
A Workflow towards the Reproducible Identification and Quantitation of Protein Carbonylation Sites in Human Plasma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
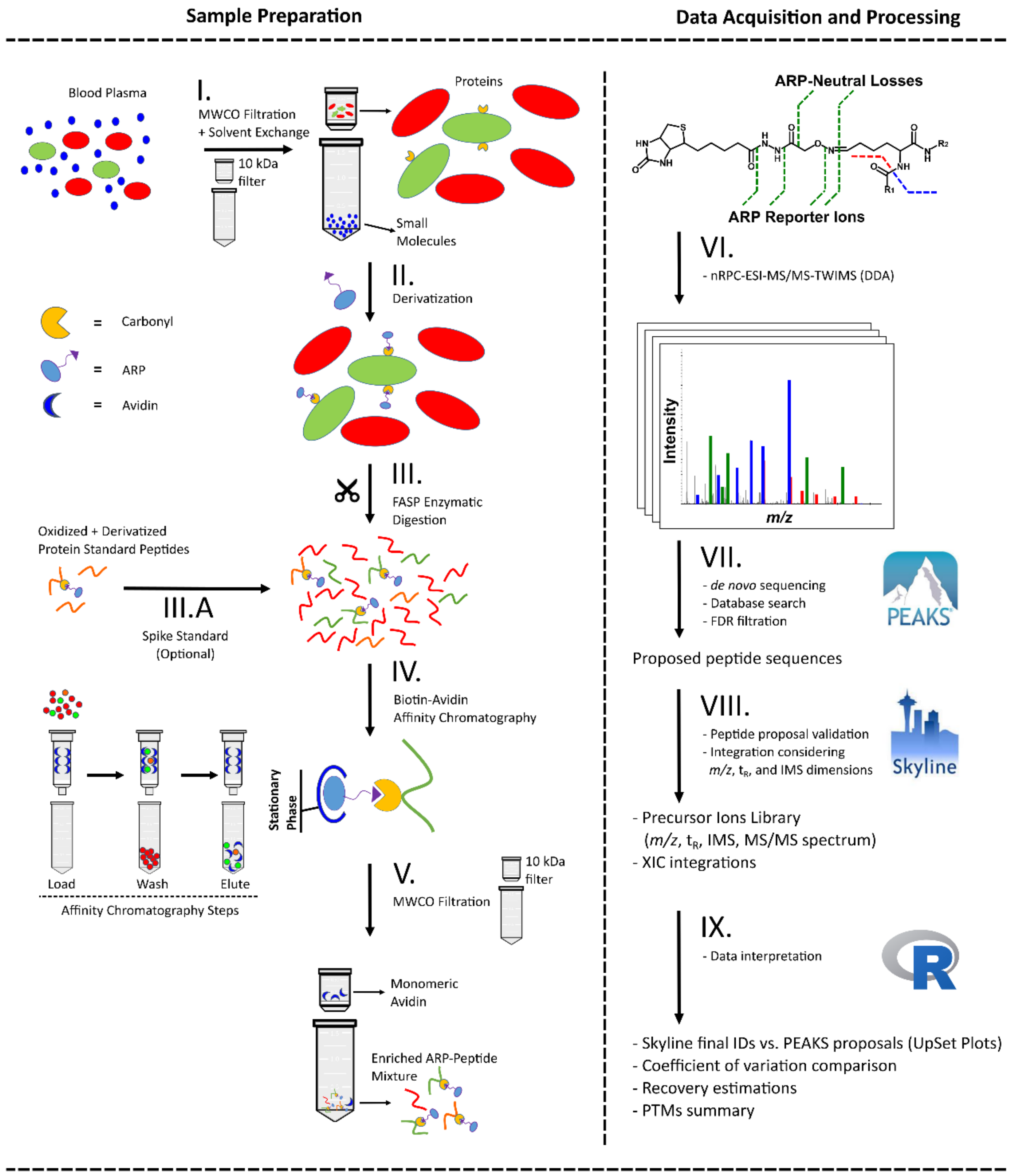
2. Materials and Methods
2.1. Materials
2.2. Human Plasma Collection
2.3. Derivatization of Reactive Carbonyl Groups
2.3.1. Oxidized Human Serum Albumin as a Protein Model
2.3.2. Plasma Samples
2.4. Protein Digestion
2.4.1. Filter-Aided Sample Preparation (FASP)
2.4.2. In-Solution Digest
2.5. Avidin Affinity Chromatography
2.6. Workflow Performance Evaluation
2.7. Mass Spectrometry Acquisition (nRPC-ESI-MS/MS-TWIMS)
2.8. Data Analysis
2.8.1. Database Search
2.8.2. LC-MS Data Integration and Filtration
2.8.3. Enrichment Profile Estimation
3. Results
3.1. Derivatization, Digestion, and Enrichment Conditions
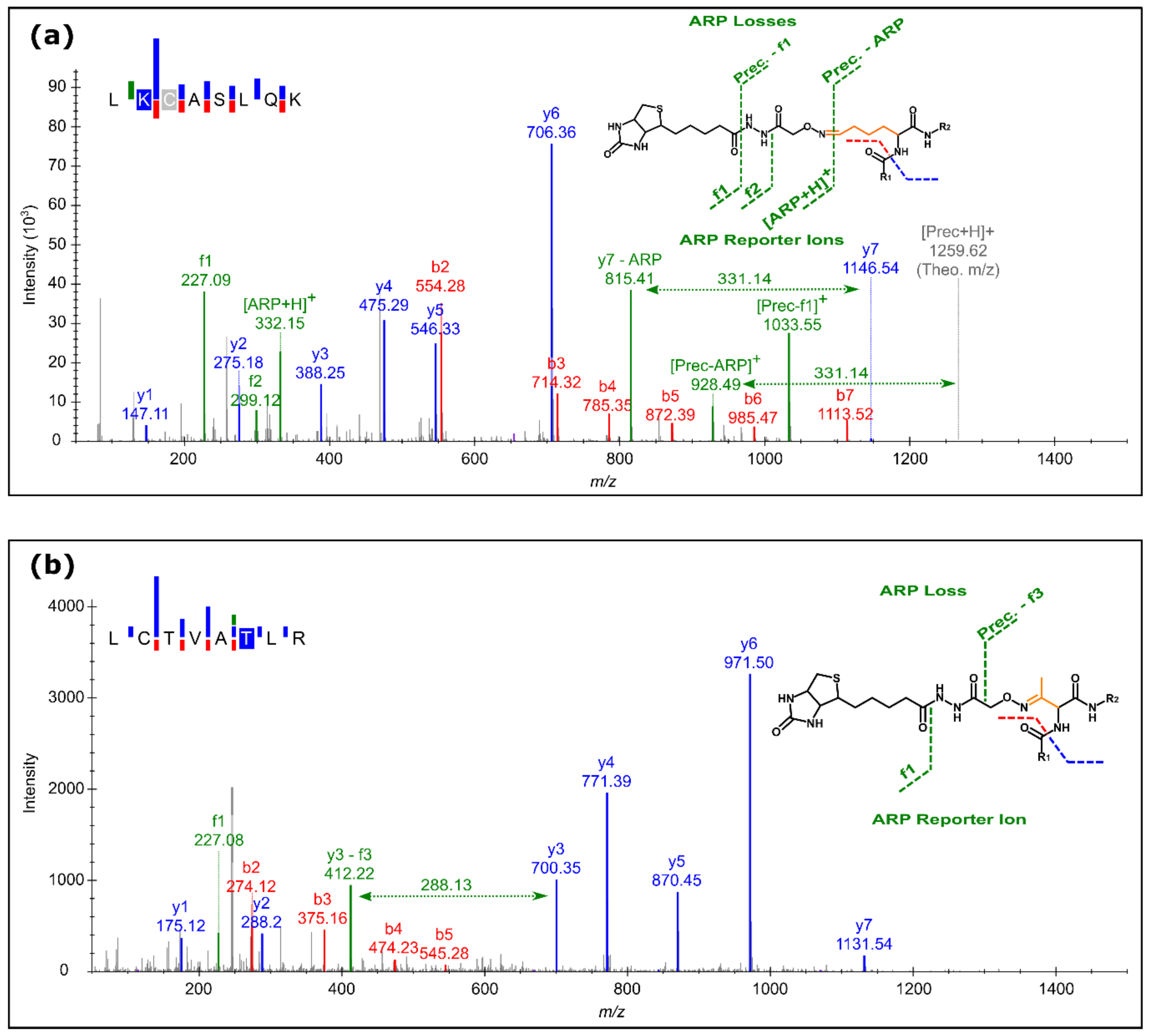
3.2. ARP Peptide Fragmentation Patterns
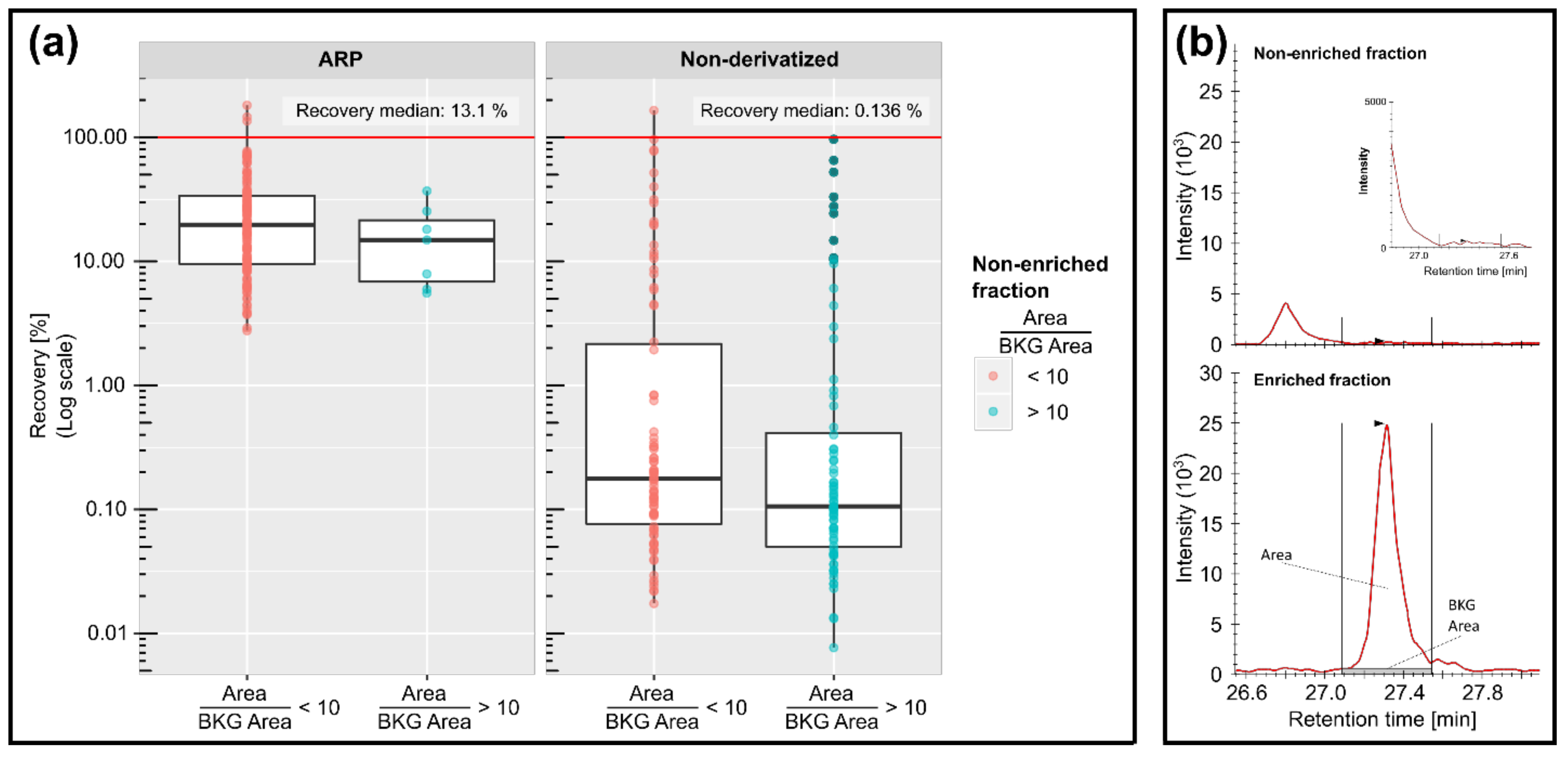
3.3. Enrichment Patterns and Matrix Interference Effects
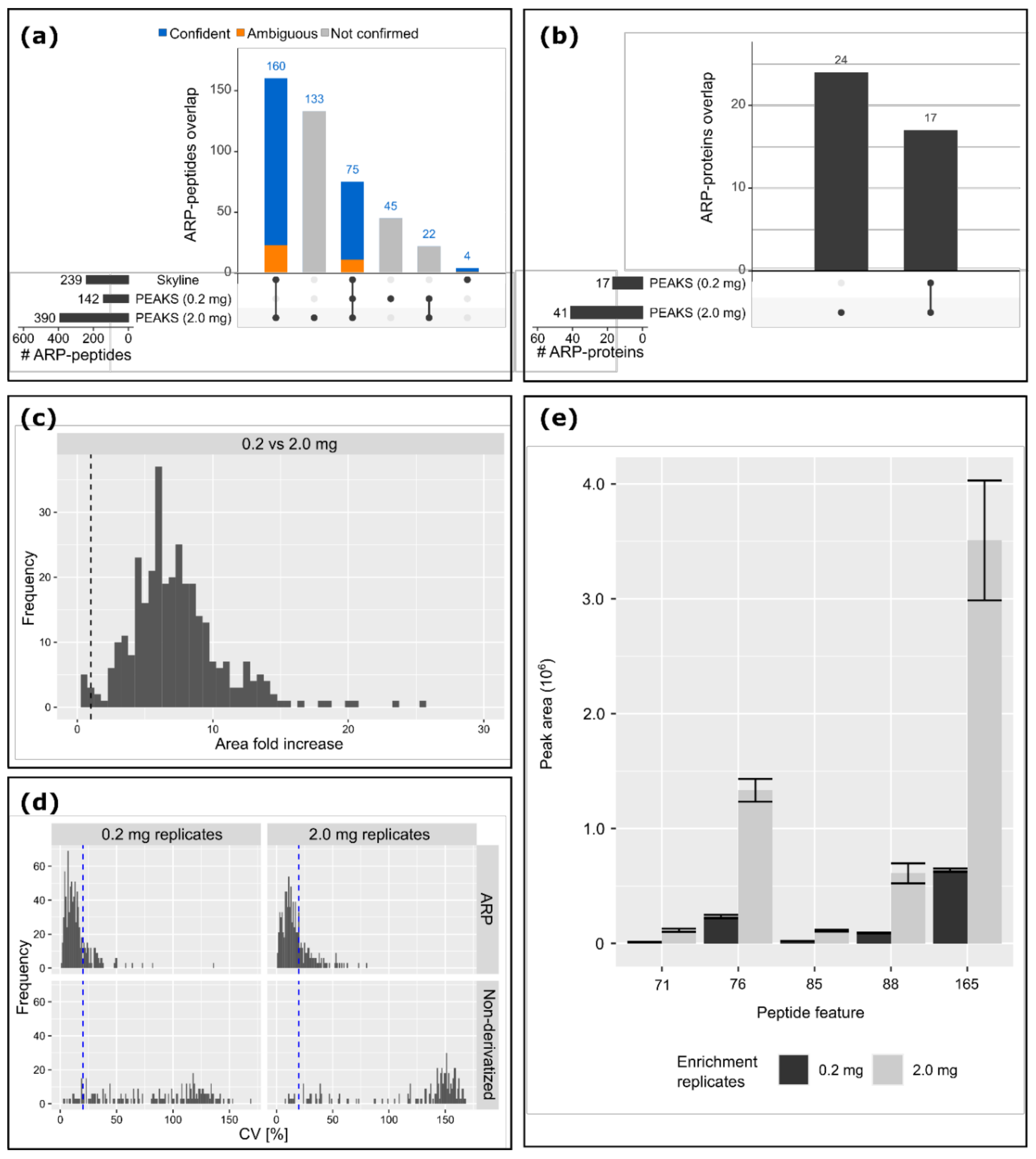
3.4. Increase in Sensitivity of ARP Peptides with Sample Preparation Upscale
4. Discussion
4.1. ARP-Specific Fragmentation Patterns Improved Identification Accuracy
4.2. Protein Derivatization Improved the Enrichment Efficiency for Target Analytes
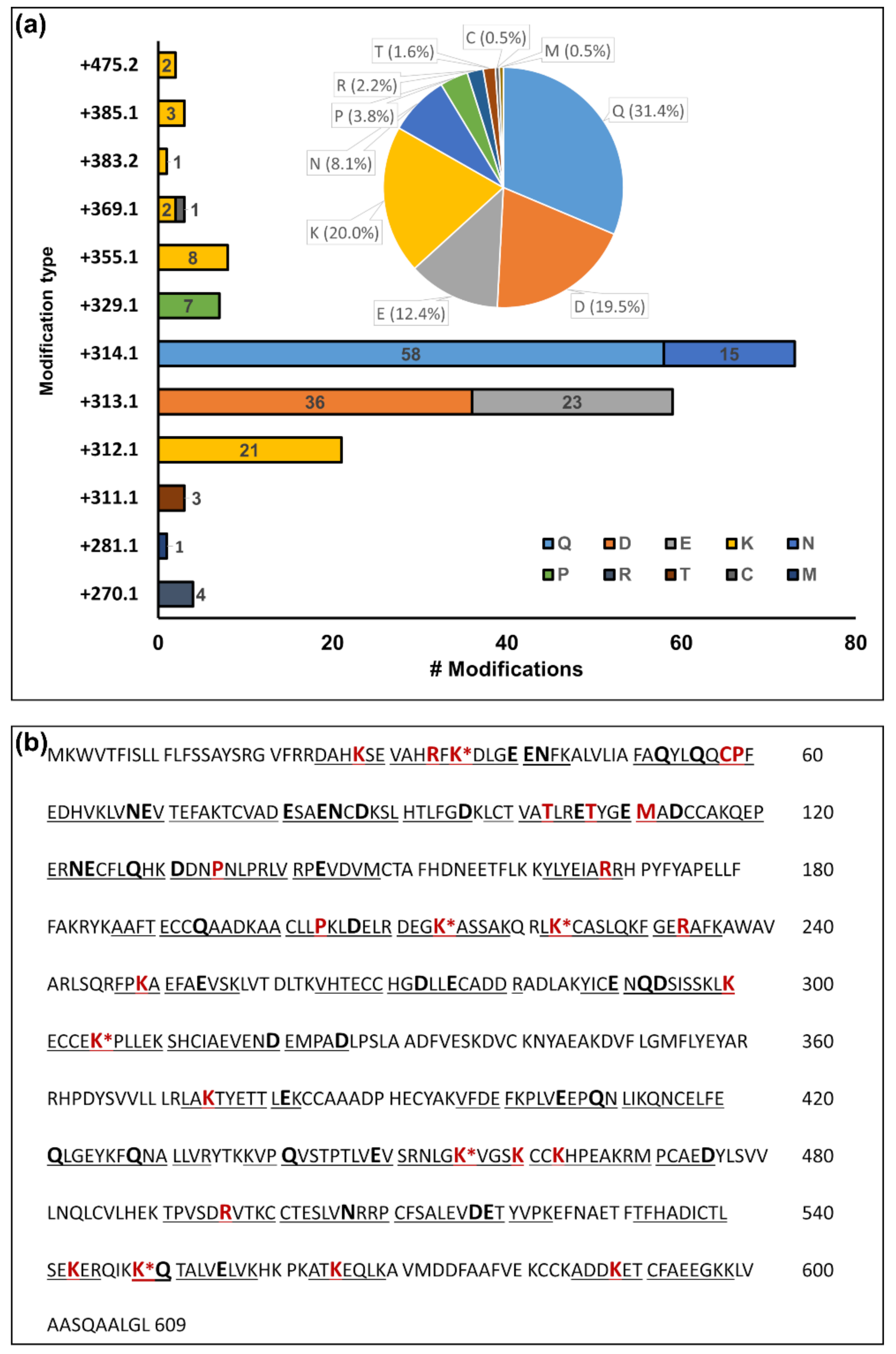
4.3. Unexpected Reactive Carbonyl PTMs
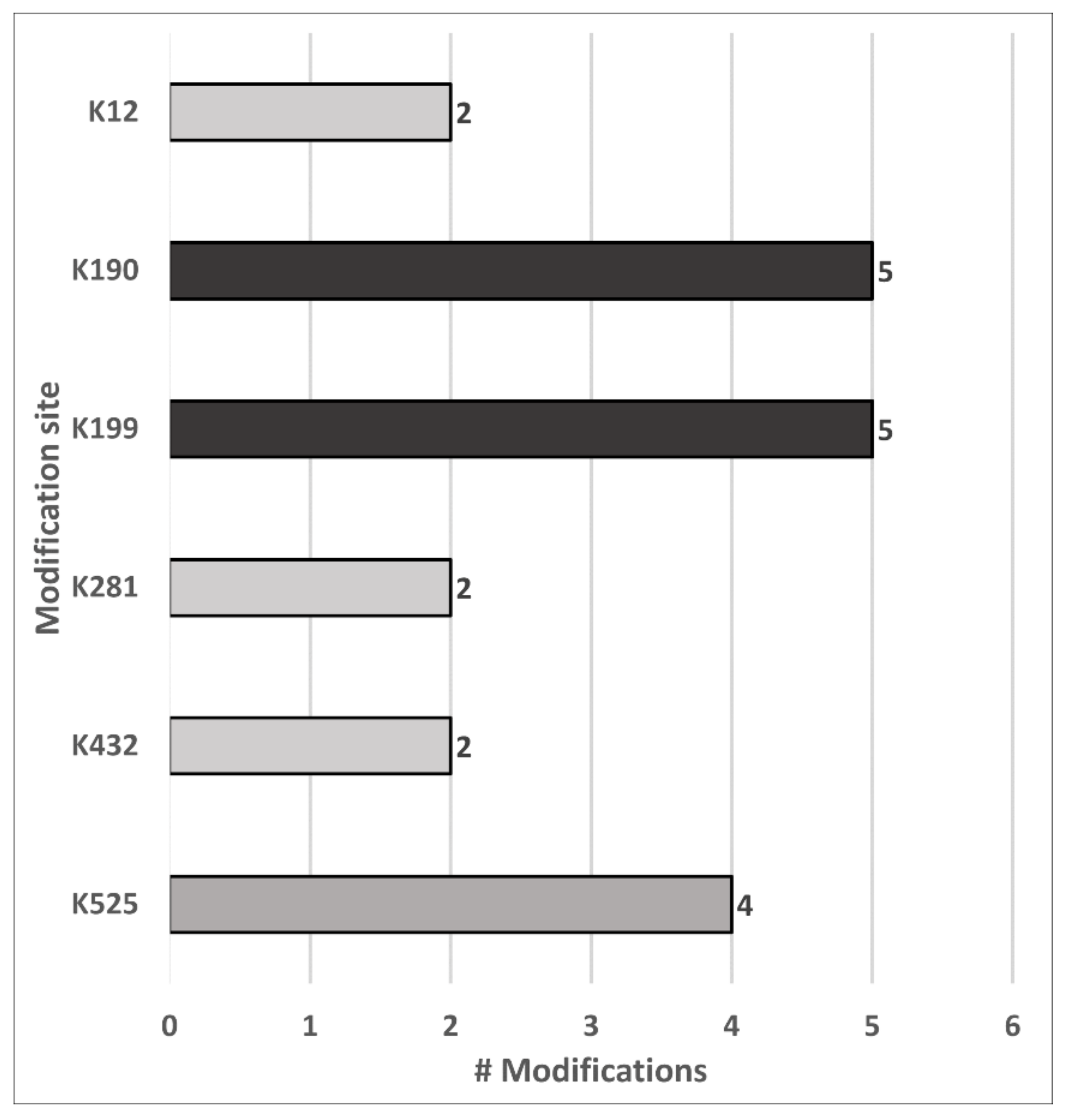
4.4. Carbonylation Site Specificity
4.5. Peptide Level Enrichment Improved the Identification Accuracy of Carbonylated Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Afonso, C.B.; Spickett, C.M. Lipoproteins as targets and markers of lipoxidation. Redox Biol. 2019, 23, 101066. [Google Scholar] [CrossRef] [PubMed]
