
Exploring Whether Iron Sequestration within the CNS of Patients with Alzheimer’s Disease Causes a Functional Iron Deficiency That Advances Neurodegeneration

Abstract
:1. Introduction
2. Materials and Methods
2.1. Human CNS Tissue
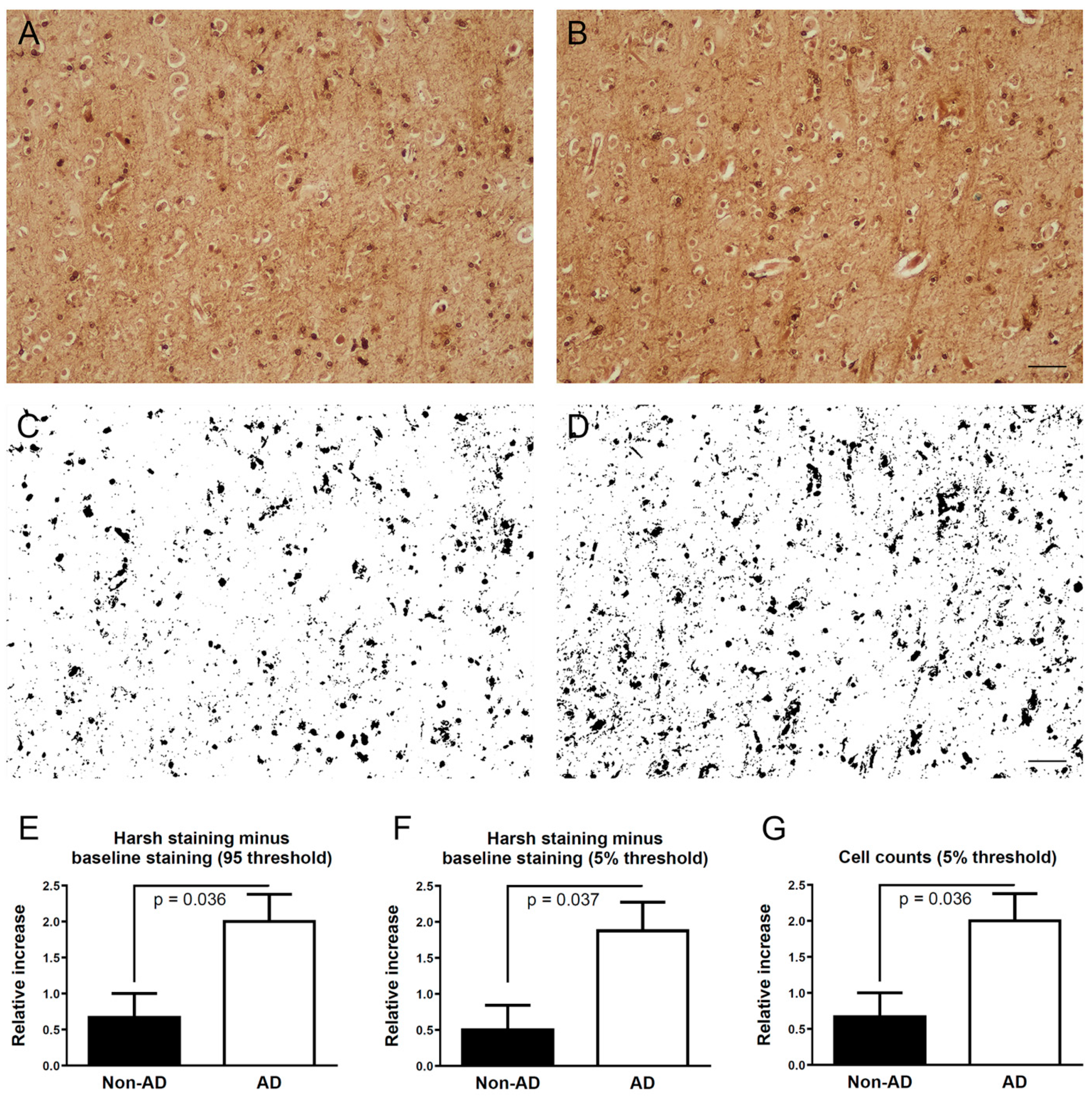
2.2. Iron Histochemical Staining
2.3. Image Analysis of Frontal Cortex Areas
2.4. Bioinformatics
3. Results and Discussion
3.1. Iron Histochemistry
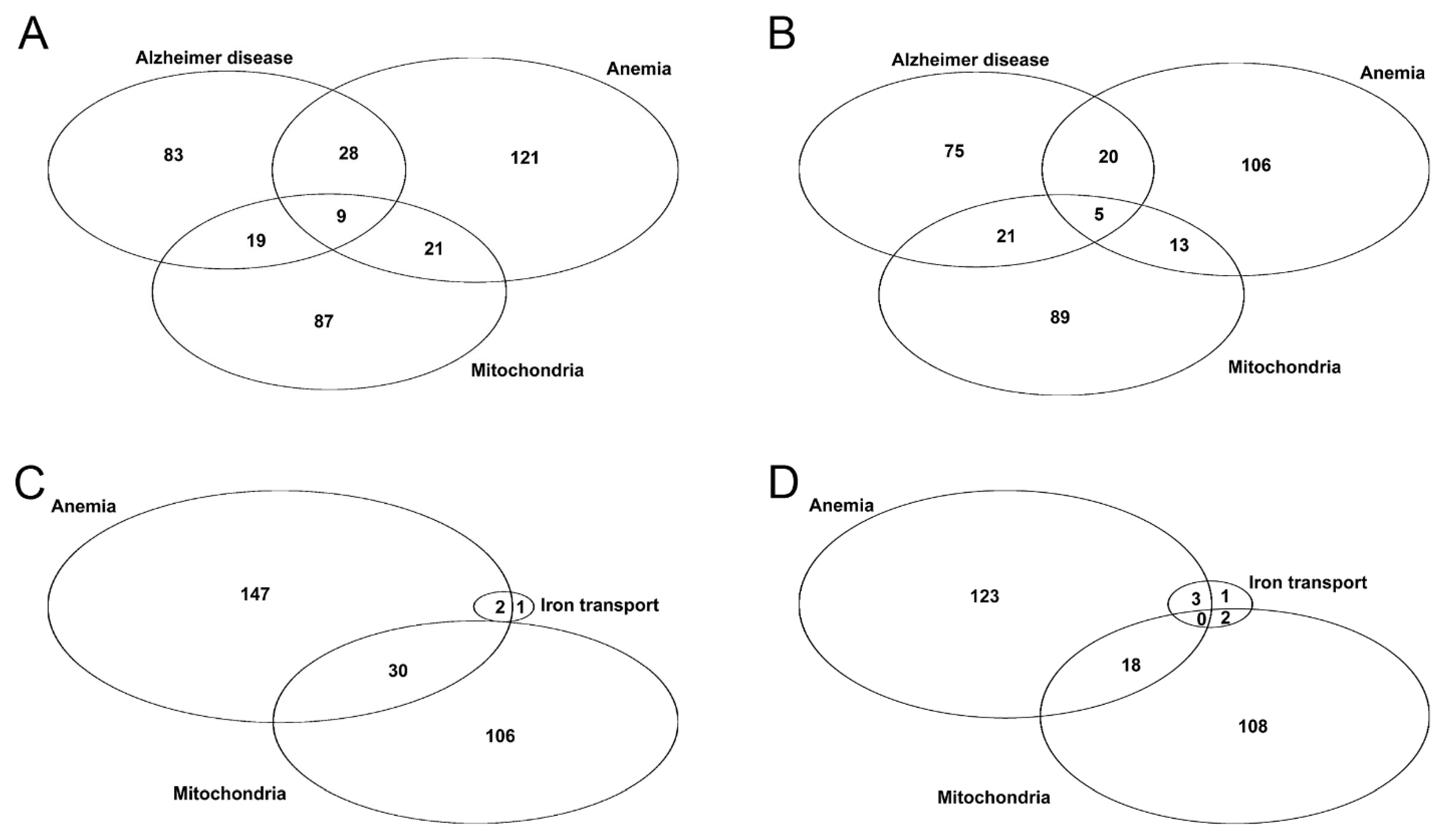
3.2. Selection of a Suitable AD Dataset
3.3. Anemia-Like Responses in AD Olfactory Bulb Dataset
3.4. Altered Expressions Related to Cellular Iron Transport in Early AD
3.5. Altered Expressions Related to Mitochondrial Iron Transport in Early AD
3.6. Changes Related to Mitochondria
3.7. Analysis of Dataset from Human Organoids with or without PITRM1
3.8. Comparison of Datasets
3.9. Hypothesis
3.10. Mechanism Leading to a Functional Deficiency
3.11. An Example of a Functional Deficiency
3.12. Senescent Cells and Impaired Ferritinophagy
3.13. Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Plascencia-Villa, G.; Perry, G. Status and future directions of clinical trials in Alzheimer’s disease. Int. Rev. Neurobiol. 2020, 154, 3–50. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M.; et al. The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurosci. 2022, 23, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.L.; Samant, N.P. Current druggable targets for therapeutic control of Alzheimer’s disease. Contemp. Clin. Trials 2021, 109, 106549. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, J.M.; Noviasky, J.; Britton, S. Aducanumab for Alzheimer’s Disease: Summarized Data From EMERGE, ENGAGE, and PRIME Studies. Sr. Care Pharm. 2022, 37, 329–334. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. The mitochondrial hypothesis: Dysfunction, bioenergetic defects, and the metabolic link to Alzheimer’s disease. Int. Rev. Neurobiol. 2020, 154, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Waiker, D.K.; Bhardwaj, B.; Saraf, P.; Shrivastava, S.K. The molecular mechanism, targets, and novel molecules in the treatment of Alzheimer’s disease. Bioorg. Chem. 2022, 119, 105562. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ding, R.; Xu, Z.; Xue, Y.; Zhang, D.; Zhang, Y.; Li, W.; Li, X. Roles and Mechanisms of the Protein Quality Control System in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 23, 345. [Google Scholar] [CrossRef] [PubMed]
- Plascencia-Villa, G.; Perry, G. Preventive and Therapeutic Strategies in Alzheimer’s Disease: Focus on Oxidative Stress, Redox Metals, and Ferroptosis. Antioxid. Redox Signal. 2021, 34, 591–610. [Google Scholar] [CrossRef] [PubMed]
- Tecalco–Cruz, A.C.; Pedraza-Chaverri, J.; Briones-Herrera, A.; Cruz-Ramos, E.; López–Canovas, L.; Zepeda–Cervantes, J. Protein degradation-associated mechanisms that are affected in Alzheimer’s disease. Mol. Cell. Biochem. 2022, 477, 915–925. [Google Scholar] [CrossRef]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef] [PubMed]
- van Bergen, J.M.G.; Li, X.; Hua, J.; Schreiner, S.J.; Steininger, S.C.; Quevenco, F.C.; Wyss, M.; Gietl, A.F.; Treyer, V.; Leh, S.E.; et al. Colocalization of cerebral iron with Amyloid beta in Mild Cognitive Impairment. Sci. Rep. 2016, 6, 35514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-G.; Park, S.; Rhee, H.Y.; Lee, K.M.; Ryu, C.-W.; Rhee, S.J.; Lee, S.Y.; Wang, Y.; Jahng, G.-H. Quantitative susceptibility mapping to evaluate the early stage of Alzheimer’s disease. NeuroImage: Clin. 2017, 16, 429–438. [Google Scholar] [CrossRef]
- Du, L.; Zhao, Z.; Cui, A.; Zhu, Y.; Zhang, L.; Liu, J.; Shi, S.; Fu, C.; Han, X.; Gao, W.; et al. Increased Iron Deposition on Brain Quantitative Susceptibility Mapping Correlates with Decreased Cognitive Function in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef] [PubMed]
