
Genetics of Neurogenic Orthostatic Hypotension in Parkinson’s Disease, Results from a Cross-Sectional In Silico Study
 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Genomic Data Processing
2.3. Linkage Disequilibrium Analysis
2.4. Expression Quantitative Trait Loci (eQTL) Analysis
2.5. Enrichment Analysis of SNPs Associated with NOH
2.6. Statistical Analysis
3. Results
3.1. Association between SNPs in PD-Related Genes and NOH
3.2. eQTL Gene Expression Mapping Analysis
3.3. Enrichment Analysis of SNPs Associated with NOH
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected Number of People with P Disease in the Most Populous Nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Espay, A.J. Disease Modification in Parkinson’s Disease: Current Approaches, Challenges, and Future Considerations. Mov. Disord. 2018, 33, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymeropoulos, M.H.; Higgins, J.J.; Golbe, L.I.; Johnson, W.G.; Ide, S.E.; Di Iorio, G.; Sanges, G.; Stenroos, E.S.; Pho, L.T.; Schaffer, A.A.; et al. Mapping of a Gene for Parkinson’s Disease to Chromosome 4q21-Q23. Science 1996, 274, 1197–1199. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. Lrrk2 in Parkinson Disease: Challenges of Clinical Trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Zhang, Y.; Shu, L.; Zhou, X.; Pan, H.; Xu, Q.; Guo, J.; Tang, B.; Sun, Q. A Meta-Analysis of Gba-Related Clinical Symptoms in Parkinson’s Disease. Parkinsons Dis. 2018, 2018, 3136415. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Goldstein, D.S. Cardiovascular Dysautonomia in Parkinson Disease: From Pathophysiology to Pathogenesis. Neurobiol. Dis. 2012, 46, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Velseboer, D.C.; de Haan, R.J.; Wieling, W.; Goldstein, D.S.; de Bie, R.M. Prevalence of Orthostatic Hypotension in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Park. Relat. Disord. 2011, 17, 724–729. [Google Scholar] [CrossRef] [Green Version]
- Quarracino, C.; Otero-Losada, M.; Capani, F.; Pérez-Lloret, S. Prevalence and Factors Related to Orthostatic Syndromes in Recently Diagnosed, Drug-Naïve Patients with Parkinson Disease. Clin. Auton. Res. 2020, 30, 265–271. [Google Scholar] [CrossRef]
- Freeman, R.; Wieling, W.; Axelrod, F.B.; Benditt, D.G.; Benarroch, E.; Biaggioni, I.; Cheshire, W.P.; Chelimsky, T.; Cortelli, P.; Gibbons, C.H.; et al. Consensus Statement on the Definition of Orthostatic Hypotension, Neurally Mediated Syncope and the Postural Tachycardia Syndrome. Clin. Auton. Res. 2011, 21, 69–72. [Google Scholar] [CrossRef]
- Pfeiffer, R. F Autonomic Dysfunction in Parkinson’s Disease. Neurotherapeutics 2020, 17, 1464–1479. [Google Scholar] [CrossRef]