- Vasil’Ev, Y.V.; Tzeng, S.-C.; Huang, L.; Maier, C.S. Protein modifications by electrophilic lipoxidation products: Adduct formation, chemical strategies and tandem mass spectrometry for their detection and identification. Mass Spectrom. Rev. 2014, 33, 157–182. [Google Scholar] [CrossRef]
- Aldini, G.; Domingues, M.R.; Spickett, C.M.; Domingues, P.; Altomare, A.; Sánchez-Gómez, F.J.; Oeste, C.L.; Pérez-Sala, D. Protein lipoxidation: Detection strategies and challenges. Redox Biol. 2015, 5, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free. Radic. Res. 2013, 47 (Suppl. 1), 3–27. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef] [Green Version]
- Weber, D.; Davies, M.J.; Grune, T. Determination of protein carbonyls in plasma, cell extracts, tissue homogenates, isolated proteins: Focus on sample preparation and derivatization conditions. Redox Biol. 2015, 5, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies. Mass Spectrom. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Kalia, J.; Raines, R.T. Hydrolytic Stability of Hydrazones and Oximes. Angew. Chem. Int. Ed. 2008, 47, 7523–7526. [Google Scholar] [CrossRef] [Green Version]
- Bollineni, R.C.; Fedorova, M.; Hoffmann, R. Qualitative and quantitative evaluation of derivatization reagents for different types of protein-bound carbonyl groups. Analyst 2013, 138, 5081–5088. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.D.; Wu, J.; Bisson, W.; Maier, C.S. Site-specific proteomic analysis of lipoxidation adducts in cardiac mitochondria reveals chemical diversity of 2-alkenal adduction. J. Proteom. 2011, 74, 2417–2429. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.; Chung, W.