- Spotorno, N.; Acosta-Cabronero, J.; Stomrud, E.; Lampinen, B.; Strandberg, O.T.; van Westen, D.; Hansson, O. Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain 2020, 143, 1341–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron Promotes the Toxicity of Amyloid β Peptide by Impeding Its Ordered Aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef] [Green Version]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Ashraf, A.; Jeandriens, J.; Parkes, H.G.; So, P.-W. Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: Evidence of ferroptosis. Redox Biol. 2020, 32, 101494. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Portbury, S.; Kalinowski, P.; Agarwal, P.; Diouf, I.; Schneider, J.A.; Morris, M.C.; Bush, A.I. Regional brain iron associated with deterioration in Alzheimer’s disease: A large cohort study and theoretical significance. Alzheimer’s Dement. 2021, 17, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J. Neurochem. 2021, 159, 804–825. [Google Scholar] [CrossRef] [PubMed]
- LeVine, S.M. Iron deposits in multiple sclerosis and Alzheimer’s disease brains. Brain Res. 1997, 760, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Perry, G.; Harris, P.L.R.; Liu, Y.; Schubert, K.A.; Smith, M.A. In Situ Oxidative Catalysis by Neurofibrillary Tangles and Senile Plaques in Alzheimer’s Disease: A Central Role for Bound Transition Metals. J. Neurochem. 2000, 74, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Moreira, P.I.; Liu, G.; Dobson, J.; Perry, G.; Smith, M.A.; Zhu, X. Iron: The Redox-active Center of Oxidative Stress in Alzheimer Disease. Neurochem. Res. 2007, 32, 1640–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, N.G.; Perry, G.; Smith, M.A.; Reddy, V.P. NMR Studies of Zinc, Copper, and Iron Binding to Histidine, the Principal Metal Ion Complexing Site of Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 20, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Bousejra-ElGarah, F.; Bijani, C.; Coppel, Y.; Faller, P.; Hureau, C. Iron(II) Binding to Amyloid-β, the Alzheimer’s Peptide. Inorg. Chem. 2011, 50, 9024–9030. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Putignano, V.; Rosato, A.; Banci, L. The human iron-proteome†. Metallomics 2018, 10, 1223–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeVine, S.M.; Zhu, H.; Tague, S.E. A Simplified Method for the Histochemical Detection of Iron in Paraffin Sections: Intracellular Iron Deposits in Central Nervous System Tissue. ASN Neuro 2021, 13, 1759091420982169. [Google Scholar] [CrossRef]
- Lachen-Montes, M.; Zelaya, M.V.; Segura, V.; Fernández-Irigoyen, J.; Santamaría, E. Progressive modulation of the human olfactory bulb transcriptome during Alzheimer’s disease evolution: Novel insights into the olfactory signaling across proteinopathies. Oncotarget 2017, 8, 69663–69679. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.-K.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 2021, 26, 5733–5750. [Google Scholar] [CrossRef] [PubMed]
- Hin, N.; Newman, M.; Pederson, S.; Lardelli, M. Iron Responsive Element-Mediated Responses to Iron Dyshomeostasis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 84, 1597–1630. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Murphy, C. Olfactory and other sensory impairments in Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Cairns, N.J.; Lantos, P.L. Olfactory centres in Alzheimer’s disease: Olfactory bulb is involved in early Braak’s stages. Neuroreport 2001, 12, 285–288. [Google Scholar] [CrossRef]
- Jobin, B.; Boller, B.; Frasnelli, J. Volumetry of Olfactory Structures in Mild Cognitive Impairment and Alzheimer’s Disease: A Systematic Review and a Meta-Analysis. Brain Sci. 2021, 11, 1010. [Google Scholar] [CrossRef]