- Perez-Lloret, S.; Rey, M.V.; Fabre, N.; Ory, F.; Spampinato, U.; Senard, J.M.; Pavy-Le Traon, A.; Montastruc, J.L.; Rascol, O. Factors Related to Orthostatic Hypotension in Parkinson’s Disease. Park. Relat. Disord. 2012, 18, 501–505. [Google Scholar] [CrossRef]
- Poon, I.O.; Braun, U. High Prevalence of Orthostatic Hypotension and Its Correlation with Potentially Causative Medications among Elderly Veterans. J. Clin. Pharm. Ther. 2005, 30, 173–178. [Google Scholar] [CrossRef]
- Farrell, M.C.; Shibao, C.A. Morbidity and Mortality in Orthostatic Hypotension. Auton. Neurosci. 2020, 229, 102717. [Google Scholar] [CrossRef]
- Feldstein, C.; Weder, A.B. Orthostatic Hypotension: A Common, Serious and Underrecognized Problem in Hospitalized Patients. J. Am. Soc. Hypertens. 2012, 6, 27–39. [Google Scholar] [CrossRef]
- Dadar, M.; Fereshtehnejad, S.M.; Zeighami, Y.; Dagher, A.; Postuma, R.B.; Collins, D.L. White Matter Hyperintensities Mediate Impact of Dysautonomia on Cognition in Parkinson’s Disease. Mov. Disord. Clin. Pract. 2020, 7, 639–647. [Google Scholar] [CrossRef]
- Derejko, M.; Sławek, J.; Wieczorek, D.; Brockhuis, B.; Dubaniewicz, M.; Lass, P. Regional Cerebral Blood Flow in Parkinson’s Disease as an Indicator of Cognitive Impairment. Nucl. Med. Commun. 2006, 27, 945–951. [Google Scholar] [CrossRef]
- Gao, Y.; Tang, H.; Nie, K.; Zhu, R.; Gao, L.; Feng, S.; Wang, L.; Zhao, J.; Huang, Z.; Zhang, Y.; et al. Hippocampal Damage and White Matter Lesions Contribute to Cognitive Impairment in Mptp-Lesioned Mice with Chronic Cerebral Hypoperfusion. Behav. Brain Res. 2019, 368, 111885. [Google Scholar] [CrossRef]
- Loureiro, D.; Bilbao, R.; Bordet, S.; Grasso, L.; Otero-Losada, M.; Capani, F.; Ponzo, O.J.; Perez-Lloret, S. A Systematic Review and Meta-Analysis on the Association between Orthostatic Hypotension and Mild Cognitive Impairment and Dementia in Parkinson’s Disease. Neurol. Sci. 2022. [Google Scholar] [CrossRef]
- Goldstein, D.S. Dysautonomia in Parkinson’s Disease: Neurocardiological Abnormalities. Lancet Neurol. 2003, 2, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Chelban, V.; Vichayanrat, E.; Schottlaende, L.; Iodice, V.; Houlden, H. Autonomic Dysfunction in Genetic Forms of Synucleinopathies. Mov. Disord. 2018, 33, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Tijero, B.; Gómez Esteban, J.C.; Somme, J.; Llorens, V.; Lezcano, E.; Martinez, A.; Rodríguez, T.; Berganzo, K.; Zarranz, J.J. Autonomic Dysfunction in Parkinsonian Lrrk2 Mutation Carriers. Park. Relat. Disord. 2013, 19, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Progression Marker Initiative. The Parkinson Progression Marker Initiative (PPMI). Prog. Neurobiol. 2011, 95, 629–635. [Google Scholar] [CrossRef]
