
Binge-like Prenatal Ethanol Exposure Causes Impaired Cellular Differentiation in the Embryonic Forebrain and Synaptic and Behavioral Defects in Adult Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Ethanol Administration
2.3. Protein Extraction, Electrophoresis, and Immunoblotting
2.4. Y-Maze Spatial Memory (SM) Task
2.5. Social Recognition Memory (SRM)
2.6. Elevated Plus Maze (EPM)
2.7. Marble Burying Test (MBT)
2.8. Nestlet Shredding Test (NST)
2.9. LTP and Long-Term Depression (LTD)
2.10. Statistical Analysis
3. Results
3.1. Effects of GDE Exposure on Body Weight and Litter Data
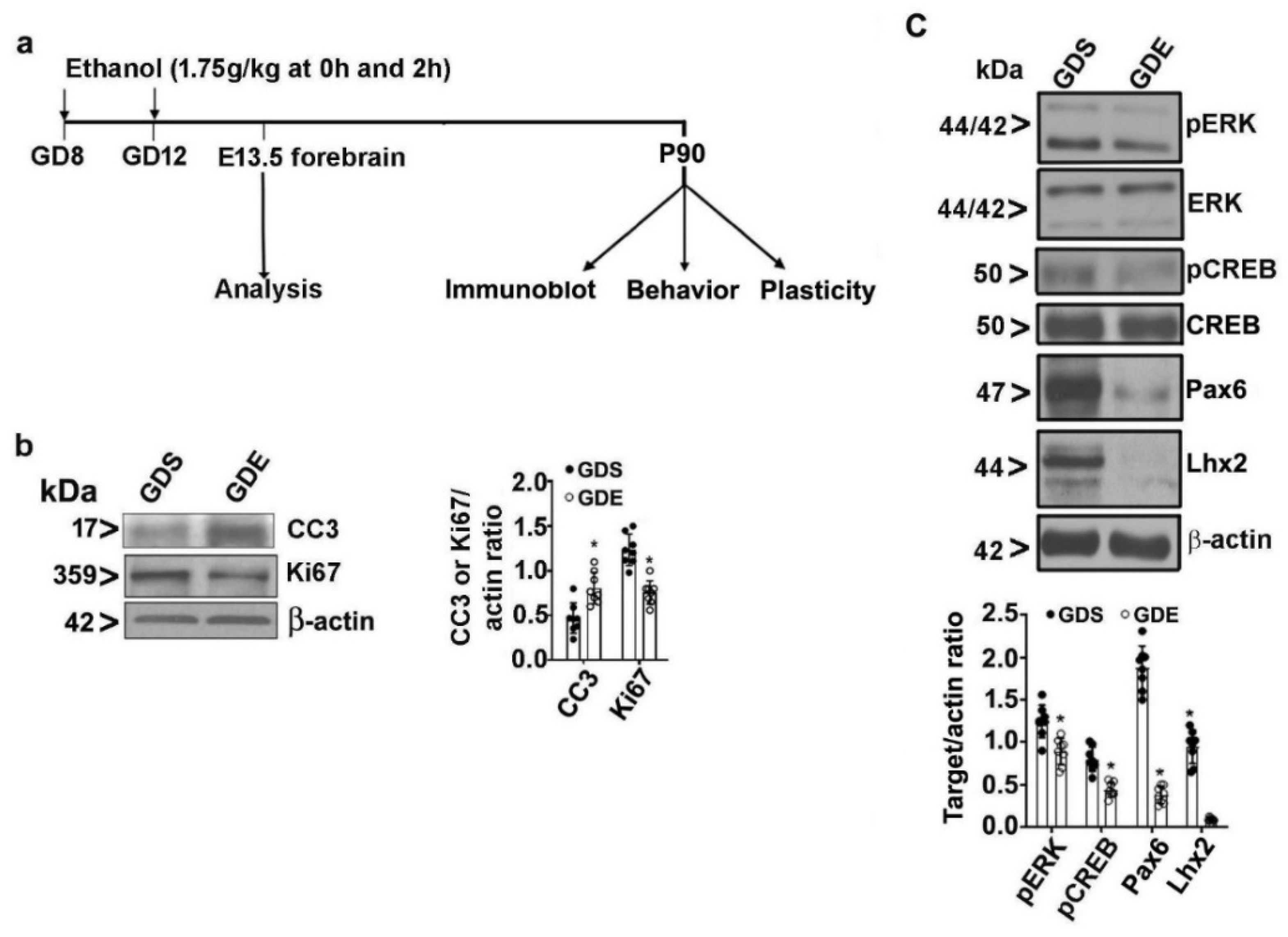
3.2. Effects of GDE Exposure on Embryonic Cell Proliferation and Survival
3.3. Effects of GDE Exposure on SM Performance