-G.; Miranda, C.L.; Singhal, M.; Stevens, J.F.; Maier, C.S. Site-Specific Protein Adducts of 4-Hydroxy-2(E)-Nonenal in Human THP-1 Monocytic Cells: Protein Carbonylation Is Diminished by Ascorbic Acid. Chem. Res. Toxicol. 2009, 23, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.D.; Bisson, W.H.; Maier, C.S. A targeted mass spectrometry-based approach for the identification and characterization of proteins containing α-aminoadipic and γ-glutamic semialdehyde residues. Anal. Bioanal. Chem. 2010, 398, 2905–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.-G.; Miranda, C.L.; Maier, C.S. Detection of carbonyl-modified proteins in interfibrillar rat mitochondria usingN′-aminooxymethylcarbonylhydrazino-D-biotin as an aldehyde/keto-reactive probe in combination with Western blot analysis and tandem mass spectrometry. Electrophoresis 2008, 29, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.-C.; Maier, C.S. Label-Free Proteomics Assisted by Affinity Enrichment for Elucidating the Chemical Reactivity of the Liver Mitochondrial Proteome toward Adduction by the Lipid Electrophile 4-hydroxy-2-nonenal (HNE). Front. Chem. 2016, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Slade, P.G.; Williams, M.V.; Chiang, A.; Iffrig, E.; Tannenbaum, S.R.; Wishnok, J.S. A Filtered Database Search Algorithm for Endogenous Serum Protein Carbonyl Modifications in a Mouse Model of Inflammation. Mol. Cell. Proteom. 2011, 10, M111-007658. [Google Scholar] [CrossRef] [Green Version]
- Bollineni, R.C.; Fedorova, M.; Blüher, M.; Hoffmann, R. Carbonylated Plasma Proteins As Potential Biomarkers of Obesity Induced Type 2 Diabetes Mellitus. J. Proteome Res. 2014, 13, 5081–5093. [Google Scholar] [CrossRef]
