. Biol. Chem. 2004, 279, 5257–5262. [Google Scholar] [CrossRef] [Green Version]
- Jessop, C.E.; Bulleid, N.J. Glutathione directly reduces an oxidoreductase in the endoplasmic reticulum of mammalian cells. J. Biol. Chem. 2004, 279, 55341–55347. [Google Scholar] [CrossRef] [Green Version]
- Molteni, S.N.; Fassio, A.; Ciriolo, M.R.; Filomeni, G.; Pasqualetto, E.; Fagioli, C.; Sitia, R. Glutathione limits Ero1-dependent oxidation in the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 32667–32673. [Google Scholar] [CrossRef] [Green Version]
- Ponsero, A.J.; Igbaria, A.; Darch, M.A.; Miled, S.; Outten, C.E.; Winther, J.R.; Palais, G.; D’Autreaux, B.; Delaunay-Moisan, A.; Toledano, M.B. Endoplasmic Reticulum Transport of Glutathione by Sec61 is Regulated by Ero1 and Bip. Mol. Cell 2017, 67, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Tsunoda, S.; Avezov, E.; Zyryanova, A.; Konno, T.; Mendes-Silva, L.; Pinho Melo, E.; Harding, H.P.; Ron, D. Intact protein folding in the glutathione-depleted endoplasmic reticulum implicates alternative protein thiol reductants. Elife 2014, 3, e03421. [Google Scholar] [CrossRef]
- Horiguchi, H.; Yurimoto, H.; Kato, N.; Sakai, Y. Antioxidant system within yeast peroxisome. Biochemical and physiological characterization of CbPmp20 in the methylotrophic yeast Candida boidinii. J. Biol. Chem. 2001, 276, 14279–14288. [Google Scholar] [CrossRef] [Green Version]
- Antonenkov, V.D.; Hiltunen, J.K. Transfer of metabolites across the peroxisomal membrane. Biochim. Biophys. Acta 2012, 1822, 1374–1386. [Google Scholar] [CrossRef] [Green Version]
- Elbaz-Alon, Y.; Morgan, B.; Clancy, A.; Amoako, T.N.; Zalckvar, E.; Dick, T.P.; Schwappach, B.; Schuldiner, M. The yeast oligopeptide transporter Opt2 is localized to peroxisomes and affects glutathione redox homeostasis. FEMS Yeast Res. 2014, 14, 1055–1067. [Google Scholar] [CrossRef] [Green Version]
- Ohdate, T.; Inoue, Y. Involvement of glutathione peroxidase 1 in growth and peroxisome formation in Saccharomyces cerevisiae in oleic acid medium. Biochim. Biophys. Acta 2012, 1821, 1295–1305. [Google Scholar] [CrossRef] [Green Version]
- Markovic, J.; Borras, C.; Ortega, A.; Sastre, J.; Vina, J.; Pallardo, F.V. Glutathione is recruited into the nucleus in early phases of cell proliferation. J. Biol. Chem. 2007, 282, 20416–20424. [Google Scholar] [CrossRef] [Green Version]
- Reichheld, J.P.; Khafif, M.; Riondet, C.; Droux, M.; Bonnard, G.; Meyer, Y. Inactivation of thioredoxin reductases reveals a complex interplay between thioredoxin and glutathione pathways in Arabidopsis development. Plant Cell 2007, 19, 1851–1865. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, G.; Palladini, G.; Vairetti, M. Intranuclear distribution, function and fate of glutathione and glutathione-S-conjugate in living rat hepatocytes studied by fluorescence microscopy. Microsc. Res. Tech. 1997, 36, 243–252. [Google Scholar] [CrossRef]
- Jeong, E.M.; Yoon, J.H.; Lim, J.; Shin, J.W.; Cho, A.Y.; Heo, J.; Lee, K.B.; Lee, J.H.; Lee, W.J.; Kim, H.J.; et al. Real-Time Monitoring of Glutathione in Living Cells Reveals that High Glutathione Levels are Required to Maintain Stem Cell Function. Stem Cell Rep. 2018, 10, 600–614. [Google Scholar] [CrossRef] [Green Version]