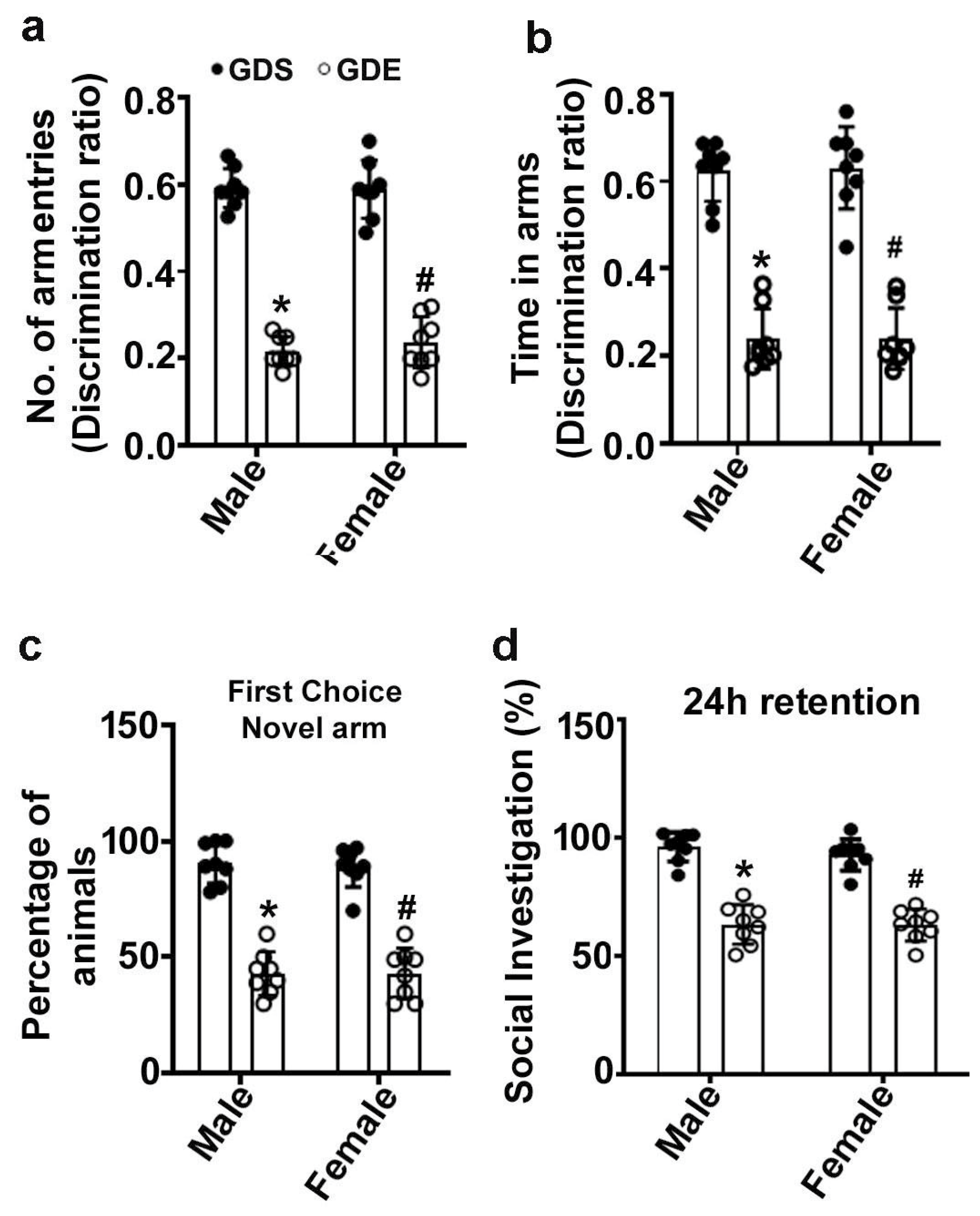
3.4. Effects of GDE Treatment on SRM
3.5. Effects of GDE Treatment on Stereotyped Repetitive Behavior in the NST and MBT
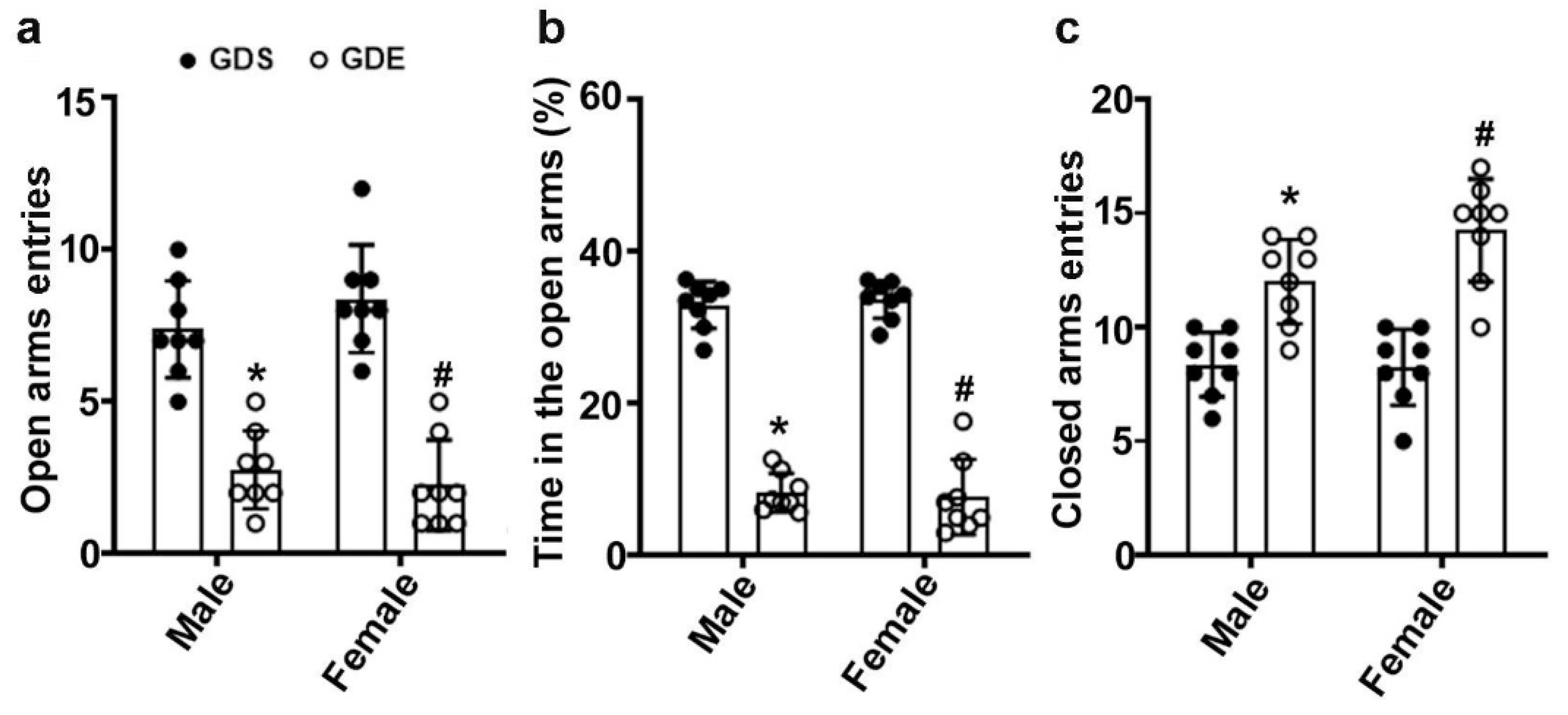
3.6. Effects of GDE Exposure on Anxiety-like Behavior in EPM
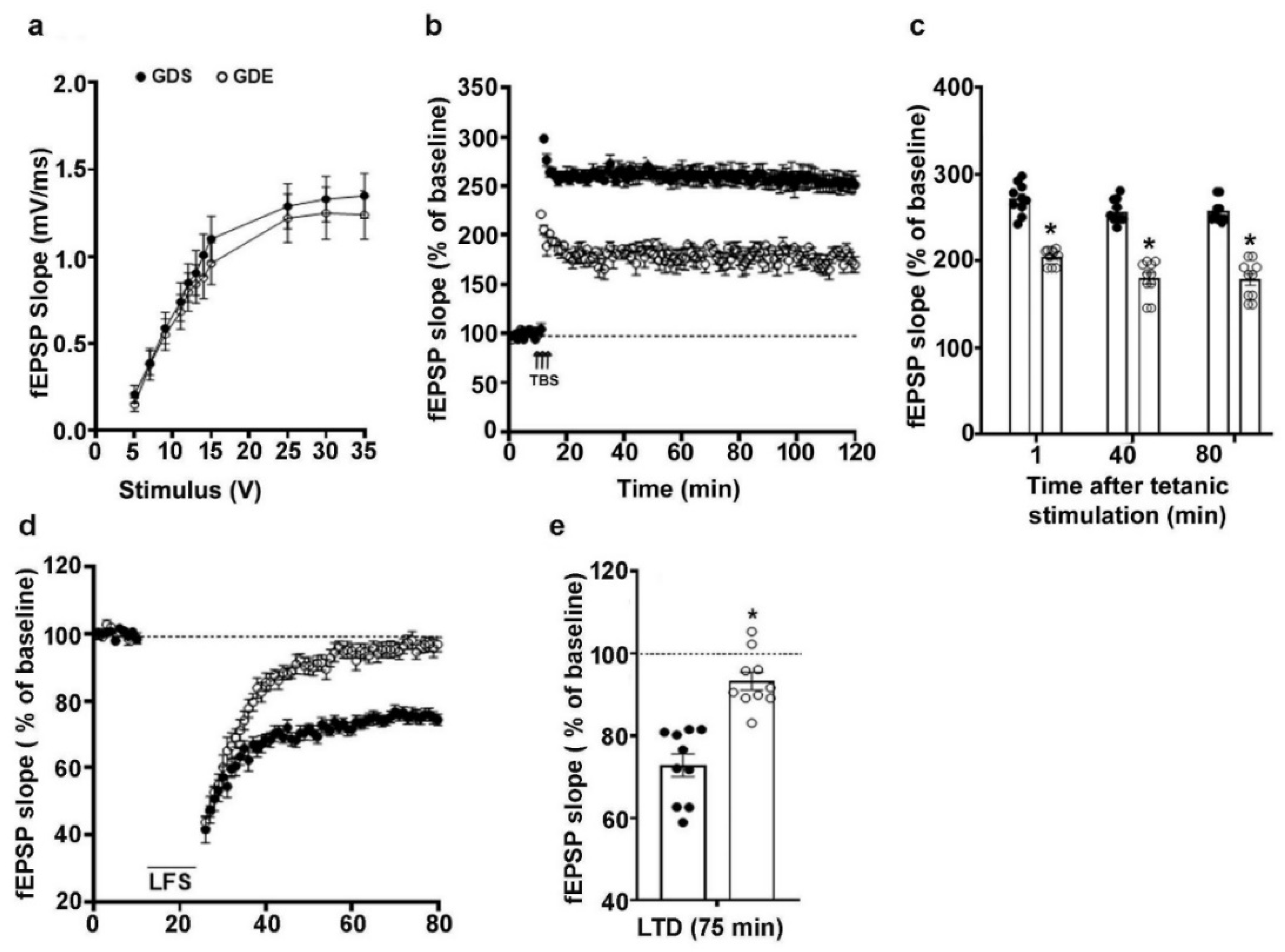
3.7. Effects of GDE Treatment on LTP and LTD
3.8. Effects of GDE on Synaptic Plasticity-Related Protein Expression in Adult Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riley, E.P.; Infante, M.A.; Warren, K.R. Fetal alcohol spectrum disorders: An overview. Neuropsychol. Rev. 2011, 21, 73–80. [Google Scholar] [CrossRef] [PubMed]
- CDC. Alcohol and Pregnancy. 2016. Available online: www.cdc.gov/vitalsigns/fasd (accessed on 15 February 2022).