- Chavez, J.; Wu, J.; Han, B.; Chung, W.-G.; Maier, C.S. New Role for an Old Probe: Affinity Labeling of Oxylipid Protein Conjugates byN‘-Aminooxymethylcarbonylhydrazinod-biotin. Anal. Chem. 2006, 78, 6847–6854. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Stevens, J.F.; Maier, C.S. Mass spectrometry-based quantification of myocardial protein adducts with acrolein in an in vivo model of oxidative stress. Mol. Nutr. Food Res. 2011, 55, 1401–1410. [Google Scholar] [CrossRef] [Green Version]
- Havelund, J.F.; Wojdyla, K.; Davies, M.J.; Jensen, O.N.; Møller, I.M.; Rogowska-Wrzesinska, A. A biotin enrichment strategy identifies novel carbonylated amino acids in proteins from human plasma. J. Proteom. 2017, 156, 40–51. [Google Scholar] [CrossRef]
- Madian, A.G.; Regnier, F.E. Profiling Carbonylated Proteins in Human Plasma. J. Proteome Res. 2010, 9, 1330–1343. [Google Scholar] [CrossRef] [PubMed]
- Madian, A.G.; Diaz-Maldonado, N.; Gao, Q.; Regnier, F.E. Oxidative stress induced carbonylation in human plasma. J. Proteom. 2011, 74, 2395–2416. [Google Scholar] [CrossRef] [Green Version]
- Neuhoff, V.; Stamm, R.; Eibl, H. Clear background and highly sensitive protein staining with Coomassie Blue dyes in polyacrylamide gels: A systematic analysis. Electrophoresis 1985, 6, 427–448. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Helm, D.; Vissers, J.P.C.; Hughes, C.J.; Hahne, H.; Ruprecht, B.; Pachl, F.; Grzyb, A.; Richardson, K.; Wildgoose, J.; Maier, S.K.; et al. Ion Mobility Tandem Mass Spectrometry Enhances Performance of Bottom-up Proteomics. Mol. Cell. Proteom. 