- Miller, V.M.; Lawrence, D.A.; Mondal, T.K.; Seegal, R.F. Reduced glutathione is highly expressed in white matter and neurons in the unperturbed mouse brain—Implications for oxidative stress associated with neurodegeneration. Brain Res. 2009, 1276, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Hyun, D.; Jenner, P.; Halliwell, B. Effect of overexpression of wild-type and mutant Cu/Zn-superoxide dismutases on oxidative damage and antioxidant defences: Relevance to Down’s syndrome and familial amyotrophic lateral sclerosis. J. Neurochem. 2001, 76, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Rizzardini, M.; Lupi, M.; Bernasconi, S.; Mangolini, A.; Cantoni, L. Mitochondrial dysfunction and death in motor neurons exposed to the glutathione-depleting agent ethacrynic acid. J. Neurol. Sci. 2003, 207, 51–58. [Google Scholar] [CrossRef]
- Rizzardini, M.; Mangolini, A.; Lupi, M.; Ubezio, P.; Bendotti, C.; Cantoni, L. Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J. Neurol. Sci. 2005, 232, 95–103. [Google Scholar] [CrossRef]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef] [Green Version]
- Muyderman, H.; Hutson, P.G.; Matusica, D.; Rogers, M.L.; Rush, R.A. The human G93A-superoxide dismutase-1 mutation, mitochondrial glutathione and apoptotic cell death. Neurochem. Res. 2009, 34, 1847–1856. [Google Scholar] [CrossRef]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 2005, 280, 33766–33774. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.K.; Kustermann, E.; Dedeoglu, A.; Jenkins, B.G. Magnetic resonance spectroscopy of regional brain metabolite markers in FALS mice and the effects of dietary creatine supplementation. Eur. J. Neurosci. 2009, 30, 2143–2150. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, G.; Calcagno, E.; Tartari, S.; Rizzardini, M.; Invernizzi, R.W.; Cantoni, L. Glutamate and glutathione interplay in a motor neuronal model of amyotrophic lateral sclerosis reveals altered energy metabolism. Neurobiol. Dis. 2011, 43, 346–355. [Google Scholar] [CrossRef]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef]
- Moujalled, D.; Grubman, A.; Acevedo, K.; Yang, S.; Ke, Y.D.; Moujalled, D.M.; Duncan, C.; Caragounis, A.; Perera, N.D.; Turner, B.J.; et al. TDP-43 mutations causing amyotrophic lateral sclerosis are associated with altered expression of RNA-binding protein hnRNP K and affect the Nrf2 antioxidant pathway. Hum. Mol. Genet. 2017, 26, 1732–1746. [Google Scholar] [CrossRef]
- Chen, T.; Turner, B.J.; Beart, P.M.; Sheehan-Hennessy, L.; Elekwachi, C.; Muyderman, H. Glutathione monoethyl ester prevents TDP-43 pathology in motor neuronal NSC-34 cells. Neurochem. Int. 2018, 112, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.R.; Pehar, M.; Cassina, P.; Beckman, J.S.; Barbeito, L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J. Neurochem. 2006, 97, 687–696. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef]
- Ross, E.K.; Winter, A.N.; Wilkins, H.M.; Sumner, W.A.; Duval, N.; Patterson, D.; Linseman, D.A. A Cystine-Rich Whey Supplement (Immunocal((R))) Delays Disease Onset and Prevents Spinal Cord Glutathione Depletion in the hSOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Antioxidants 2014, 3, 843–865. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1(G93A) mice model. Neurobiol. Dis. 2011, 43, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Killoy, K.M.; Harlan, B.A.; Pehar, M.; Helke, K.L.; Johnson, J.A.; Vargas, M.R. Decreased glutathione levels cause overt motor neuron degeneration in hSOD1(WT) over-expressing mice. Exp. Neurol. 2018, 302, 129–135. [Google Scholar] [CrossRef]
- Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Stipanuk, M.H.; Dominy, J.E., Jr.; Lee, J.I.; Coloso, R.M. Mammalian cysteine metabolism: New insights into regulation of cysteine metabolism. J. Nutr. 2006, 136, 1652S–1659S. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Okuno, S.; Sato, K.; Keino-Masu, K.; Masu, M.; Bannai, S. Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J. Neurosci. 2002, 22, 8028–8033. [Google Scholar] [CrossRef] [Green Version]