- Rossi, M.; Perez-Lloret, S.; Merello, M. How Much Time Is Needed in Clinical Practice to Reach a Diagnosis of Clinically Established Parkinson’s Disease? Park. Relat. Disord. 2021, 92, 53–58. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. Plink: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Machiela, M.J.; Chanock, S.J. Ldlink: A Web-Based Application for Exploring Population-Specific Haplotype Structure and Linking Correlated Alleles of Possible Functional Variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef] [Green Version]
- Oscanoa, J.; Sivapalan, L.; Gadaleta, E.; Dayem Ullah, A.Z.; Lemoine, N.R.; Chelala, C. Snpnexus: A Web Server for Functional Annotation of Human Genome Sequence Variation (2020 Update). Nucleic Acids Res. 2020, 48, W185–W192. [Google Scholar] [CrossRef]
- Rafanelli, M.; Walsh, K.; Hamdan, M.H.; Buyan-Dent, L. Autonomic Dysfunction: Diagnosis and Management. Handb. Clin. Neurol. 2019, 167, 123–137. [Google Scholar]
- Yuan, J.; Matsuura, E.; Higuchi, Y.; Hashiguchi, A.; Nakamura, T.; Nozuma, S.; Sakiyama, Y.; Yoshimura, A.; Izumo, S.; Takashima, H. Hereditary Sensory and Autonomic Neuropathy Type Iid Caused by an Scn9a Mutation. Neurology 2013, 80, 1641–1649. [Google Scholar] [CrossRef]
- Fuchs, J.; Nilsson, C.; Kachergus, J.; Munz, M.; Larsson, E.M.; Schüle, B.; Langston, J.W.; Middleton, F.A.; Ross, O.A.; Hulihan, M.; et al. Phenotypic Variation in a Large Swedish Pedigree Due to Snca Duplication and Triplication. Neurology 2007, 68, 916–922. [Google Scholar] [CrossRef]
- Ruffle, J.K.; Hyare, H.; Howard, M.A.; Farmer, A.D.; Apkarian, A.V.; Williams, R.S.C.; Aziz, Q.; Nachev, P. The Autonomic Brain: Multi-Dimensional Generative Hierarchical Modelling of the Autonomic Connectome. Cortex 2021, 143, 164–179. [Google Scholar] [CrossRef]
- Bony, C.; Roche, S.; Shuichi, U.; Sasaki, T.; Crackower, M.A.; Penninger, J.; Mano, H.; Pucéat, M. A Specific Role of Phosphatidylinositol 3-Kinase Gamma. A Regulation of Autonomic Ca(2)+ Oscillations in Cardiac Cells. J. Cell Biol. 2001, 152, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.Z.; Chen, J.; Hu, Z.W.; Hoffman, B.B. Phosphorylation of the Camp Response Element-Binding Protein and Activation of Transcription by Alpha1 Adrenergic Receptors. J. Biol. Chem. 1998, 273, 30033–30038. [Google Scholar] [CrossRef] [Green Version]
- Nie, L.; Pascoa, T.C.; Pike, W.A.C.; Bushell, S.R.; Quigley, A.; Ruda, G.F.; Chu, A.; Cole, V.; Speedman, D.; Moreira, T.; et al. The Structural Basis of Fatty Acid Elongation by the Elovl Elongases. Nat. Struct. Mol. Biol. 2021, 28, 512–520. [Google Scholar] [CrossRef]
- Clark, C.M.; Monahan, K.D.; Drew, R.C. Omega-3 Polyunsaturated Fatty Acid Supplementation Reduces Blood Pressure but Not Renal Vasoconstrictor Response to Orthostatic Stress in Healthy Older Adults. Physiol. Rep. 2018, 6, e13674. [Google Scholar] [CrossRef]