- Floyd, R.L.; Weber, M.K.; Denny, C.; O’Connor, M.J. Prevention of fetal alcohol spectrum disorders. Dev. Disabil. Res. Rev. 2009, 15, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling. Brain Sci. 2015, 5, 456–493. [Google Scholar] [CrossRef] [Green Version]
- Coles, C.D.; Goldstein, F.C.; Lynch, M.E.; Chen, X.; Kable, J.A.; Johnson, K.C.; Hu, X. Memory and brain volume in adults prenatally exposed to alcohol. Brain Cogn. 2011, 75, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.M.; Crocker, N.; Deweese, B.N.; Roesch, S.C.; Coles, C.D.; Kable, J.A.; May, P.A.; Kalberg, W.O.; Sowell, E.R.; Jones, K.L.; et al. Prenatal alcohol exposure, attention-deficit/hyperactivity disorder, and sluggish cognitive tempo. Alcohol. Clin. Exp. Res. 2013, 37 (Suppl. S1), E338–E346. [Google Scholar] [CrossRef]
- Green, C.R.; Mihic, A.M.; Nikkel, S.M.; Stade, B.C.; Rasmussen, C.; Munoz, D.P.; Reynolds, J.N. Executive function deficits in children with fetal alcohol spectrum disorders (FASD) measured using the Cambridge Neuropsychological Tests Automated Battery (CANTAB). J. Child. Psychol. Psychiatry 2009, 50, 688–697. [Google Scholar] [CrossRef]
- Jacobson, S.W.; Jacobson, J.L.; Stanton, M.E.; Meintjes, E.M.; Molteno, C.D. Biobehavioral markers of adverse effect in fetal alcohol spectrum disorders. Neuropsychol. Rev. 2011, 21, 148–166. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.L.; Hoyme, H.E.; Robinson, L.K.; del Campo, M.; Manning, M.A.; Prewitt, L.M.; Chambers, C.D. Fetal alcohol spectrum disorders: Extending the range of structural defects. Am. J. Med. Genet. A 2010, 152, 2731–2735. [Google Scholar] [CrossRef] [Green Version]
- Lebel, C.; Mattson, S.N.; Riley, E.P.; Jones, K.L.; Adnams, C.M.; May, P.A.; Bookheimer, S.Y.; O’Connor, M.J.; Narr, K.L.; Kan, E.Z.; et al. Sowell, A longitudinal study of the long-term consequences of drinking during pregnancy: Heavy In Utero alcohol exposure disrupts the normal processes of brain development. J. Neurosci. 2012, 32, 15243–15251. [Google Scholar] [CrossRef]
- Bhatara, V.; Loudenberg, R.; Ellis, R. Association of attention deficit hyperactivity disorder and gestational alcohol exposure: An exploratory study. J. Atten. Disord. 2006, 9, 515–522. [Google Scholar] [CrossRef]
- Burden, M.J.; Jacobson, S.W.; Sokol, R.J.; Jacobson, J.L. Effects of prenatal alcohol exposure on attention and working memory at 7.5 years of age. Alcohol. Clin. Exp. Res. 2005, 29, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Coles, C.D.; Platzman, K.A.; Raskind-Hood, C.L.; Brown, R.T.; Falek, A.; Smith, I.E. A comparison of children affected by prenatal alcohol exposure and attention deficit, hyperactivity disorder. Alcohol. Clin. Exp. Res. 1997, 21, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Kaemingk, K.L.; Halverson, P.T. Spatial memory following prenatal alcohol exposure: More than a material specific memory deficit. Child. Neuropsychol. 2000, 6, 115–128. [Google Scholar] [CrossRef]
- Kaemingk, K.L.; Mulvaney, S.; Halverson, P.T. Learning following prenatal alcohol exposure: Performance on verbal and visual multitrial tasks. Arch. Clin. Neuropsychol. 2003, 18, 33–47. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, K.D.; Nanson, J. Clinical implications of a link between fetal alcohol spectrum disorder and attention-deficit hyperactivity disorder. Can. J. Psychiatry 2002, 47, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Streissguth, A.P.; O’Malley, K. Neuropsychiatric implications and long-term consequences of fetal alcohol spectrum disorders. Semin. Clin. Neuropsychiatry 2000, 5, 177–190. [Google Scholar] [CrossRef]
- Streissguth, A.P.; Sampson, P.D.; Olson, H.C.; Bookstein, F.L.; Barr, H.M.; Scott, M.; Feldman, J.; Mirsky, A.F. Maternal drinking during pregnancy: Attention and short-term memory in 14-year-old offspring—A longitudinal prospective study. Alcohol. Clin. Exp. Res. 1994, 18, 202–218. [Google Scholar] [CrossRef] [Green Version]
- Uecker, A.; Nadel, L. Spatial locations gone awry: Object and spatial memory deficits in children with fetal alcohol syndrome. Neuropsychologia 1996, 34, 209–223. [Google Scholar] [CrossRef]
- Uecker, A.; Nadel, L. Spatial but not object memory impairments in children with fetal alcohol syndrome. Am. J. Ment. Retard. 1998, 103, 12–18. [Google Scholar] [CrossRef]
- Willford, J.A.; Richardson, G.A.; Leech, S.L.; Day, N.L. Verbal and visuospatial learning and memory function in children with moderate prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2004, 28, 497–507. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, C.M.; Nassar, N.; Kurinczuk, J.J.; de Klerk, N.; Geelhoed, E.; Elliott, E.J.; Bower, C. Prenatal alcohol exposure and risk of birth defects. Pediatrics 2010, 126, e843–e850. [Google Scholar] [CrossRef] [PubMed]
- Finer, L.B.; Zolna, M.R. Unintended pregnancy in the United States: Incidence and disparities, 2006. Contraception 2011, 84, 478–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, D.A.; Sileanu, F.E.; Zhao, X.; Mor, M.K.; Judge-Golden, C.; Callegari, L.S.; Borrero, S. History of unintended pregnancy and patterns of contraceptive use among racial and ethnic minority women veterans. Am. J. Obstet. Gynecol. 2020, 223, 564.e1–564.e13. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.C.; Wilsnack, S.C.; Foster, D.G.; Delucchi, K.L. Alcohol use before and during unwanted pregnancy. Alcohol. Clin. Exp. Res. 2014, 38, 2844–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strandberg-Larsen, K.; Nielsen, N.R.; Andersen, A.M.N.; Olsen, J.; Gronbaek, M. Characteristics of women who binge drink before and after they become aware of their pregnancy. Eur. J. Epidemiol. 2008, 23, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Tough, S.; Tofflemire, K.; Clarke, M.; Newburn-Cook, C. Do women change their drinking behaviors while trying to conceive? An opportunity for preconception counseling. Clin. Med. Res. 2006, 4, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Berman, R.F.; Hannigan, J.H. Effects of prenatal alcohol exposure on the hippocampus: Spatial behavior, electrophysiology, and neuroanatomy. Hippocampus 2000, 10, 94–110. [Google Scholar] [CrossRef]
- Brown, K.L.; Calizo, L.H.; Goodlett, C.R.; Stanton, M.E. Neonatal alcohol exposure impairs acquisition of eyeblink conditioned responses during discrimination learning and reversal in weanling rats. Dev. Psychobiol. 2007, 49, 243–257. [Google Scholar] [CrossRef]
- Ieraci, A.; Herrera, D.G. Single alcohol exposure in early life damages hippocampal stem/progenitor cells and reduces adult neurogenesis. Neurobiol. Dis. 2007, 26, 597–605. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Horster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef]