- Albano, R.; Liu, X.; Lobner, D. Regulation of system x(c)- in the SOD1-G93A mouse model of ALS. Exp. Neurol. 2013, 250, 69–73. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Johnson, R.J. Uric acid: The oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619. [Google Scholar] [CrossRef] [Green Version]
- Keizman, D.; Ish-Shalom, M.; Berliner, S.; Maimon, N.; Vered, Y.; Artamonov, I.; Tsehori, J.; Nefussy, B.; Drory, V.E. Low uric acid levels in serum of patients with ALS: Further evidence for oxidative stress? J. Neurol. Sci. 2009, 285, 95–99. [Google Scholar] [CrossRef]
- Paganoni, S.; Nicholson, K.; Chan, J.; Shui, A.; Schoenfeld, D.; Sherman, A.; Berry, J.; Cudkowicz, M.; Atassi, N.; Pooled Resource Open-Access, A.L.S.C.T.C. Urate levels predict survival in amyotrophic lateral sclerosis: Analysis of the expanded Pooled Resource Open-Access ALS clinical trials database. Muscle Nerve 2018, 57, 430–434. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Liang, W.; Wang, T.; Wang, S.; Wang, X.; Wang, Y.; Jiang, H.; Feng, H. Neuroprotection by urate on the mutant hSOD1-related cellular and Drosophila models of amyotrophic lateral sclerosis: Implication for GSH synthesis via activating Akt/GSK3beta/Nrf2/GCLC pathways. Brain Res. Bull. 2019, 146, 287–301. [Google Scholar] [CrossRef]
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Shariff, M.; Bartlett, S.E. The role of the glucocorticoids in developing resilience to stress and addiction. Front. Psychiatry 2013, 4, 68. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, L.J.; Sapolsky, R.M. Glucocorticoids increase the accumulation of reactive oxygen species and enhance adriamycin-induced toxicity in neuronal culture. Exp. Neurol. 1996, 141, 201–206. [Google Scholar] [CrossRef]
- Behl, C.; Lezoualc’h, F.; Trapp, T.; Widmann, M.; Skutella, T.; Holsboer, F. Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurons in vitro. Endocrinology 1997, 138, 101–106. [Google Scholar] [CrossRef]
- Caccamo, A.; Medina, D.X.; Oddo, S. Glucocorticoids exacerbate cognitive deficits in TDP-25 transgenic mice via a glutathione-mediated mechanism: Implications for aging, stress and TDP-43 proteinopathies. J. Neurosci. 2013, 33, 906–913. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Konig, J.; Keppler, D.; Lindenau, J.; Schulz, J.B.; Dringen, R. The multidrug resistance protein MRP1 mediates the release of glutathione disulfide from rat astrocytes during oxidative stress. J. Neurochem. 2001, 76, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Lee, S.G.; Kegelman, T.P.; Su, Z.Z.; Das, S.K.; Dash, R.; Dasgupta, S.; Barral, P.M.; Hedvat, M.; Diaz, P.; et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: Opportunities for developing novel therapeutics. J. Cell Physiol. 2011, 226, 2484–2493. [Google Scholar] [CrossRef] [Green Version]
- Shibasaki, K.; Hosoi, N.; Kaneko, R.; Tominaga, M.; Yamada, K. Glycine release from astrocytes via functional reversal of GlyT1. J. Neurochem. 2017, 140, 395–403. [Google Scholar] [CrossRef]
- Kranich, O.; Hamprecht, B.; Dringen, R. Different preferences in the utilization of amino acids for glutathione synthesis in cultured neurons and astroglial cells derived from rat brain. Neurosci. Lett. 1996, 219, 211–214. [Google Scholar] [CrossRef]