- Monahan, K.D.; Wilson, T.E.; Ray, C.A. Omega-3 Fatty Acid Supplementation Augments Sympathetic Nerve Activity Responses to Physiological Stressors in Humans. Hypertension 2004, 44, 732–738. [Google Scholar] [CrossRef] [Green Version]
- Moritoh, Y.; Abe, S.I.; Akiyama, H.; Kobayashi, A.; Koyama, R.; Hara, R.; Kasai, S.; Watanabe, M. The Enzymatic Activity of Inositol Hexakisphosphate Kinase Controls Circulating Phosphate in Mammals. Nat. Commun. 2021, 12, 4847. [Google Scholar] [CrossRef]
- Gilani, A.; Ramsay, S.E.; Juraschek, S.P.; Papacosta, O.; Lennon, L.T.; Whincup, P.H.; Wannamethee, S.G. Associations of the Systolic and Diastolic Components of Orthostatic Hypotension with Markers of Cardiovascular Risk in Older Men: A Cross-Sectional Analysis from the British Regional Heart Study. J. Clin. Hypertens. 2020, 22, 1892–1901. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, X.; Eaton, M.; Wu, J.; Ma, Z.; Lai, S.; Park, A.; Ahmad, T.S.; Que, Z.; Lee, J.H.; et al. Severe Deficiency of the Voltage-Gated Sodium Channel Na(V)1.2 Elevates Neuronal Excitability in Adult Mice. Cell Rep. 2021, 36, 109495. [Google Scholar] [CrossRef]
- Wolff, M.; Brunklaus, A.; Zuberi, S.M. Phenotypic Spectrum and Genetics of Scn2a-Related Disorders, Treatment Options, and Outcomes in Epilepsy and Beyond. Epilepsia 2019, 60 (Suppl. S3), S59–S67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, F.Z.; Campain, A.E.; Yang, Y.H.; Morris, B.J. Meta-Analysis of Genome-Wide Gene Expression Differences in Onset and Maintenance Phases of Genetic Hypertension. Hypertension 2010, 56, 319–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Palmieri, F.; Indiveri, C. The Mitochondrial Carnitine Acyl-Carnitine Carrier (Slc25a20): Molecular Mechanisms of Transport, Role in Redox Sensing and Interaction with Drugs. Biomolecules 2021, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Nunes, J.; Taipa, R.; Melo Pires, M.; Pinto Basto, J.; Barros, P. Adult Polyglucosan Body Disease—An Atypical Compound Heterozygous with a Novel Gbe1 Mutation. Neurol. Sci. 2021, 42, 2955–2959. [Google Scholar] [CrossRef]
- Chen, S.; Zou, J.L.; He, S.; Li, W.; Zhang, J.W.; Li, S.J. Adult-Onset Autosomal Dominant Leukodystrophy and Neuronal Intranuclear Inclusion Disease: Lessons from Two New Chinese Families. Neurol. Sci. 2022, 43, 1–9. [Google Scholar] [CrossRef]
- Shibao, C.A.; Joos, K.; Phillips, J.A., 3rd; Cogan, J.; Newman, J.H.; Hamid, R.; Meiler, J.; Capra, J.; Sheehan, J.; Vetrini, F.; et al. Familial Autonomic Ganglionopathy Caused by Rare Chrna3 Genetic Variants. Neurology 2021, 97, e145–e155. [Google Scholar] [CrossRef]