- Joshi, V.; Subbanna, S.; Shivakumar, M.; Basavarajappa, B.S. CB1R regulates CDK5 signaling and epigenetically controls Rac1 expression contributing to neurobehavioral abnormalities in mice postnatally exposed to ethanol. Neuropsychopharmacology 2019, 44, 514–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulik, K.K.; Johnston, M.C.; Webb, M.A. Fetal alcohol syndrome: Embryogenesis in a mouse model. Science 1981, 214, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Webster, W.S.; Walsh, D.A.; Lipson, A.H.; McEwen, S.E. Teratogenesis after acute alcohol exposure in inbred and outbred mice. Neurobehav. Toxicol. 1980, 2, 227–234. [Google Scholar]
- Ashwell, K.W.; Zhang, L.L. Forebrain hypoplasia following acute prenatal ethanol exposure: Quantitative analysis of effects on specific forebrain nuclei. Pathology 1996, 28, 161–166. [Google Scholar] [CrossRef]
- Dunty, W.C., Jr.; Zucker, R.M.; Sulik, K.K. Hindbrain and cranial nerve dysmorphogenesis result from acute maternal ethanol administration. Dev. Neurosci. 2002, 24, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Godin, E.A.; O’Leary-Moore, S.K.; Khan, A.A.; Parnell, S.E.; Ament, J.J.; Dehart, D.B.; Johnson, B.W.; Johnson, G.A.; Styner, M.A.; Sulik, K.K. Magnetic resonance microscopy defines ethanol-induced brain abnormalities in prenatal mice: Effects of acute insult on gestational day 7. Alcohol. Clin. Exp. Res. 2010, 34, 98–111. [Google Scholar] [CrossRef]
- Kotch, L.E.; Sulik, K.K. Experimental fetal alcohol syndrome: Proposed pathogenic basis for a variety of associated facial and brain anomalies. Am. J. Med. Genet. 1992, 44, 168–176. [Google Scholar] [CrossRef]
- Lipinski, R.J.; Hammond, P.; O’Leary-Moore, S.K.; Ament, J.J.; Pecevich, S.J.; Jiang, Y.; Budin, F.; Parnell, S.E.; Suttie, M.; Godin, E.A.; et al. Ethanol-induced face-brain dysmorphology patterns are correlative and exposure-stage dependent. PLoS ONE 2012, 7, e43067. [Google Scholar] [CrossRef] [Green Version]
- Schambra, U.B.; Lauder, J.M.; Petrusz, P.; Sulik, K.K. Development of neurotransmitter systems in the mouse embryo following acute ethanol exposure: A histological and immunocytochemical study. Int. J. Dev. Neurosci. 1990, 8, 507–522. [Google Scholar] [CrossRef]
- Sulik, K.K. Craniofacial defects from genetic and teratogen-induced deficiencies in presomite embryos. Birth. Defects. Orig. Artic. Ser. 1984, 20, 79–98. [Google Scholar]
- Sulik, K.K. Genesis of alcohol-induced craniofacial dysmorphism. Exp. Biol. Med. 2005, 230, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Sulik, K.K.; Johnston, M.C.; Daft, P.A.; Russell, W.E.; Dehart, D.B. Fetal alcohol syndrome and DiGeorge anomaly: Critical ethanol exposure periods for craniofacial malformations as illustrated in an animal model. Am. J. Med. Genet. Suppl. 1986, 2, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Dumas, R.M.; Rabe, A. Augmented memory loss in aging mice after one embryonic exposure to alcohol. Neurotoxicol. Teratol. 1994, 16, 605–612. [Google Scholar] [CrossRef]
- Endres, M.; Toso, L.; Roberson, R.; Park, J.; Abebe, D.; Poggi, S.; Spong, C.Y. Prevention of alcohol-induced developmental delays and learning abnormalities in a model of fetal alcohol syndrome. Am. J. Obstet. Gynecol. 2005, 193, 1028–1034. [Google Scholar] [CrossRef]
- Summers, B.L.; Henry, C.M.; Rofe, A.M.; Coyle, P. Dietary zinc supplementation during pregnancy prevents spatial and object recognition memory impairments caused by early prenatal ethanol exposure. Behav. Brain Res. 2008, 186, 230–238. [Google Scholar] [CrossRef]
- Schambra, U.B.; Lewis, C.N.; Harrison, T.A. Deficits in spatial learning and memory in adult mice following acute, low or moderate levels of prenatal ethanol exposure during gastrulation or neurulation. Neurotoxicol. Teratol. 2017, 62, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.C.; Moyano, H.F.; Spear, L.P.; Spear, N.E. Acute alcohol exposure during gestational day 8 in the rat: Effects upon physical and behavioral parameters. Alcohol 1984, 1, 459–464. [Google Scholar] [CrossRef]
- Sadrian, B.; Lopez-Guzman, M.; Wilson, D.A.; Saito, M. Distinct neurobehavioral dysfunction based on the timing of developmental binge-like alcohol exposure. Neuroscience 2014, 280, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Rouzer, S.K.; Cole, J.M.; Johnson, J.M.; Varlinskaya, E.I.; Diaz, M.R. Moderate Maternal Alcohol Exposure on Gestational Day 12 Impacts Anxiety-Like Behavior in Offspring. Front. Behav. Neurosci. 2017, 11, 183. [Google Scholar] [CrossRef] [Green Version]
- Diaz, M.R.; Mooney, S.M.; Varlinskaya, E.I. Acute prenatal exposure to ethanol on gestational day 12 elicits opposing deficits in social behaviors and anxiety-like behaviors in Sprague Dawley rats. Behav. Brain Res. 2016, 310, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Webster, W.S.; Walsh, D.A.; McEwen, S.E.; Lipson, A.H. Some teratogenic properties of ethanol and acetaldehyde in C57BL/6J mice: Implications for the study of the fetal alcohol syndrome. Teratology 1983, 27, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Perissi, V.; Dasen, J.S.; Kurokawa, R.; Wang, Z.; Korzus, E.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. Factor-specific modulation of CREB-binding protein acetyltransferase activity. Proc. Natl. Acad Sci. USA 1999, 96, 3652–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbanna, S.; Joshi, V.; Basavarajappa, B.S. Activity-dependent Signaling and Epigenetic Abnormalities in Mice Exposed to Postnatal Ethanol. Neuroscience 2018, 392, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Nagre, N.N.; Umapathy, N.S.; Pace, B.S.; Basavarajappa, B.S. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int. J. Neuro Psychopharmacol. 2015, 18, 1–15. [Google Scholar] [CrossRef]
- Bito, H.; Deisseroth, K.; Tsien, R.W. CREB phosphorylation and dephosphorylation: A Ca (2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 1996, 87, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.G.; Groth, R.D.; Ma, H.; Barrett, C.F.; Owen, S.F.; Safa, P.; Tsien, R.W. Ca (V) 1 and Ca (V) 2 channels engage distinct modes of Ca (2+) signaling to control CREB-dependent gene expression. Cell 2012, 149, 1112–1124. [Google Scholar] [CrossRef] [Green Version]
- Krahe, T.E.; Wang, W.; Medina, A.E. Phosphodiesterase inhibition increases CREB phosphorylation and restores orientation selectivity in a model of fetal alcohol spectrum disorders. PLoS ONE 2009, 4, e6643. [Google Scholar] [CrossRef]
- Subbanna, S.; Nagre, N.N.; Shivakumar, M.; Joshi, V.; Psychoyos, D.; Kutlar, A.; Umapathy, N.S.; Basavarajappa, B.S. CB1R-Mediated Activation of Caspase-3 Causes Epigenetic and Neurobehavioral Abnormalities in Postnatal Ethanol-Exposed Mice. Front. Mol. Neurosci. 2018, 11, 45. [Google Scholar] [CrossRef] [Green Version]
- Heroux, N.A.; Horgan, C.J.; Rosen, J.B.; Stanton, M.E. Cholinergic rescue of neurocognitive insult following third-trimester equivalent alcohol exposure in rats. Neurobiol. Learn. Mem. 2019, 163, 107030. [Google Scholar] [CrossRef]