- Lledo, P.-M.; Valley, M. Adult Olfactory Bulb Neurogenesis. Cold Spring Harb. Perspect. Biol. 2016, 8, a018945. [Google Scholar] [CrossRef]
- Raha, A.A.; Vaishnav, R.A.; Friedland, R.P.; Bomford, A.; Raha-Chowdhury, R. The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 55. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.-D.; Pang, P.; Zhou, X.-T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.-Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Qian, Z.M.; Ke, Y. Rethinking the role of ceruloplasmin in brain iron metabolism. Brain Res. Rev. 2001, 35, 287–294. [Google Scholar] [CrossRef]
- Wan, L.; Nie, G.; Zhang, J.; Zhao, B. Overexpression of Human Wild-Type Amyloid-β Protein Precursor Decreases the Iron Content and Increases the Oxidative Stress of Neuroblastoma SH-SY5Y Cells. J. Alzheimer’s Dis. 2012, 30, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Tsatsanis, A.; Wong, B.X.; Gunn, A.P.; Ayton, S.; Bush, A.I.; Devos, D.; Duce, J.A. Amyloidogenic processing of Alzheimer’s disease β-amyloid precursor protein induces cellular iron retention. Mol. Psychiatry 2020, 25, 1958–1966. [Google Scholar] [CrossRef]
- Wang, Z.; Li, M.; Liu, Y.; Qiao, Z.; Bai, T.; Yang, L.; Liu, B. Dihydroartemisinin triggers ferroptosis in primary liver cancer cells by promoting and unfolded protein response-induced upregulation of CHAC1 expression. Oncol. Rep. 2021, 46, 240. [Google Scholar] [CrossRef]
- Ripa, R.; Dolfi, L.; Terrigno, M.; Pandolfini, L.; Savino, A.; Arcucci, V.; Groth, M.; Tozzini, E.T.; Baumgart, M.; Cellerino, A. MicroRNA miR-29 controls a compensatory response to limit neuronal iron accumulation during adult life and aging. BMC Biol. 2017, 15, 9. [Google Scholar] [CrossRef] [Green Version]
- Terzi, E.M.; Sviderskiy, V.O.; Alvarez, S.W.; Whiten, G.C.; Possemato, R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci. Adv. 2021, 7, eabg4302. [Google Scholar] [CrossRef]
- Yi, X.; Tang, X. Exosomes From miR-19b-3p-Modified ADSCs Inhibit Ferroptosis in Intracerebral Hemorrhage Mice. Front. Cell Dev. Biol. 2021, 9, 661317. [Google Scholar] [CrossRef]
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and Copper in Mitochondrial Diseases. Cell Metab. 2013, 17, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.; Dhorajia, V.V.; Kim, J.; Kim, Y. Mitochondrial iron metabolism and neurodegenerative diseases. Neurotoxicology 2022, 88, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Bayeva, M.; Ghanefar, M.; Potini, V.; Sun, L.; Mutharasan, R.K.; Wu, R.; Khechaduri, A.; Naik, T.J.; Ardehali, H. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl. Acad. Sci. USA 2012, 109, 4152–4157. [Google Scholar] [CrossRef] [Green Version]
- Pearson, S.A.; Cowan, J.A. Evolution of the human mitochondrial ABCB7 [2Fe–2S](GS)4 cluster exporter and the molecular mechanism of an E433K disease-causing mutation. Arch. Biochem. Biophys. 2021, 697, 108661. [Google Scholar] [CrossRef]
- Pearson, S.A.; Cowan, J.A. Glutathione-coordinated metal complexes as substrates for cellular transporters. Metallomics 2021, 13, mfab015. [Google Scholar] [CrossRef]
- Schmucker, S.; Argentini, M.; Calmels, N.; Martelli, A.; Puccio, H. The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 2008, 17, 3521–3531. [Google Scholar] [CrossRef] [Green Version]
- Zoccarato, F.; Cavallini, L.; Alexandre, A. Respiration-dependent Removal of Exogenous H2O2 in Brain Mitochondria: Inhibition by Ca2+. J. Biol. Chem. 2004, 279, 4166–4174. [Google Scholar] [CrossRef] [Green Version]
- Goto, S.; Kawakatsu, M.; Izumi, S.-I.; Urata, Y.; Kageyama, K.; Ihara, Y.; Koji, T.; Kondo, T. Glutathione S-transferase π localizes in mitochondria and protects against oxidative stress. Free Radic. Biol. Med. 2009, 46, 1392–1403. [Google Scholar] [CrossRef] [Green Version]