- Wang, X.F.; Cynader, M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem. 2000, 74, 1434–1442. [Google Scholar] [CrossRef]
- Ogita, K.; Yoneda, Y. Selective potentiation by L-cysteine of apparent binding activity of [3H]glutathione in synaptic membranes of rat brain. Biochem. Pharmacol. 1989, 38, 1499–1505. [Google Scholar] [CrossRef]
- Ogita, K.; Yoneda, Y. Possible presence of [3H]glutathione (GSH) binding sites in synaptic membranes from rat brain. Neurosci. Res. 1987, 4, 486–496. [Google Scholar] [CrossRef]
- Lanius, R.A.; Krieger, C.; Wagey, R.; Shaw, C.A. Increased [35S]glutathione binding sites in spinal cords from patients with sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 1993, 163, 89–92. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- McLean, C.W.; Mirochnitchenko, O.; Claus, C.P.; Noble-Haeusslein, L.J.; Ferriero, D.M. Overexpression of glutathione peroxidase protects immature murine neurons from oxidative stress. Dev. Neurosci. 2005, 27, 169–175. [Google Scholar] [CrossRef]
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.J.; Mitsumoto, H.; Shungu, D.C. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107. [Google Scholar] [CrossRef]
- Weerasekera, A.; Peeters, R.; Sima, D.; Dresselaers, T.; Sunaert, S.; de Vocht, J.; Claeys, K.; van Huffel, S.; van Damme, P.; Himmelreich, U. Motor cortex metabolite alterations in amyotrophic lateral sclerosis assessed in vivo using edited and non-edited magnetic resonance spectroscopy. Brain Res. 2019, 1718, 22–31. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Nicholson, K.; Jafari-Khouzani, K.; Bogner, W.; Wang, J.; Chan, J.; Macklin, E.A.; Levine-Weinberg, M.; Breen, C.; Schwarzschild, M.A.; et al. Imaging Neurochemistry and Brain Structure Tracks Clinical Decline and Mechanisms of ALS in Patients. Front. Neurol. 2020, 11, 590573. [Google Scholar] [CrossRef]
- Yang, L.; Lv, X.; Du, H.; Wu, D.; Wang, M. Causal effects of serum metabolites on amyotrophic lateral sclerosis: A Mendelian randomization study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 97, 109771. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the gamma-glutamyl cycle. IUBMB Life 2018, 70, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Chio, A.; Cucatto, A.; Terreni, A.A.; Schiffer, D. Reduced glutathione in amyotrophic lateral sclerosis: An open, crossover, randomized trial. Ital. J. Neurol. Sci. 1998, 19, 363–366. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; Sexton, P.M.; Ellis, T.; Hayden, D.L.; Gwilt, P.R.; Whalen, J.; Brown, R.H., Jr. The pharmacokinetics and pharmaco-dynamics of Procysteine in amyotrophic lateral sclerosis. Neurology 1999, 52, 1492–1494. [Google Scholar] [CrossRef]
- Louwerse, E.S.; Weverling, G.J.; Bossuyt, P.M.; Meyjes, F.E.; de Jong, J.M. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch. Neurol. 1995, 52, 559–564. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K. Glutathione in the Nervous System as a Potential Therapeutic Target to Control the Development and Progression of Amyotrophic Lateral Sclerosis. Antioxidants 2021, 10, 1011. https://doi.org/10.3390/antiox10071011
Kim K. Glutathione in the Nervous System as a Potential Therapeutic Target to Control the Development and Progression of Amyotrophic Lateral Sclerosis. Antioxidants. 2021; 10(7):1011. https://doi.org/10.3390/antiox10071011
Chicago/Turabian StyleKim, Kiyoung. 2021. "Glutathione in the Nervous System as a Potential Therapeutic Target to Control the Development and Progression of Amyotrophic Lateral Sclerosis" Antioxidants 10, no. 7: 1011. https://doi.org/10.3390/antiox10071011