- Wassenberg, T.; Deinum, J.; van Ittersum, F.J.; Kamsteeg, E.J.; Pennings, M.; Verbeek, M.M.; Wevers, R.A.; van Albada, M.E.; Kema, I.P.; Versmissen, J.; et al. Clinical Presentation and Long-Term Follow-up of Dopamine Beta Hydroxylase Deficiency. J. Inherit. Metab. Dis. 2021, 44, 554–565. [Google Scholar] [CrossRef]


{kind=link}
| NOH (n = 35) | Without NOH (n = 269) | p-Value | |
|---|---|---|---|
| Age (years) | 61.10 ± 9.7 | 58.40 ± 9.90 | 0.122 |
| Males | 21 (60.00%) | 157 (58.15%) | 0.034 |
| Age at PD onset (years) | 60.40 ± 9.95 | 57.45 ± 9.89 | 0.106 |
| PD duration (months) * | 7 (1–64) | 5 (1–84) | 0.276 |
| Hoehn and Yahr | 1.67 ± 0.53 | 1.63 ± 0.53 | 0.616 |
| MDS-UPDRS Total score | 34.14 ± 20.39 | 32.76 ± 16.20 | 0.706 |
| Part I score | 6.05 ± 4.99 | 6.14 ± 4.45 | 0.876 |
| Part II score | 6.52 ± 5.51 | 6.33 ± 4.96 | 0.840 |
| Part III score | 21.15 ± 12.23 | 19.77 ± 10.16 | 0.530 |
| Part IV score | 2.03 ± 3.21 | 1.47 ± 2.61 | 0.660 |
| Anti-Parkinsonian drugs | 8 (23.53%) | 77 (28.52%) | 0.779 |
| L-DOPA | 5 (14.7%) | 52 (19.26%) | 0.881 |
| Dopamine Agonist | 3 (8.82%) | 41(15.18%) | 0.359 |
| Other drugs | 4(11.76%) | 56 (20.74%) | 0.523 |
| Levodopa-equivalent daily dose (mg/day) | 920 ± 499 | 620 ± 828 | 0.169 |
| CSF Biomarkers | |||
| tTau | 172.3 ± 52.91 | 165.56 ± 59.73 | 0.500 |
| pTau | 14.45 ± 5.35 | 13.95 ± 5.22 | 0.640 |
| a-Synuclein | 95.595 ± 32.5 | 104.71 ± 43.9 | 0.390 |
| Neurofilament Light Chain (NFLC) | 120.13 ± 68.67 | 100.24 ± 60.8 | 0.290 |
| SNP (Gene) | Whole Sample (n = 304) | NOH (n = 35) | Without NOH (n = 269) | OR (95% CI) | p-Value | Adjusted p-Value |
|---|---|---|---|---|---|---|
| rs104893877 (SNCA) | CC:292 (96.10%) CT:12 (3.90%) TT:0 | CC:31 (88.57%) CT:4 (11.43%) TT:0 | CC:261 (97.03%) CT:8 (2.97%) TT:0 | 58.03 (2.95–190) | 0.012 | 0.031 |
| rs34311866 (TMEM175) | TT:180 (59.21%) TC:105 (34.54%) CC:19 (6.25%) | TT:17 (48.57%) TC:13 (37.14%) CC:5 (14.29%) | TT:163 (60.59%) TC:92 (34.20%) CC:14 (5.21%) | 8.46 (2.98–29.81) | <0.001 | 0.002 |
| rs6812193 (FAM47E- STBD1) | CC:132 (43.42%) CT:133 (43.75%) TT:39 (12.83%) | CC:14 (40.00%) CT:15 (42.86%) TT:6 (17.14%) | CC:118 (43.87%) CT:118 (43.87%) TT: 33 (12.27%) | 3.67 (1.44–11.00) | 0.010 | 0.031 |
| rs11060180 (CCDC62) | AA:101 (33.22%) AG:141 (46.38%) GG:62 (20.39%) | AA:8 (22.86%) AG:16 (45.71%) GG:11 (31.43%) | AA:93 (34.57%) AG:125 (46.46%) GG:51 (18.95%) | 3.54 (1.53–9.67) | 0.007 | 0.031 |
| rs353116 (SCN3A) | CC:119 (39.15%) CT:140 (46.10%) TT:45 (14.80%) | CC:12 (34.28%) CT:14 (40.00%) TT:9 (25.71%) | CC:107 (39.77%) CT:126 (46.84%) TT:36 (13.38%) | 3.48 (1.39–10.10) | 0.012 | 0.031 |
| rs3910105 (SNCA) | AA:101 (33.22%) AG:146 (48.03%) GG:57 (18.75%) | AA:5 (14.28%) AG:22 (62.86%) GG:8 (22.86%) | AA:96 (35.68%) AG:124 (46.1%) GG:49 (18.22%) | 3.48 (1.31–10.46) | 0.017 | 0.034 |