- Alikhani, N.; Guo, L.; Yan, S.; Du, H.; Pinho, C.M.; Chen, J.X.; Glaser, E.; Yan, S.S. Decreased Proteolytic Activity of the Mitochondrial Amyloid-β Degrading Enzyme, PreP Peptidasome, in Alzheimer’s Disease Brain Mitochondria. J. Alzheimer’s Dis. 2011, 27, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, S.; McDonald, J.; Cuyugan, L.; Aldrich, J.; Kurdoglu, A.; Adkins, J.; Serrano, G.; Beach, T.G.; Craig, D.W.; Valla, J.; et al. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol. Aging 2015, 36, 583–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, C.M.; Teixeira, P.F.; Glaser, E. Mitochondrial import and degradation of amyloid-β peptide. Biochim. Biophys. Acta 2014, 1837, 1069–1074. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, D.; Catania, A.; Viscomi, C.; Deleidi, M.; Bindoff, L.; Ghezzi, D.; Zeviani, M. Role of PITRM1 in Mitochondrial Dysfunction and Neurodegeneration. Biomedicines 2021, 9, 833. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, D.; Torsvik, J.; Dallabona, C.; Teixeira, P.; Sztromwasser, P.; Fernandez-Vizarra, E.; Cerutti, R.; Reyes, A.; Preziuso, C.; D’Amati, G.; et al. Defective PITRM 1 mitochondrial peptidase is associated with Aβ amyloidotic neurodegeneration. EMBO Mol. Med. 2016, 8, 176–190. [Google Scholar] [CrossRef]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Ebralidze, I.I.; She, Z.; Kraatz, H.-B. Electrochemical studies of tau protein-iron interactions—Potential implications for Alzheimer’s Disease. Electrochim. Acta 2017, 236, 384–393. [Google Scholar] [CrossRef]
- Ahmadi, S.; Wu, B.; Song, R.; Zhu, S.; Simpson, A.; Wilson, D.J.; Kraatz, H.-B. Exploring the interactions of iron and zinc with the microtubule binding repeats R1 and R4. J. Inorg. Biochem. 2020, 205, 110987. [Google Scholar] [CrossRef]
- Rao, S.S.; Adlard, P.A. Untangling Tau and Iron: Exploring the Interaction Between Iron and Tau in Neurodegeneration. Front. Mol. Neurosci. 2018, 11, 276. [Google Scholar] [CrossRef] [Green Version]
- Madsen, S.J.; DiGiacomo, P.S.; Zeng, Y.; Goubran, M.; Chen, Y.; Rutt, B.K.; Born, D.; Vogel, H.; Sinclair, R.; Zeineh, M.M. Correlative Microscopy to Localize and Characterize Iron Deposition in Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2020, 4, 525–536. [Google Scholar] [CrossRef]
- Yamamoto, A.; Shin, R.-W.; Hasegawa, K.; Naiki, H.; Sato, H.; Yoshimasu, F.; Kitamoto, T. Iron (III) induces aggregation of hyperphosphorylated τ and its reduction to iron (II) reverses the aggregation: Implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 2002, 82, 1137–1147. [Google Scholar] [CrossRef]
- Liu, J.-L.; Fan, Y.-G.; Yang, Z.-S.; Wang, Z.-Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef] [Green Version]
- Sands, S.A.; Leung-Toung, R.; Wang, Y.; Connelly, J.; LeVine, S.M. Enhanced Histochemical Detection of Iron in Paraffin Sections of Mouse Central Nervous System Tissue: Application in the APP/PS1 Mouse Model of Alzheimer’s Disease. ASN Neuro 2016, 8, 1759091416670978. [Google Scholar] [CrossRef] [Green Version]
- Schrag, M.; Crofton, A.; Zabel, M.; Jiffry, A.; Kirsch, D.; Dickson, A.; Mao, X.W.; Vinters, H.V.; Domaille, D.W.; Chang, C.J.; et al. Effect of Cerebral Amyloid Angiopathy on Brain Iron, Copper, and Zinc in Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 24, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Bulk, M.; Moursel, L.G.; van der Graaf, L.M.; van Veluw, S.J.; Greenberg, S.M.; van Duinen, S.G.; van Buchem, M.A.; van Rooden, S.; van der Weerd, L. Cerebral Amyloid Angiopathy with Vascular Iron Accumulation and Calcification. Stroke 2018, 49, 2081–2087. [Google Scholar] [CrossRef]