2014, 13, 3709–3715. [Google Scholar] [CrossRef] [Green Version]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Rardin, M.J.; MacLean, B.X.; Zawadzka, A.M.; Frewen, B.E.; Cusack, M.P.; Sorensen, D.J.; Bereman, M.S.; Jing, E.; Wu, C.C.; et al. Platform-independent and Label-free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline. Mol. Cell. Proteom. 2012, 11, 202–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLean, B.X.; Pratt, B.S.; Egertson, J.D.; MacCoss, M.J.; Smith, R.D.; Baker, E.S. Using Skyline to Analyze Data-Containing Liquid Chromatography, Ion Mobility Spectrometry, and Mass Spectrometry Dimensions. J. Am. Soc. Mass Spectrom. 2018, 29, 2182–2188. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zielinska, D.F.; Mann, M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Anal. Biochem. 2011, 410, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S.; Lebl, M. Streptavidin and avidin recognize peptide ligands with different motifs. ImmunoMethods 1992, 1, 11–15. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, A.B. Molecular Clocks: Deamidation of Asparaginyl and Glutaminyl Residues in Peptides and Proteins; Althouse Press: Cave Junction, OR, USA, 2004; ISBN 1-59087-250-0. [Google Scholar]
- Athmer, L.; Kindrachuk, J.; Georges, F.; Napper, S.; Moelling, K.; Schad, K.; Bosse, M.; Zimmermann, S.; Schweneker, M. The Influence of Protein Structure on the Products Emerging from Succinimide Hydrolysis. J. Biol. Chem. 2002, 277, 30502–30507. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.Y.; Yang, X. Proteases for Cell Suicide: Functions and Regulation of Caspases. Microbiol. Mol. Biol. Rev. 2000, 64, 821–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmelfarb, J.; McMonagle, E. Albumin is the major plasma protein target of oxidant stress in uremia. Kidney Int. 2001, 60, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Aksenov, M.; Aksenova, M.; Butterfield, D.A.; Markesbery, W.R. Oxidative Modification of Creatine Kinase BB in Alzheimer’s Disease Brain. J. Neurochem. 