| rs329648 (MIR4696) | CC:113 (37.17%) TC:148 (48.68%) TT:43 (14.14%) | CC:8 (22.86%) TC:24 (68.57%) TT:3 (8.57%) | CC:105 (39.03%) TC:124 (46.09%) TT:40 (14.87%) | 3.47 (1.27–11.54) | 0.023 | 0.034 |
| rs13294100 (SH3GL2) | GG:137 (45.07%) GT:137 (45.07%) TT:30 (9.86%) | GG:11 (31.43%) GT:20 (57.14%) TT:4 (11.43%) | GG:126 (46.84%) GT:117 (43.49%) TT:26 (9.66%) | 3.34 (1.27–10.27) | 0.021 | 0.034 |
| rs55785911 (LZTS3/DDRGK1) | GG:117 (38.48%) GA:143 (47.04%) AA:44 (14.47%) | GG:10 (28.57%) GA:18 (51.42%) AA:7 (20.00%) | GG:107 (39.41%) GA:125 (46.47%) AA:37 (13.75%) | 2.52 (1.03–6.67) | 0.048 | 0.048 |
| rs10906923 (ITGA8) | AA:140 (46.1%) AC:124 (40.79%) CC:40 (13.16%) | AA:18 (51.43%) AC:14 (40.03%) CC:3 (8.57%) | AA:122 (45.35%) AC:110 (40.89%) CC:37 (13.75%) | 0.37 (0.13–0.91) | 0.042 | 0.048 |
| rs12497850 (IP6K2) | TT:138 (45.39%) TG:140 (46.05%) GG:26 (8.55%) | TT:16 (45.71%) TG:19 (54.28%) GG: 0 | TT:122 (45.35%) TG:121 (44.98%) GG:26 (9.66%) | 0.29 (0.09–0.80) | 0.024 | 0.035 |
| rs10797576 (SIPA1L2) | CC:206 (67.76%) CT:91 (29.93%) TT:7 (2.30%) | CC: 25 (71.43%) CT:10 (28.57%) TT: 0 | CC:181 (67.28%) CT:81 (30.11%) TT:7 (2.61%) | 0.19 (0.04–0.74) | 0.027 | 0.035 |
| rs2694528 (NDUFAF2) | CC:242 (79.60%) CA:57 (18.75%) AA:5 (1.64%) | CC:30 (85.71%) CA:5 (14.28%) | CC:212 (78.81%) CA:52 (19.33%) AA:5 (1.86%) | 0.13 (0.01–0.88) | 0.048 | 0.048 |
| SNP | Chr | Position (bp) | Ref | ALT | MAF SNPnexus | MAF in the Study | Gene | Gene Position | HWE |
|---|---|---|---|---|---|---|---|---|---|
| rs10797576 | 1 | 232,664,611 | C | T | 0.127396 | 0.1728 | SIPA1L2 | Intronic | 0.802 |
| rs10906923 | 10 | 15,569,598 | C | A | 0.460863 | 0.3389 | ITGA8 | Intronic | 0.194 |
| rs329648 | 11 | 133,765,367 | T | T | 0.464457 | 0.3854 | MIR4696 | Intronic | 0.746 |
| rs34637584 | 12 | 40,734,202 | G | A | 0.000400 | 0.1096 | LRRK2 | 3-UTR. coding nonsyn | 0.489 |
| rs11060180 | 12 | 123,303,586 | A | G | 0.251597 | 0.4369 | CCDC62 | Intronic | 0.327 |
| rs11158026 | 14 | 55,348,869 | C | C | 0.489816 | 0.3317 | GCH1 | Non-coding. Intronic | 0.496 |
| rs353116 | 2 | 166,133,632 | C | T | 0.442692 | 0.3787 | SCN2A | Non-coding. Intronic | 0.9105 |
| rs55785911 | 20 | 3,153,503 | G | A | 0.39976 | 0.3833 | LZTS3 | Intronic | 0.937 |
| rs12497850 | 3 | 48,748,989 | G | G | 0.26857 | 0.3173 | IP6K2 | Non-coding. Intronic | 0.298 |
| rs34311866 | 4 | 951,947 | T | C | 0.139976 | 0.2342 | TMEM175 | 3-UTR | 0.503 |
| rs6812193 | 4 | 77,198,986 | C | T | 0.313299 | 0.3422 | FAM47E-STBD1 | Intronic | 0.725 |
| rs3910105 | 4 | 90,682,571 | A | G | 0.307708 | 0.4302 | SNCA | Intronic | 0.829 |
| rs104893877 | 4 | 90,749,300 | C | T | - | 0.02 | SNCA | Coding non syn | 0.270 |