- Heroux, N.A.; Robinson-Drummer, P.A.; Kawan, M.; Rosen, J.B.; Stanton, M.E. Neonatal ethanol exposure impairs long-term context memory formation and prefrontal immediate early gene expression in adolescent rats. Behav. Brain Res. 2019, 359, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, S.A.; Robinson-Drummer, P.A.; Schreiber, W.B.; Asok, A.; Rosen, J.B.; Stanton, M.E. Impairment of the context preexposure facilitation effect in juvenile rats by neonatal alcohol exposure is associated with decreased Egr-1 mRNA expression in the prefrontal cortex. Behav. Neurosci. 2018, 132, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, M.; Subbanna, S.; Joshi, V.; Basavarajappa, B.S. Postnatal Ethanol Exposure Activates HDAC-Mediated Histone Deacetylation, Impairs Synaptic Plasticity Gene Expression and Behavior in Mice. Int. J. Neuropsychopharmacol. 2020, 23, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Fish, E.W.; Wieczorek, L.A.; Rumple, A.; Suttie, M.; Moy, S.S.; Hammond, P.; Parnell, S.E. The enduring impact of neurulation stage alcohol exposure: A combined behavioral and structural neuroimaging study in adult male and female C57BL/6J mice. Behav. Brain Res. 2018, 338, 173–184. [Google Scholar] [CrossRef]
- White, S.A.; Weber, J.N.; Howard, C.D.; Favero, C.B. Effects of binge ethanol exposure during first-trimester equivalent on corticothalamic neurons in Swiss Webster outbred mice. Neuroreport 2015, 26, 1083–1088. [Google Scholar] [CrossRef]
- Lundquist, F. The determination of ethyl alcohol in blood and tissue. Meth. Biochem. Analy. 1959, 7, 217–251. [Google Scholar]
- Subbanna, S.; Basavarajappa, B.S. Pre-administration of G9a/GLP inhibitor during Synaptogenesis Prevents Postnatal Ethanol-induced LTP Deficits and Neurobehavioral Abnormalities in Adult Mice. Exp. Neurol. 2014, 261, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Kogan, J.H.; Frankland, P.W.; Silva, A.J. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus 2000, 10, 47–56. [Google Scholar] [CrossRef]
- Thor, D.H.; Wainwright, K.L.; Holloway, W.R. Persistence of attention to a novel conspecific: Some developmental variables in laboratory rats. Dev. Psychobiol. 1982, 15, 1–8. [Google Scholar] [CrossRef]
- Bahi, A. Individual differences in elevated plus-maze exploration predicted higher ethanol consumption and preference in outbred mice. Pharmacol. Biochem. Behav. 2013, 105, 83–88. [Google Scholar] [CrossRef]
- Bahi, A.; Dreyer, J.L. Chronic psychosocial stress causes delayed extinction and exacerbates reinstatement of ethanol-induced conditioned place preference in mice. Psychopharmacology 2014, 231, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Angoa-Perez, M.; Kane, M.J.; Briggs, D.I.; Francescutti, D.M.; Kuhn, D.M. Marble burying and nestlet shredding as tests of repetitive, compulsive-like behaviors in mice. J. Vis. Exp. 2013, 82, 50978. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Burant, A.; Bui, N.; Graham, D.; Yuva-Paylor, L.A.; Paylor, R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology 2009, 204, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Eissa, N.; Jayaprakash, P.; Azimullah, S.; Ojha, S.K.; Al-Houqani, M.; Jalal, F.Y.; Lazewska, D.; Kiec-Kononowicz, K.; Sadek, B. The histamine H3R antagonist DL77 attenuates autistic behaviors in a prenatal valproic acid-induced mouse model of autism. Sci. Rep. 2018, 8, 13077. [Google Scholar] [CrossRef]
- Pensalfini, A.; Kim, S.; Subbanna, S.; Bleiwas, C.; Goulbourne, C.N.; Stavrides, P.H.; Jiang, Y.; Lee, J.H.; Darji, S.; Pawlik, M.; et al. Endosomal Dysfunction Induced by Directly Overactivating Rab5 Recapitulates Prodromal and Neurodegenerative Features of Alzheimer’s Disease. Cell Rep. 2020, 33, 108420. [Google Scholar] [CrossRef]
- Burd, L.; Blair, J.; Dropps, K. Prenatal alcohol exposure, blood alcohol concentrations and alcohol elimination rates for the mother, fetus and newborn. J. Perinatol. 2012, 32, 652–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, C.L.; Burne, T.H.; Lavidis, N.A.; Moritz, K.M. Low dose prenatal ethanol exposure induces anxiety-like behaviour and alters dendritic morphology in the basolateral amygdala of rat offspring. PLoS ONE 2013, 8, e54924. [Google Scholar] [CrossRef] [Green Version]
- Komada, M.; Hara, N.; Kawachi, S.; Kawachi, K.; Kagawa, N.; Nagao, T.; Ikeda, Y. Mechanisms underlying neuro-inflammation and neurodevelopmental toxicity in the mouse neocortex following prenatal exposure to ethanol. Sci. Rep. 2017, 7, 4934. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lunde-Young, R.; Naik, V.; Ramirez, J.; Orzabal, M.; Ramadoss, J. Chronic Binge Alcohol Exposure during Pregnancy Alters mTOR System in Rat Fetal Hippocampus. Alcohol. Clin. Exp. Res. 2020, 44, 1329–1336. [Google Scholar] [CrossRef]
- Tyler, C.R.; Allan, A.M. Prenatal alcohol exposure alters expression of neurogenesis-related genes in an Ex Vivo cell culture model. Alcohol 2014, 48, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, C.F.; Morton, R.A.; Diaz, M.R.; Topper, L. Does moderate drinking harm the fetal brain? Insights from animal models. Trends. Neurosci. 2012, 35, 284–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbier, E.; Houchi, H.; Warnault, V.; Pierrefiche, O.; Daoust, M.; Naassila, M. Effects of prenatal and postnatal maternal ethanol on offspring response to alcohol and psychostimulants in long evans rats. Neuroscience 2009, 161, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Parnell, S.E.; Holloway, H.E.; Baker, L.K.; Styner, M.A.; Sulik, K.K. Dysmorphogenic effects of first trimester-equivalent ethanol exposure in mice: A magnetic resonance microscopy-based study. Alcohol. Clin. Exp. Res. 2014, 38, 2008–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schambra, U.B.; Nunley, K.; Harrison, T.A.; Lewis, C.N. Consequences of low or moderate prenatal ethanol exposures during gastrulation or neurulation for open field activity and emotionality in mice. Neurotoxicol. Teratol. 2016, 57, 39–53. [Google Scholar] [CrossRef]
- Sulik, K.K.; Johnston, M.C. Sequence of developmental alterations following acute ethanol exposure in mice: Craniofacial features of the fetal alcohol syndrome. Am. J. Anat. 1983, 166, 257–269. [Google Scholar] [CrossRef]
- Otis, E.M.; Brent, R. Equivalent ages in mouse and human embryos. Anat. Rec. 1954, 120, 33–63. [Google Scholar] [CrossRef]
- Schambra, U.B.; Goldsmith, J.; Nunley, K.; Liu, Y.; Harirforoosh, S.; Schambra, H.M. Low and moderate prenatal ethanol exposures of mice during gastrulation or neurulation delays neurobehavioral development. Neurotoxicol. Teratol. 2015, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Aglawe, M.M.; Kale, M.B.; Rahangdale, S.R.; Kotagale, N.R.; Umekar, M.J.; Taksande, B.G. Agmatine improves the behavioral and cognitive impairments associated with chronic gestational ethanol exposure in rats. Brain Res. Bull. 2021, 167, 37–47. [Google Scholar] [CrossRef]