2002, 74, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of Oxidative Damage in Human Disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Fedorova, M. Diversity of Protein Carbonylation Pathways. In Protein Carbonylation; Wiley: Hoboken, NJ, USA, 2017; pp. 48–82. [Google Scholar]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, H.; Regnier, F. Affinity Chromatographic Selection of Carbonylated Proteins Followed by Identification of Oxidation Sites Using Tandem Mass Spectrometry. Anal. Chem. 2005, 77, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Madian, A.G.; Myracle, A.D.; Diaz-Maldonado, N.; Rochelle, N.S.; Janle, E.M.; Regnier, F.E. Differential Carbonylation of Proteins as a Function ofin vivoOxidative Stress. J. Proteome Res. 2011, 10, 3959–3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Stevens, J.F.; Maier, C.S. Design, Synthesis, and Application of a Hydrazide-Functionalized Isotope-Coded Affinity Tag for the Quantification of Oxylipid−Protein Conjugates. Anal. Chem. 2007, 79, 3342–3354. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Regnier, F. Identification of yeast oxidized proteins. J. Chromatogr. A 2007, 1141, 22–31. [Google Scholar] [CrossRef]
- Shahrokh, Z.; Eberlein, G.; Buckley, D.; Paranandi, M.V.; Aswad, D.W.; Stratton, P.; Mischak, R.; Wang, Y.J. Major Degradation Products of Basic Fibroblast Growth Factor: Detection of Succinimide and Iso-aspartate in Place of Aspartate15. Pharm. Res. 1994, 11, 936–944. [Google Scholar] [CrossRef]