| rs2694528 | 5 | 60,273,923 | C | C | 0.134585 | 0.108 | NDUFAF2 | Intronic | 0.949 |
| rs13294100 | 9 | 17,579,690 | T | G | 0.457268 | 0.322 | SH3GL2 | Non-coding. Intronic | 0.621 |
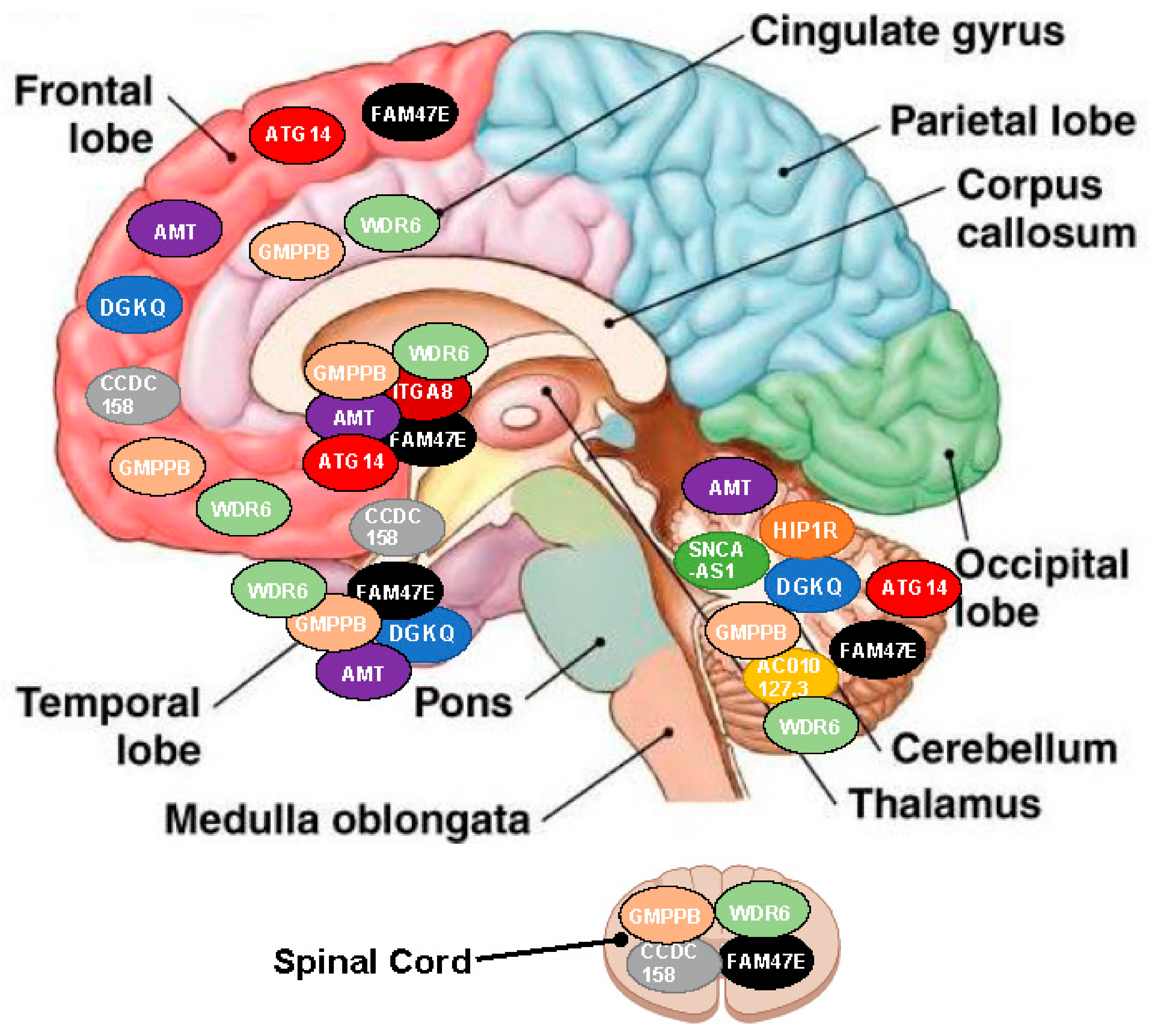
| SNP (Gene) | SNP in LD | Chr | Influence on Gene | Gene Expressed | Effect Size | p-Values | R2 | D´ |
|---|---|---|---|---|---|---|---|---|
| rs34311866 (TMEM175) | 27 | 4 | DGKQ | BHPC, BCRB, BFC | −0.47/ −0.19 | 5.90 × 10−7 | 0.65/1.00 | 0.86/1.00 |
| rs34311866 (TMEM175) | 1 | 12 | HIP1R | BCRB | 0.22 | 3.90 × 10−6 | 1.00 | 1.00 |
| Rs3910105 (SNCA) | 61 | 4 | SNCA-AS1 | BCRB | −0.63/ −0.39 | 1.66 × 10−12/ 2.11 × 10−5 | 0.62/1.00 | 0.86/1.00 |
| Rs353116 (SCN2A) | 2 | 2 | AC010127.3 | BCRB | 0.59 | 9.00 × 10−6 | 0.17 | 0.88 |
| Rs6812193 (FAM47E-STBD) | 14 | 4 | CCDC158 | BC, BFC, BSC | 0.32/0.48 | 1.20 × 10−5 | 0.5/1.00 | 0.97/1.00 |
| Rs6812193 (FAM47E-STBD) | 99 | 4 | FAM47E | BCB, BCRB, BC, BFC, BHPC, BNA, PB, BSC | 0.26/0.45 | 7.59 × 10−5/ 8.78 × 10−9 | 0.91/1.00 | 0.99/1.00 |
| Rs10906923 (FAM47E-STBD) | 200 | 10 | ITGA8 | BCB, BC, BHY, BNA | −0.41/ −0.31 | 4.49 × 10−5/ 7.67 × 10−8 | 0.53/ 1.00 | 0.84/ 1.00 |
| Rs12497850 (IP6K2) | 1232 | 3 | AMT | BCRB, BC, BCB, BNA, BFC, BHPC, BP | 0.19/ 0.67 | 2.02 × 10−4/ 5.28 × 10−27 | 0.57/ 1.00 | 0.82/ 1.00 |
| Rs12497850 (IP6K2) | 351 | 3 | GMPPB | BAMY, BAC, BCRB, BC, BFC, BHPC, BP, BSC, BSN | −0.51/ −0.51 | 1.48 × 10−10/ 4.20 × 10−4 | 0.57/ 0.88 | 0.82/0.95 |
| Rs12497850 (IP6K2) | 1602 | 3 | WDR6 | BAMY, BCRB, BCB, BC, BFC, BHPC, BNA, BP, BSC, BSN | −0.45/ 0.40 | 2.03 × 10−27/ 1.89 × 10−4 | 0.50/ 1.00 | 0.88/ 1.00 |
| Rs11158026 (GCH1) | 39 | 14 | ATG14 | BFC, BP, BCRB | −0.43/ 0.35 | 1.62 × 10−8/ 3.20 × 10−5 | 0.50/ 1.00 | 0.82/1.00 |
| NOH-Associated SNP | Targeted Genes | Localization and Effect Direction (Arrow) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hippocampus | Frontal Cortex | Anterior Cingulate Cortex | Amygdala | Cerebellum | Spinal cord | Caudate | Accumbens | Putamen | Hypothalamus | ||
| rs34311866 | DGKQ | ↓ | ↓ | - | - | ↓ | - | - | - | - | - |
| HIP1R | - | - | - | - | ↑ | - | - | - | - | - | |
| rs3910105 | SNCA-AS1 | - | - | - | - | ↓ | - | - | - | - | - |
| rs353116 | AC010127.3 | - | - | - | - | ↑ | - | - | - | - | - |
| rs6812193 | CCDC 158 | - | ↑ | - | - | - | ↑ | - | - | - | ↑ |
| FAM47E | ↑ | ↑ | - | - | ↑ | ↑ | ↑ | ↑ | ↑ | - | |
| rs10906923 | ITGA8 | - | - | - | - | - | - | ↓ | ↓ | - | - |
| rs12497850 | AMT | ↑ | ↑ | - | - | ↑ | - | ↑ | ↑ | ↑ | - |
| GMPPB | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | - | - | ↓ | - | |
| WDR6 | ↓/↑ * | ↓/↑ * | ↓/↑ * | - | ↓/↑ * | ↓/↑ * | ↓/↑ * | ↓/↑ * | ↓/↑ * | - | |