- Dandekar, M.P.; Bharne, A.P.; Borkar, P.D.; Subhedar, N.K.; Kokare, D.M. Maternal ethanol exposure reshapes CART system in the rat brain: Correlation with development of anxiety, depression and memory deficits. Neuroscience 2019, 406, 126–139. [Google Scholar] [CrossRef]
- Dunty, W.C., Jr.; Chen, S.Y.; Zucker, R.M.; Dehart, D.B.; Sulik, K.K. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: Implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol. Clin. Exp. Res. 2001, 25, 1523–1535. [Google Scholar] [CrossRef]
- Sulik, K.K.; Lauder, J.M.; Dehart, D.B. Brain malformations in prenatal mice following acute maternal ethanol administration. J. Devl. Neurosci. 1984, 2, 203–214. [Google Scholar] [CrossRef]
- Kotch, L.E.; Sulik, K.K. Patterns of ethanol-induced cell death in the developing nervous system of mice; neural fold states through the time of anterior neural tube closure. Int. J. Dev. Neurosci. 1992, 10, 273–279. [Google Scholar] [CrossRef]
- Louis, L.K.; Gopurappilly, R.; Surendran, H.; Dutta, S.; Pal, R. Transcriptional profiling of human neural precursors post alcohol exposure reveals impaired neurogenesis via dysregulation of ERK signaling and miR-145. J. Neurochem. 2017, 146, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Wu, Z.; Xu, L.; Fang, Y.; Xu, Y. Maternal supplementation of nucleotides improves the behavioral development of prenatal ethanol-exposed mice. Cogn. Affect. Behav. Neurosci. 2014, 14, 879–890. [Google Scholar] [CrossRef]
- Naseer, M.I.; Lee, H.Y.; Ullah, N.; Ullah, I.; Park, M.S.; Kim, M.O. siRNA-mediated GABA (B) receptor at early fetal rat brain upon acute and chronic ethanol exposure: Down regulation of PKA and p-CREB expression. Synapse 2011, 65, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Huttner, W.B.; Calegari, F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem. Cell. 2009, 5, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.L.; Zhang, Z.G.; Roberts, C.; LeTourneau, Y.; Lu, M.; Zhang, L.; Wang, Y.; Chopp, M. Lengthening the G (1) phase of neural progenitor cells is concurrent with an increase of symmetric neuron generating division after stroke. J. Cereb. Blood Flow Metab. 2008, 28, 602–611. [Google Scholar] [CrossRef] [Green Version]
- Aronne, M.P.; Guadagnoli, T.; Fontanet, P.; Evrard, S.G.; Brusco, A. Effects of prenatal ethanol exposure on rat brain radial glia and neuroblast migration. Exp. Neurol. 2011, 229, 364–371. [Google Scholar] [CrossRef]
- Aronne, M.P.; Evrard, S.G.; Mirochnic, S.; Brusco, A. Prenatal ethanol exposure reduces the expression of the transcriptional factor Pax6 in the developing rat brain. Ann. N. Y. Acad. Sci. 2008, 1139, 478–498. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, P.H.; Ng, S.S.; Wong, O.G.; Liu, J.; He, M.L.; Kung, H.F.; Lin, M.C. A critical role of Pax6 in alcohol-induced fetal microcephaly. Neurobiol. Dis. 2004, 16, 370–376. [Google Scholar] [CrossRef]
- Wentzel, P.; Eriksson, U.J. Genetic influence on dysmorphogenesis in embryos from different rat strains exposed to ethanol in vivo and In Vitro. Alcohol. Clin. Exp. Res. 2008, 32, 874–887. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Milivojevic, V.; Zecevic, N. Enforced Pax6 expression rescues alcohol-induced defects of neuronal differentiation in cultures of human cortical progenitor cells. Alcohol. Clin. Exp. Res. 2012, 36, 1374–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.C.; Go, H.S.; Bak, H.R.; Choi, C.S.; Choi, I.; Kim, P.; Han, S.H.; Han, S.M.; Shin, C.Y.; Ko, K.H. Prenatal exposure of ethanol induces increased glutamatergic neuronal differentiation of neural progenitor cells. J. Biomed. Sci. 2010, 17, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulchand, S.; Grove, E.A.; Porter, F.D.; Tole, S. LIM-homeodomain gene Lhx2 regulates the formation of the cortical hem. Mech. Dev. 2001, 100, 165–175. [Google Scholar] [CrossRef]
- Chou, S.J.; Perez-Garcia, C.G.; Kroll, T.T.; O’Leary, D.D. Lhx2 specifies regional fate in Emx1 lineage of telencephalic progenitors generating cerebral cortex. Nat. Neurosci. 2009, 12, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto-Torii, K.; Kawasawa, Y.I.; Kuhn, A.; Rakic, P. Combined transcriptome analysis of fetal human and mouse cerebral cortex exposed to alcohol. Proc. Natl. Acad Sci. USA 2011, 108, 4212–4217. [Google Scholar] [CrossRef] [Green Version]
- Abbott, C.W.; Rohac, D.J.; Bottom, R.T.; Patadia, S.; Huffman, K.J. Prenatal Ethanol Exposure and Neocortical Development: A Transgenerational Model of FASD. Cereb. Cortex. 2018, 28, 2908–2921. [Google Scholar] [CrossRef]
- Cantacorps, L.; Alfonso-Loeches, S.; Moscoso-Castro, M.; Cuitavi, J.; Gracia-Rubio, I.; Lopez-Arnau, R.; Escubedo, E.; Guerri, C.; Valverde, O. Maternal alcohol binge drinking induces persistent neuroinflammation associated with myelin damage and behavioural dysfunctions in offspring mice. Neuropharmacology 2017, 123, 368–384. [Google Scholar] [CrossRef] [Green Version]
- Gil-Mohapel, J.; Boehme, F.; Kainer, L.; Christie, B.R. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: Insights from different rodent models. Brain Res. Rev. 2010, 64, 283–303. [Google Scholar] [CrossRef]
- Wagner, J.L.; Zhou, F.C.; Goodlett, C.R. Effects of one- and three-day binge alcohol exposure in neonatal C57BL/6 mice on spatial learning and memory in adolescence and adulthood. Alcohol 2014, 48, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Almeida, L.; Andreu-Fernandez, V.; Navarro-Tapia, E.; Aras-Lopez, R.; Serra-Delgado, M.; Martinez, L.; Garcia-Algar, O.; Gomez-Roig, M.D. Murine Models for the Study of Fetal Alcohol Spectrum Disorders: An Overview. Front. Pediatr. 2020, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S.; Subbanna, S. Epigenetic Mechanisms in Developmental Alcohol-Induced Neurobehavioral Deficits. Brain Sci. 2016, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noor, S.; Milligan, E.D. Lifelong Impacts of Moderate Prenatal Alcohol Exposure on Neuroimmune Function. Front Immunol. 2018, 9, 1107. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, K.; Brigman, J.L. The impact of prenatal alcohol exposure on social, cognitive and affective behavioral domains: Insights from rodent models. Alcohol 2016, 51, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Finlay, B.L.; Darlington, R.B. Linked regularities in the development and evolution of mammalian brains. Science 1995, 268, 1578–1584. [Google Scholar] [CrossRef]