- Levine, R.L.; Garland, D.; Oliver, C.N.; Amici, A.; Climent, I.; Lenz, A.-G.; Ahn, B.-W.; Shaltiel, S.; Stadtman, E.R. Determination of carbonyl content in oxidatively modified proteins. In Methods in Enzymology; Elsevier BV: Amsterdam, The Netherlands, 1990; Volume 186, pp. 464–478. [Google Scholar]
- Dalle-Donne, I.; Carini, M.; Orioli, M.; Vistoli, G.; Regazzoni, L.; Colombo, G.; Rossi, R.; Milzani, A.; Aldini, G. Protein carbonylation: 2,4-dinitrophenylhydrazine reacts with both aldehydes/ketones and sulfenic acids. Free Radic. Biol. Med. 2009, 46, 1411–1419. [Google Scholar] [CrossRef]
- Campos-Pinto, I.; Méndez, L.; Schouten, J.; Wilkins, J.; Fedorova, M.; Pitt, A.R.; Davis, P.; Spickett, C.M. Epitope mapping and characterization of 4-hydroxy-2-nonenal modified-human serum albumin using two different polyclonal antibodies. Free Radic. Biol. Med. 2019, 144, 234–244. [Google Scholar] [CrossRef]
- Anguizola, J.; Matsuda, R.; Barnaby, O.S.; Hoy, K.; Wa, C.; DeBolt, E.; Koke, M.; Hage, D.S. Review: Glycation of human serum albumin. Clin. Chim. Acta 2013, 425, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Aldini, G.; Vistoli, G.; Regazzoni, L.; Gamberoni, L.; Facino, R.M.; Yamaguchi, S.; Uchida, K.; Carini, M. Albumin Is the Main Nucleophilic Target of Human Plasma: A Protective Role Against Pro-atherogenic Electrophilic Reactive Carbonyl Species? Chem. Res. Toxicol. 2008, 21, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Gamberoni, L.; Orioli, M.; Beretta, G.; Regazzoni, L.; Facino, R.M.; Carini, M. Mass spectrometric characterization of covalent modification of human serum albumin by 4-hydroxy-trans-2-nonenal. J. Mass Spectrom. 2006, 41, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, H.; Edmands, W.; Luca, R.; Yano, Y.; Regazzoni, L.; Iavarone, A.T.; Williams, E.R.; Rappaport, S.M. Adductomics Pipeline for Untargeted Analysis of Modifications to Cys34 of Human Serum Albumin. Anal. Chem. 2016, 88, 10504–10512. [Google Scholar] [CrossRef] [Green Version]