- Atamna, H. Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev. 2004, 3, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H. Heme binding to Amyloid-β peptide: Mechanistic role in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 10, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Boyle, K. Amyloid-β peptide binds with heme to form a peroxidase: Relationship to the cytopathologies of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 3381–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139 Suppl 1, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Suragani, R.N.V.S.; Zachariah, R.S.; Velazquez, J.G.; Liu, S.; Sun, C.-W.; Townes, T.M.; Chen, J.-J. Heme-regulated eIF2α kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood 2012, 119, 5276–5284. [Google Scholar] [CrossRef]
- Krukowski, K.; Nolan, A.; Frias, E.S.; Boone, M.; Ureta, G.; Grue, K.; Paladini, M.-S.; Elizarraras, E.; Delgado, L.; Bernales, S.; et al. Small molecule cognitive enhancer reverses age-related memory decline in mice. eLife 2020, 9, e62048. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Li, Y.; Du, J.; Liu, H.; Kanazawa, K.; Nemoto, T.; Nakanishi, H.; Zhao, Y. Copper binding properties of a tau peptide associated with Alzheimer’s disease studied by CD, NMR, and MALDI-TOF MS. Peptides 2006, 27, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Hung, V.W.-S.; Masoom, H.; Kerman, K. Label-free electrochemical detection of Amyloid beta aggregation in the presence of iron, copper and zinc. J. Electroanal. Chem. 2012, 681, 89–95. [Google Scholar] [CrossRef]
- Constantinidis, J. Hypothesis Regarding Amyloid and Zinc in the Pathogenesis of Alzheimer Disease: Potential for Preventive Intervention. Alzheimer Dis. Assoc. Disord. 1991, 5, 31–35. [Google Scholar] [CrossRef]
- Klevay, L.M. Alzheimer’s disease as copper deficiency. Med. Hypotheses 2008, 70, 802–807. [Google Scholar] [CrossRef]
- Hole, M.; Underhaug, J.; Diez, H.; Ying, M.; Røhr, Å.K.; Jorge-Finnigan, A.; Fernàndez-Castillo, N.; García-Cazorla, A.; Andersson, K.K.; Teigen, K.; et al. Discovery of compounds that protect tyrosine hydroxylase activity through different mechanisms. Biochim. Biophys. Acta 2015, 1854, 1078–1089. [Google Scholar] [CrossRef]
- Acevedo, K.; Masaldan, S.; Opazo, C.M.; Bush, A.I. Redox active metals in neurodegenerative diseases. J. Biol. Inorg. Chem. 2019, 24, 1141–1157. [Google Scholar] [CrossRef]
- Uauy, R.; Olivares, M.; Gonzalez, M. Essentiality of copper in humans. Am. J. Clin. Nutr. 1998, 67, S952–S959. [Google Scholar] [CrossRef] [Green Version]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19. [Google Scholar] [CrossRef] [Green Version]
- Vigani, G.; Maffi, D.; Zocchi, G. Iron availability affects the function of mitochondria in cucumber roots. New Phytol. 2009, 182, 127–136. [Google Scholar] [CrossRef]
- Chung, Y.J.; Swietach, P.; Curtis, M.K.; Ball, V.; Robbins, P.A.; Lakhal-Littleton, S. Iron-Deficiency Anemia Results in Transcriptional and Metabolic Remodeling in the Heart Toward a Glycolytic Phenotype. Front. Cardiovasc. Med. 2021, 7, 616920. [Google Scholar] [CrossRef]
- Simonian, N.A.; Hyman, B.T. Functional alterations in Alzheimer’s disease: Diminution of cytochrome oxidase in the hippocampal formation. J. Neuropathol. Exp. Neurol. 1993, 52, 580–585. [Google Scholar] [CrossRef]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical Cytochrome Oxidase Activity Is Reduced in Alzheimer’s Disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Wong-Riley, M.; Antuono, P.; Ho, K.-C.; Egan, R.; Hevner, R.; Liebl, W.; Huang, Z.; Rachel, R.; Jones, J. Cytochrome oxidase in Alzheimer’s disease: Biochemical, histochemical, and immunohistochemical analyses of the visual and other systems. Vis. Res. 1997, 37, 3593–3608. [Google Scholar] [CrossRef] [Green Version]
- Maurer, I.; Zierz, S.; Möller, H.J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Morais, F.M.; Ribeiro, A.M.; Moreira, F.A.; Silva, P.V.G. Systematic review and meta-analysis on the role of mitochondrial cytochrome c oxidase in Alzheimer’s disease. Acta Neuropsychiatr. 2021, 33, 55–64. [Google Scholar] [CrossRef]