- Schneider, M.L.; Moore, C.F.; Adkins, M.M. The effects of prenatal alcohol exposure on behavior: Rodent and primate studies. Neuropsychol. Rev. 2011, 21, 186–203. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lebel, C. Evaluation of Brain Alterations and Behavior in Children with Low Levels of Prenatal Alcohol Exposure. JAMA Netw. Open 2022, 5, e225972. [Google Scholar] [CrossRef]
- Kelly, S.J.; Day, N.; Streissguth, A.P. Effects of prenatal alcohol exposure on social behavior in humans and other species. Neurotoxicol. Teratol. 2000, 22, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Olson, H.C.; Feldman, J.J.; Streissguth, A.P.; Sampson, P.D.; Bookstein, F.L. Neuropsychological deficits in adolescents with fetal alcohol syndrome: Clinical findings. Alcohol. Clin. Exp. Res. 1998, 22, 1998–2012. [Google Scholar] [CrossRef]
- Thomas, S.E.; Kelly, S.J.; Mattson, S.N.; Riley, E.P. Comparison of social abilities of children with fetal alcohol syndrome to those of children with similar IQ scores and normal controls. Alcohol. Clin. Exp. Res. 1998, 22, 528–533. [Google Scholar] [CrossRef]
- Hamilton, D.A.; Akers, K.G.; Rice, J.P.; Johnson, T.E.; Candelaria-Cook, F.T.; Maes, L.I.; Rosenberg, M.; Valenzuela, C.F.; Savage, D.D. Prenatal exposure to moderate levels of ethanol alters social behavior in adult rats: Relationship to structural plasticity and immediate early gene expression in frontal cortex. Behav. Brain Res. 2010, 207, 290–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, D.A.; Barto, D.; Rodriguez, C.I.; Magcalas, C.M.; Fink, B.C.; Rice, J.P.; Bird, C.W.; Davies, S.; Savage, D.D. Effects of moderate prenatal ethanol exposure and age on social behavior, spatial response perseveration errors and motor behavior. Behav. Brain Res. 2014, 269, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, M.S.C.; Chong, S.; Burne, T.H. Early gestational exposure to moderate concentrations of ethanol alters adult behaviour in C57BL/6J mice. Behav. Brain Res. 2013, 252, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Shukla, P.K.; Meena, A.S.; Rao, R.; Rao, R. Deletion of TLR-4 attenuates fetal alcohol exposure-induced gene expression and social interaction deficits. Alcohol 2018, 73, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Varlinskaya, E.I.; Mooney, S.M. Acute exposure to ethanol on gestational day 15 affects social motivation of female offspring. Behav. Brain Res. 2014, 261, 106–109. [Google Scholar] [CrossRef] [Green Version]
- Boschen, K.E.; Hamilton, G.F.; Delorme, J.E.; Klintsova, A.Y. Activity and social behavior in a complex environment in rats neonatally exposed to alcohol. Alcohol 2014, 48, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.J.; Dillingham, R.R. Sexually dimorphic effects of perinatal alcohol exposure on social interactions and amygdala DNA and DOPAC concentrations. Neurotoxicol. Teratol. 1994, 16, 377–384. [Google Scholar] [CrossRef]
- Lugo, J.N., Jr.; Marino, M.D.; Cronise, K.; Kelly, S.J. Effects of alcohol exposure during development on social behavior in rats. Physiol. Behav. 2003, 78, 185–194. [Google Scholar] [CrossRef]
- Mooney, S.M.; Varlinskaya, E.I. Acute prenatal exposure to ethanol and social behavior: Effects of age, sex, and timing of exposure. Behav. Brain Res. 2011, 216, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Hayes, N.; Moritz, K.M.; Reid, N. Parent-reported sleep problems in school-aged children with fetal alcohol spectrum disorder: Association with child behaviour, caregiver, and family functioning. Sleep Med. 2020, 74, 307–314. [Google Scholar] [CrossRef]
- O’Connor, M.J.; Paley, B. Psychiatric conditions associated with prenatal alcohol exposure. Dev. Disabil. Res. Rev. 2009, 15, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Popova, S.; Temple, V.; Dozet, D.; O’Hanlon, G.; Toews, C.; Rehm, J. Health, social and legal outcomes of individuals with diagnosed or at risk for fetal alcohol spectrum disorder: Canadian example. Drug Alcohol Depend. 2021, 219, 108487. [Google Scholar] [CrossRef]
- Schmidt, N.B.; Buckner, J.D.; Keough, M.E. Anxiety sensitivity as a prospective predictor of alcohol use disorders. Behav. Modif. 2007, 31, 202–219. [Google Scholar] [CrossRef]
- Brocardo, P.S.; Boehme, F.; Patten, A.; Cox, A.; Gil-Mohapel, J.; Christie, B.R. Anxiety- and depression-like behaviors are accompanied by an increase in oxidative stress in a rat model of fetal alcohol spectrum disorders: Protective effects of voluntary physical exercise. Neuropharmacology 2012, 62, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- He, F. The relationship of prenatal ethanol exposure and anxiety-related behaviors and central androgen receptor and vasopressin expression in adult male mandarin voles. Neuroscience 2014, 266, 224–234. [Google Scholar] [CrossRef]
- Liang, J.; Shen, Y.; Shao, X.M.; Scott, M.B.; Ly, E.; Wong, S.; Nguyen, A.; Tan, K.; Kwon, B.; Olsen, R.W.; et al. Dihydromyricetin prevents fetal alcohol exposure-induced behavioral and physiological deficits: The roles of GABAA receptors in adolescence. Neurochem. Res. 2014, 39, 1147–1161. [Google Scholar] [CrossRef] [Green Version]
- Wille-Bille, A.; Miranda-Morales, R.S.; Pucci, M.; Bellia, F.; D’Addario, C.; Pautassi, R.M. Prenatal ethanol induces an anxiety phenotype and alters expression of dynorphin & nociceptin/orphanin FQ genes. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 85, 77–88. [Google Scholar]
- Carneiro, L.M.; Diogenes, J.P.; Vasconcelos, S.M.; Aragao, G.F.; Noronha, E.C.; Gomes, P.B.; Viana, G.S. Behavioral and neurochemical effects on rat offspring after prenatal exposure to ethanol. Neurotoxicol. Teratol. 2005, 27, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Osborn, J.A.; Kim, C.K.; Steiger, J.; Weinberg, J. Prenatal ethanol exposure differentially alters behavior in males and females on the elevated plus maze. Alcohol. Clin. Exp. Res. 1998, 22, 685–696. [Google Scholar] [CrossRef]
- Osborn, J.A.; Yu, C.; Gabriel, K.; Weinberg, J. Fetal ethanol effects on benzodiazepine sensitivity measured by behavior on the elevated plus-maze. Pharmacol. Biochem. Behav. 1998, 60, 625–633. [Google Scholar] [CrossRef]
- Wieczorek, L.; Fish, E.W.; O’Leary-Moore, S.K.; Parnell, S.E.; Sulik, K.K. Hypothalamic-pituitary-adrenal axis and behavioral dysfunction following early binge-like prenatal alcohol exposure in mice. Alcohol 2015, 49, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popova, N.K.; Morozova, M.V.; Naumenko, V.S. Ameliorative effect of BDNF on prenatal ethanol and stress exposure-induced behavioral disorders. Neurosci. Lett. 2011, 505, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Baculis, B.C.; Diaz, M.R.; Valenzuela, C.F. Third trimester-equivalent ethanol exposure increases anxiety-like behavior and glutamatergic transmission in the basolateral amygdala. Pharmacol. Biochem. Behav. 2015, 137, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, M.R.; Jotty, K.; Locke, J.L.; Jones, S.R.; Valenzuela, C.F. Moderate Alcohol Exposure during the Rat Equivalent to the Third Trimester of Human Pregnancy Alters Regulation of GABAA Receptor-Mediated Synaptic Transmission by Dopamine in the Basolateral Amygdala. Front. Pediatr. 2014, 2, 46. [Google Scholar] [CrossRef] [Green Version]