| rs11158026 | ATG14 | - | ↓/↑ * | - | - | ↓/↑ * | - | - | - | ↓/↑ * | - |
| Genes Involved | Metabolic Pathway | p-Value | Adjusted p-Values |
|---|---|---|---|
| IP6K2 | Synthesis of inositol phosphate in the nucleus | 0.003 | 0.047 |
| Inositol phosphate metabolism | 0.039 | 0.047 | |
| SH3GL2 | Retrograde neurotrophin signaling | 0.012 | 0.047 |
| InlB-mediated entry of listeria monocytogenes into a cell | 0.013 | 0.047 | |
| Listeria monocytogenes entry into host cells | 0.017 | 0.047 | |
| Negative regulation of MET activity | 0.017 | 0.047 | |
| EGFR downregulation | 0.026 | 0.047 | |
| Signaling by EGFR | 0.042 | 0.047 | |
| Lysosome vesicle biogenesis | 0.029 | 0.047 | |
| Recycling pathway of L1 | 0.040 | 0.047 | |
| Golgi-associated vesicle biogenesis | 0.046 | 0.047 | |
| SCN2A | Interaction between L1 and ankyrins | 0.026 | 0.047 |
| Sodium channel | 0.035 | 0.047 | |
| SH3GL2-SCN2A | L1CAM interactions | 0.004 | 0.047 |
| ITGA8 | Molecules associated with elastic fibers | 0.032 | 0.047 |
| Elastic fiber formation | 0.037 | 0.047 | |
| NDUFAF2 | Mitochondrial complex I biogenesis | 0.041 | 0.047 |
| PRKAR2A | ROBO receptors bind AKAP5 | 0.013 | 0.047 |
| AC-mediated CREB1 phosphorylation | 0.018 | 0.047 | |
| PKA activation in glucagon signaling | 0.025 | 0.047 | |
| PKA activation | 0.027 | 0.047 | |
| PKA-mediated phosphorylation of CREB | 0.029 | 0.047 | |
| DARPP-32 events | 0.035 | 0.047 | |
| Glucagon signaling in metabolic regulation | 0.048 | 0.048 | |
| SLC25A20 | Carnitine metabolism | 0.019 | 0.047 |
| ELOVL7 | Synthesis of very-long-chain fatty acyl-CoA | 0.035 | 0.047 |
| Fatty acid metabolism | 0.028 | 0.047 | |
| QARS | Mitochondrial tRNA aminoacylation | 0.031 | 0.047 |
| Cytosolic tRNA aminoacylation | 0.035 | 0.047 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chevalier, G.; Udovin, L.; Otero-Losada, M.; Bordet, S.; Capani, F.; Luo, S.; Goetz, C.G.; Perez-Lloret, S. Genetics of Neurogenic Orthostatic Hypotension in Parkinson’s Disease, Results from a Cross-Sectional In Silico Study. Brain Sci. 2023, 13, 506. https://doi.org/10.3390/brainsci13030506
Chevalier G, Udovin L, Otero-Losada M, Bordet S, Capani F, Luo S, Goetz CG, Perez-Lloret S. Genetics of Neurogenic Orthostatic Hypotension in Parkinson’s Disease, Results from a Cross-Sectional In Silico Study. Brain Sciences. 2023; 13(3):506. https://doi.org/10.3390/brainsci13030506
Chicago/Turabian StyleChevalier, Guenson, Lucas Udovin, Matilde Otero-Losada, Sofia Bordet, Francisco Capani, Sheng Luo, Christopher G. Goetz, and Santiago Perez-Lloret. 2023. "Genetics of Neurogenic Orthostatic Hypotension in Parkinson’s Disease, Results from a Cross-Sectional In Silico Study" Brain Sciences 13, no. 3: 506. https://doi.org/10.3390/brainsci13030506