- Li, Y.; Park, J.-S.; Deng, J.-H.; Bai, Y. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J. Bioenerg. Biomembr. 2006, 38, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Cavallucci, V.; Ferraina, C.; D’Amelio, M. Key Role of Mitochondria in Alzheimer’s Disease Synaptic Dysfunction. Curr. Pharm. Des. 2013, 19, 6440–6450. [Google Scholar] [CrossRef]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The Whole Structure of the 13-Subunit Oxidized Cytochrome c Oxidase at 2.8 Å. Science 1996, 272, 1136–1144. [Google Scholar] [CrossRef]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadenbach, B. Complex IV—The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 58, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Liddell, J.R. Targeting mitochondrial metal dyshomeostasis for the treatment of neurodegeneration. Neurodegener. Dis. Manag. 2015, 5, 345–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, L.; Lippe, G.; Marchese, E.; De Martino, A.; Mavelli, I.; Rotilio, G.; Ciriolo, M.R. Decrease of cytochrome c oxidase protein in heart mitochondria of copper-deficient rats. Biometals 1998, 11, 207–212. [Google Scholar] [CrossRef] [PubMed]
- de Ungria, M.; Rao, R.; Wobken, J.D.; Luciana, M.; Nelson, C.A.; Georgieff, M.K. Perinatal Iron Deficiency Decreases Cytochrome c Oxidase (CytOx) Activity in Selected Regions of Neonatal Rat Brain. Pediatr. Res. 2000, 48, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, L.M.; Libedinsky, A.; Elorza, A.A. Role of Copper on Mitochondrial Function and Metabolism. Front. Mol. Biosci. 2021, 8, 711227. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.; Filley, C.M.; Kleinschmidt-DeMasters, B.K. Electron transport chain defects in Alzheimer’s disease brain. Neurology 1994, 44, 1090–1096. [Google Scholar] [CrossRef]
- Parker, W.D.; Parks, J.K. Cytochrome C Oxidase in Alzheimer’s Disease Brain: Purification and Characterization. Neurology 1995, 45 Pt 1, 482–486. [Google Scholar] [CrossRef]
- Atamna, H.; Frey, W.H., 2nd. A role for heme in Alzheimer’s disease: Heme binds amyloid and has altered metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 11153–11158. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, B.E.; Stone, M.L.; Gorman, N.; Sinclair, P.R.; Perry, G.; Smith, M.A.; Zhu, X. Heme-a, the heme prosthetic group of cytochrome c oxidase, is increased in Alzheimer’s disease. Neurosci. Lett. 2009, 461, 302–305. [Google Scholar] [CrossRef] [Green Version]
- Vitali, M.; Venturelli, E.; Galimberti, D.; Gatta, L.B.; Scarpini, E.; Finazzi, D. Analysis of the genes coding for subunit 10 and 15 of cytochrome c oxidase in Alzheimer’s disease. J. Neural Transm. 2009, 116, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Garcia, S.; Padgett, K.R.; Moraes, C.T. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum. Mol. Genet. 2012, 21, 5066–5077. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Diaz, F.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 14163–14168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atamna, H.; Frey, W.H., 2nd. Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 2007, 7, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; De Strooper, B.; Arancibia-Cárcamo, I.L. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 2021, 44, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Thadathil, N.; Delotterie, D.F.; Xiao, J.; Hori, R.; McDonald, M.P.; Khan, M.M. DNA Double-Strand Break Accumulation in Alzheimer’s Disease: Evidence from Experimental Models and Postmortem Human Brains. Mol. Neurobiol. 2021, 58, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-M. Aging, Cellular Senescence, and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 1989. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.