- Sayre, L.M.; Lin, D.; Yuan, Q.; Zhu, X.; Tang, X. Protein Adducts Generated from Products of Lipid Oxidation: Focus on HNE and ONE. Drug Metab. Rev. 2006, 38, 651–675. [Google Scholar] [CrossRef] [PubMed]
- Spiller, S.; Li, Y.; Blüher, M.; Welch, L.; Hoffmann, R. Glycated lysine-141 in haptoglobin improves the diagnostic accuracy for type 2 diabetes mellitus in combination with glycated hemoglobin HbA1c and fasting plasma glucose. Clin. Proteom. 2017, 14, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiller, S.; Frolov, A.; Hoffmann, R. Quantification of Specific Glycation Sites in Human Serum Albumin as Prospective Type 2 Diabetes Mellitus Biomarkers. Protein Pept. Lett. 2018, 24, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas Echeverri, J.C.; Milkovska-Stamenova, S.; Hoffmann, R. A Workflow towards the Reproducible Identification and Quantitation of Protein Carbonylation Sites in Human Plasma. Antioxidants 2021, 10, 369. https://doi.org/10.3390/antiox10030369
Rojas Echeverri JC, Milkovska-Stamenova S, Hoffmann R. A Workflow towards the Reproducible Identification and Quantitation of Protein Carbonylation Sites in Human Plasma. Antioxidants. 2021; 10(3):369. https://doi.org/10.3390/antiox10030369
Chicago/Turabian StyleRojas Echeverri, Juan Camilo, Sanja Milkovska-Stamenova, and Ralf Hoffmann. 2021. "A Workflow towards the Reproducible Identification and Quantitation of Protein Carbonylation Sites in Human Plasma" Antioxidants 10, no. 3: 369. https://doi.org/10.3390/antiox10030369