- Patten, A.R.; Brocardo, P.S.; Sakiyama, C.; Wortman, R.C.; Noonan, A.; Gil-Mohapel, J.; Christie, B.R. Impairments in hippocampal synaptic plasticity following prenatal ethanol exposure are dependent on glutathione levels. Hippocampus 2013, 23, 1463–1475. [Google Scholar] [CrossRef]
- Puglia, M.P.; Valenzuela, C.F. Repeated third trimester-equivalent ethanol exposure inhibits long-term potentiation in the hippocampal CA1 region of neonatal rats. Alcohol 2010, 44, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Xie, S.; Basavarajappa, B.S. Anandamide-CB1 Receptor Signaling Contributes to Postnatal Ethanol-Induced Neonatal Neurodegeneration, Adult Synaptic and Memory Deficits. J. Neuoscience 2013, 33, 6350–6366. [Google Scholar] [CrossRef] [Green Version]
- Patten, A.R.; Fontaine, C.J.; Christie, B.R. A comparison of the different animal models of fetal alcohol spectrum disorders and their use in studying complex behaviors. Front. Pediatr. 2014, 2, 93. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.P.; Byrnes, M.L.; Brien, J.F.; Reynolds, J.N.; Dringenberg, H.C. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur. J. Neurosci. 2002, 16, 1593–1598. [Google Scholar] [CrossRef]
- Fontaine, C.J.; Pinar, C.; Yang, W.; Pang, A.F.; Suesser, K.E.; Choi, J.S.J.; Christie, B.R. Impaired Bidirectional Synaptic Plasticity in Juvenile Offspring Following Prenatal Ethanol Exposure. Alcohol. Clin. Exp. Res. 2019, 43, 2153–2166. [Google Scholar] [CrossRef]
- Kervern, M.; de Ferron, B.S.; Alaux-Cantin, S.; Fedorenko, O.; Antol, J.; Naassila, M.; Pierrefiche, O. Aberrant NMDA-dependent LTD after perinatal ethanol exposure in young adult rat hippocampus. Hippocampus 2015, 25, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Honse, Y.; Nixon, K.M.; Browning, M.D.; Leslie, S.W. Cell surface expression of NR1 splice variants and NR2 subunits is modified by prenatal ethanol exposure. Neuroscience 2003, 122, 689–698. [Google Scholar] [CrossRef]
- Nixon, K.; Hughes, P.D.; Amsel, A.; Leslie, S.W. NMDA receptor subunit expression following early postnatal exposure to ethanol. Brain Res. Dev. Brain Res. 2002, 139, 295–299. [Google Scholar] [CrossRef]
- Nixon, K.; Hughes, P.D.; Amsel, A.; Leslie, S.W. NMDA receptor subunit expression after combined prenatal and postnatal exposure to ethanol. Alcohol. Clin. Exp. Res. 2004, 28, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Samudio-Ruiz, S.L.; Allan, A.M.; Sheema, S.; Caldwell, K.K. Hippocampal N-methyl-D-aspartate receptor subunit expression profiles in a mouse model of prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2010, 34, 342–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Savage, D.D.; Swartzwelder, H.S. Effects of prenatal ethanol exposure on hippocampal ionotropic-quisqualate and kainate receptors. Alcohol. Clin. Exp. Res. 1992, 16, 816–821. [Google Scholar] [CrossRef]
- Montagud-Romero, S.; Cantacorps, L.; Fernandez-Gomez, F.J.; Nunez, C.; Minarro, J.; Rodriguez-Arias, M.; Milanes, M.V.; Valverde, O. Unraveling the molecular mechanisms involved in alcohol intake and withdrawal in adolescent mice exposed to alcohol during early life stages. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2021, 104, 110025. [Google Scholar] [CrossRef]
- Vaglenova, J.; Pandiella, N.; Wijayawardhane, N.; Vaithianathan, T.; Birru, S.; Breese, C.; Suppiramaniam, V.; Randal, C. Aniracetam reversed learning and memory deficits following prenatal ethanol exposure by modulating functions of synaptic AMPA receptors. Neuropsychopharmacology 2008, 33, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Wijayawardhane, N.; Shonesy, B.C.; Vaithianathan, T.; Pandiella, N.; Vaglenova, J.; Breese, C.R.; Dityatev, A.; Suppiramaniam, V. Ameliorating effects of preadolescent aniracetam treatment on prenatal ethanol-induced impairment in AMPA receptor activity. Neurobiol. Dis. 2008, 29, 81–91. [Google Scholar] [CrossRef]
- Anggono, V.; Huganir, R.L. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 2012, 22, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.F.; Sun, L.L.; Cui, C.L.; Han, J.S. NAc Shell Arc/Arg3.1 Protein Mediates Reconsolidation of Morphine CPP by Increased GluR1 Cell Surface Expression: Activation of ERK-Coupled CREB is Required. Int. J. Neuro Psychopharmacol. 2015, 18, pyv030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.J.; Zukin, R.S. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends. Neurosci. 2007, 30, 126–134. [Google Scholar] [CrossRef]
- Wang, X.; Carlson, V.C.C.; Studholme, C.; Newman, N.; Ford, M.M.; Grant, K.A.; Kroenke, C.D. In Utero MRI identifies consequences of early-gestation alcohol drinking on fetal brain development in rhesus macaques. Proc. Natl. Acad Sci. USA 2020, 117, 10035–10044. [Google Scholar] [CrossRef]
- Uchida, S.; Teubner, B.J.; Hevi, C.; Hara, K.; Kobayashi, A.; Dave, R.M.; Shintaku, T.; Jaikhan, P.; Yamagata, H.; Suzuki, T.; et al. CRTC1 Nuclear Translocation Following Learning Modulates Memory Strength via Exchange of Chromatin Remodeling Complexes on the Fgf1 Gene. Cell Rep. 2017, 18, 352–366. [Google Scholar] [CrossRef]
- Manji, S.; Pei, J.; Loomes, C.; Rasmussen, C. A review of the verbal and visual memory impairments in children with foetal alcohol spectrum disorders. Dev. Neurorehabil. 2009, 12, 239–247. [Google Scholar] [CrossRef]
- Rasmussen, C.; Andrew, G.; Zwaigenbaum, L.; Tough, S. Neurobehavioural outcomes of children with fetal alcohol spectrum disorders: A Canadian perspective. Paediatr. Child Health 2008, 13, 185–191. [Google Scholar]
- Rasmussen, C.; Horne, K.; Witol, A. Neurobehavioral functioning in children with fetal alcohol spectrum disorder. Child Neuropsychol. 2006, 12, 453–468. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subbanna, S.; Basavarajappa, B.S. Binge-like Prenatal Ethanol Exposure Causes Impaired Cellular Differentiation in the Embryonic Forebrain and Synaptic and Behavioral Defects in Adult Mice. Brain Sci. 2022, 12, 793. https://doi.org/10.3390/brainsci12060793
Subbanna S, Basavarajappa BS. Binge-like Prenatal Ethanol Exposure Causes Impaired Cellular Differentiation in the Embryonic Forebrain and Synaptic and Behavioral Defects in Adult Mice. Brain Sciences. 2022; 12(6):793. https://doi.org/10.3390/brainsci12060793
Chicago/Turabian StyleSubbanna, Shivakumar, and Balapal S. Basavarajappa. 2022. "Binge-like Prenatal Ethanol Exposure Causes Impaired Cellular Differentiation in the Embryonic Forebrain and Synaptic and Behavioral Defects in Adult Mice" Brain Sciences 12, no. 6: 793. https://doi.org/10.3390/brainsci12060793