-T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, M.; Guan, H.; Liu, Z.; Zhao, S.; Takeuchi, S.; Yanagisawa, D.; Tooyama, I. Mitochondrial ferritin in neurodegenerative diseases. Neurosci. Res. 2013, 77, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borquez, D.A.; Urrutia, P.J.; Núñez, M.T. Iron, the endolysosomal system and neuroinflammation: A matter of balance. Neural Regen. Res. 2022, 17, 1003–1004. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.-S.; Breuer, W.; Munnich, A.; Cabantchik, Z.I. Redistribution of accumulated cell iron: A modality of chelation with therapeutic implications. Blood 2008, 111, 1690–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakhlon, O.; Breuer, W.; Munnich, A.; Cabantchik, Z.I. Iron redistribution as a therapeutic strategy for treating diseases of localized iron accumulation. Can. J. Physiol. Pharmacol. 2010, 88, 187–196. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Total Number of Genes Analyzed: 30,259 | |||||
|---|---|---|---|---|---|
| Number of Genes with ≥ 1.5 Fold Expression: 2860 | |||||
| Number of Genes with ≤ 1.5 Fold Expression: 3307 | |||||
| BioProfiler Category | Number (#) of Genes in Category | # of Genes with ≥1.5× Expression (p ≤ 0.05) | Hypergeometric p Value | # of Genes with ≤1.5× Expression (p ≤ 0.05) | Hypergeometric p Value |
| Alzheimer’s Disease | 829 | 139 | 1.58e-11 | 121 | 5.79e-4 |
| Anemia (primary inquiry) | 909 | 179 | 9.81e-22 | 144 | 3.02e-6 |
| Mitochondria | 864 | 136 | 1.96e-9 | 128 | 2.21e-4 |
| Hypoxia | 122 | 10 | 7.27e-1 | 20 | 4.21e-2 |
| Iron | 194 | 25 | 6.92e-2 | 25 | 2.19e-1 |
| Iron transport | 24 | 3 | 3.99e-1 | 6 | 4.05e-2 |
| Heme synthesis | 31 | 3 | 5.72e-1 | 5 | 2.47e-1 |
| Iron responsive elements [38] | # of genes in category | # of genes with ≥1.5× expression (p ≤ 0.05) | Hypergeometric p value | # of genes with ≤1.5× expression (p ≤ 0.05) | Hypergeometric p value |
| 3′ IRE | 1885 | 262 | 7.04e-11 | 382 | 1.01e-34 |
| 5′ IRE | 697 | 106 | 6.49e-7 | 139 | 1.30e-12 |
| HQ 3′ IRE | 189 | 24 | 8.39e-2 | 43 | 2.30e-6 |
| HQ 5′ IRE | 66 | 11 | 4.43e-2 | 9 | 2.93e-1 |
| Human iron-proteome category | # of genes in category | # of genes with ≥1.5× expression (p ≤ 0.05) | Hypergeometric p value | # of genes with ≤1.5× expression (p ≤ 0.05) | Hypergeometric p value |
| Proteins binding individual iron ions | 137 | 18 | 9.54e-2 | 21 | 6.95e-2 |
| Heme-binding proteins | 173 | 25 | 2.14e-2 | 22 | 2.57e-1 |
| Iron–sulfur proteins | 68 | 12 | 2.46e-2 | 14 | 1.41e-2 |
| UniProt keyword | # of genes in category | # of genes with ≥1.5× expression (p ≤ 0.05) | Hypergeometric p value | # of genes with ≤1.5× expression (p ≤ 0.05) | Hypergeometric p value |
| Iron KW-0408 | 344 | 48 | 4.13e-3 | 45 | 1.17e-1 |
| Total Number of Genes Analyzed: 19,561 | |||||
|---|---|---|---|---|---|
| Number of Genes with ≥ 1.5 Fold Expression: 222 | |||||
| Number of Genes with ≤ 1.5 Fold Expression: 228 | |||||
| BioProfiler Category | Number (#) of Genes in Category | # of Genes with ≥1.5× Expression (FDR ≤ 0.05) | Hypergeometric p Value | # of Genes with ≤1.5× Expression (FDR ≤ 0.05) | Hypergeometric p Value |
| Alzheimer’s Disease | 789 | 10 | 4.06e-1 | 23 | 5.26e-5 |
| Anemia (primary inquiry) | 879 | 16 | 4.31e-2 | 21 | 1.51e-3 |
| Mitochondria | 846 | 10 | 4.93e-1 | 18 | 1.04e-2 |
| Hypoxia | 117 | 0 | 1.00 | 2 | 3.97e-1 |
| Iron | 194 | 6 | 2.36e-2 | 4 | 1.91e-1 |
| Iron transport | 26 | 1 | 2.57e-1 | 0 | 1.00 |
| Heme synthesis | 32 | 0 | 1.00 | 1 | 3.13e-1 |
| Iron-responsive elements [38] | # of genes in category | # of genes with ≥1.5× expression (FDR ≤ 0.05) | Hypergeometric p value | # of genes with ≤1.5× expression (FDR ≤ 0.05) | Hypergeometric p value |
| 3′ IRE | 1762 | 17 | 7.92e-1 | 23 | 3.15e-1 |
| 5′ IRE | 638 | 14 | 1.45e-2 | 15 | 8.07e-3 |
| HQ 3′ IRE | 165 | 2 | 5.61e-1 | 5 | 4.43e-2 |
| HQ 5′ IRE | 57 | 2 | 1.37e-1 | 3 | 2.88e-2 |
| Human iron-proteome category | # of genes in category | # of genes with ≥1.5× expression (FDR ≤ 0.05) | Hypergeometric p value | # of genes with ≤1.5× expression (FDR ≤ 0.05) | Hypergeometric p value |
| Proteins binding individual iron ions | 135 | 2 | 4.54e-1 | 2 | 4.68e-1 |
| Heme-binding proteins | 163 | 1 | 8.46e-1 | 1 | 8.53e-1 |
| Iron–sulfur proteins | 65 | 0 | 1.00 | 0 | 1.00 |
| UniProt keyword | # of genes in category | # of genes with ≥1.5× expression (FDR ≤ 0.05) | Hypergeometric p value | # of genes with ≤1.5× expression (FDR ≤ 0.05) | Hypergeometric p value |
| Iron KW-0408 | 317 | 1 | 9.74e-1 | 3 | 7.17e-1 |
| Cluster | Cell Type | Class of Genes Upregulated by ≥1.5× with Hypergeometric p ≤ 0.05 | Class of Genes Downregulated by ≤1.5× with Hypergeometric p ≤ 0.05 |
|---|---|---|---|
| 0 | Neurons | Anemia | Alzheimer’s; Anemia |
| 1 | Neurons | Anemia | - |
| 2 | Neurons | 5′-IRE | - |
| 3 | Glia | - | Alzheimer’s; iron; 5′-IRE; Mitochondria; Proteins binding individual iron |
| 4 | Progenitor cells | - | - |
| 5 | Neurons | Anemia | - |
| 6 | Progenitor cells | - | Mitochondria |
| 7 | Neurons | Anemia | - |
| 8 | Neurons | Anemia | 5′-IRE; HQ 5′-IRE |
| 9 | Astrocytes | - | - |
| 10 | Progenitor cells | Anemia | - |
| 11 | Glia | Anemia | - |
| 12 | Neurons | - | - |
| 13 | Astrocytes | HQ 3′-IRE | Alzheimer’s; Anemia; Mitochondria |
| 14 | Neurons | - | - |
| 15 | Neurons | - | - |
| 16 | Progenitor cells | Anemia | - |
| 17 | Neurons | - | 5′-IRE; HQ 5′-IRE |
| 18 | Neurons | HQ 5′-IRE | 3′-IRE; HQ 3′-IRE; Mitochondria; Heme synthesis |
| 19 | Neurons | - | - |
| 20 | Neurons | - | - |
| 21 | Neurons | - | - |
| 22 | Microglia | - | - |
| 23 | Glia | - | Alzheimer’s; Mitochondria |
| 24 | Progenitor cells | Anemia; Iron | 5′-IRE |
| 25 | Microglia | - | - |
| BioProfiler Category | Genes with ≥1.5× in Common between Datasets | Hyper-Geometric p Value | Genes with ≤1.5× in Common between Datasets | Hyper-Geometric p Value | Genes with ≥1.5× or ≤1.5× in Common between Datasets | Hyper-Geometric p Value | |
|---|---|---|---|---|---|---|---|
| (Initial Stage AD Olfactory Bulb vs. Control Olfactory Bulb) vs. (PITRM1 KO vs. WT Cerebral Organoids) | Anemia | ARG2, RPS29, SHMT2 | 3.16e-1 | CTNNB1, FSTL1, HMGB1, MEIS2, SRGAP3 | 4.96e-2 | ARG2, CTNNB1, FSTL1, GPI, HMGB1, MAPK10, MEIS2, RPS29, SHMT2, SRGAP3, THY1 | 1.02e-1 |
| (Middle Stage AD Olfactory Bulb vs. Control Olfactory Bulb) vs. (PITRM1 KO vs. WT Cerebral Organoids) | Anemia | ARG2, SHMT2, THY1 | 2.13e-1 | HMGB1, TUBA1A, TUBA1C | 1.94 e-1 | ARG2, GPI, HMGB1, KMT2C, MAPK10, PBX1, PGM3, RPS29, SHMT2, SRGAP3, THY1, TUBA1A, TUBA1C | 3.40e-3 |
| (Advanced Stage AD Olfactory Bulb vs. Control Olfactory Bulb) vs. (PITRM1 KO vs. WT Cerebral Organoids) | Anemia | SHMT2 | 8.69e-1 | GPI, PBX1, SRGAP3 | 3.23e-1 | CALR, FSTL1, GPI, PBX1, RPS10, RPS29, SHMT2, SRGAP3, TUBA1C | 2.93e-1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
LeVine, S.M.; Tsau, S.; Gunewardena, S. Exploring Whether Iron Sequestration within the CNS of Patients with Alzheimer’s Disease Causes a Functional Iron Deficiency That Advances Neurodegeneration. Brain Sci. 2023, 13, 511. https://doi.org/10.3390/brainsci13030511
LeVine SM, Tsau S, Gunewardena S. Exploring Whether Iron Sequestration within the CNS of Patients with Alzheimer’s Disease Causes a Functional Iron Deficiency That Advances Neurodegeneration. Brain Sciences. 2023; 13(3):511. https://doi.org/10.3390/brainsci13030511
Chicago/Turabian StyleLeVine, Steven M., Sheila Tsau, and Sumedha Gunewardena. 2023. "Exploring Whether Iron Sequestration within the CNS of Patients with Alzheimer’s Disease Causes a Functional Iron Deficiency That Advances Neurodegeneration" Brain Sciences 13, no. 3: 511. https://doi.org/10.3390/brainsci13030511