
Mechanisms Underlying Cognitive Impairment Induced by Prenatal Alcohol Exposure

Department of Pharmacology and Toxicology, College of Pharmacy, Qassim University, Al Qassim 51452, Saudi Arabia
Brain Sci. 2022, 12(12), 1667; https://doi.org/10.3390/brainsci12121667
Submission received: 8 November 2022
/
Revised: 29 November 2022
/
Accepted: 2 December 2022
/
Published: 4 December 2022
(This article belongs to the Special Issue New Advances in Neurobiology and Behavioral Disturbance in Alcohol Use Disorder (AUD) including Fetal Alcohol Spectrum Disorder (FASD))
Abstract
:Alcohol is one of the most commonly used illicit substances among pregnant women. Clinical and experimental studies have revealed that prenatal alcohol exposure affects fetal brain development and ultimately results in the persistent impairment of the offspring’s cognitive functions. Despite this, the rate of alcohol use among pregnant women has been progressively increasing. Various aspects of human and animal behavior, including learning and memory, are dependent on complex interactions between multiple mechanisms, such as receptor function, mitochondrial function, and protein kinase activation, which are especially vulnerable to alterations during the developmental period. Thus, the exploration of the mechanisms that are altered in response to prenatal alcohol exposure is necessary to develop an understanding of how homeostatic imbalance and various long-term neurobehavioral impairments manifest following alcohol abuse during pregnancy. There is evidence that prenatal alcohol exposure results in vast alterations in mechanisms such as long-term potentiation, mitochondrial function, and protein kinase activation in the brain of offspring. However, to the best of our knowledge, there are very few recent reviews that focus on the cognitive effects of prenatal alcohol exposure and the associated mechanisms. Therefore, in this review, we aim to provide a comprehensive summary of the recently reported alterations to various mechanisms following alcohol exposure during pregnancy, and to draw potential associations with behavioral changes in affected offspring.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Alcohol (ethanol) is perhaps the most commonly used and socially accepted psychoactive substance. Alcohol consumption is highly addictive, and evidence indicates that it can cause serious systemic side effects, such as heart and lung diseases, and increase the risk of cancer and susceptibility to some infectious diseases [1,2]. More recently, alcohol use was also identified as a risk factor for dementia and cognitive decline [3,4]. Recent studies showed that 10% of pregnant women are exposed to alcohol worldwide [5,6]. Alcohol consumption is not recommended during pregnancy due to the risk of cognitive dysfunction in offspring [7], and studies provided undeniable evidence that alcohol exposure during pregnancy can lead to mental retardation in children [8,9]. Fetal alcohol spectrum disorders (FASDs) are an umbrella term used to refer to the neurobehavioral and physical impacts of prenatal alcohol exposure, and fetal alcohol syndrome (FAS) is one of the most severe consequences in this spectrum of disorders. FASD is a serious global public health concern, and its effects can persist through to adulthood. Its incidence ranges from 1% to 20% among children [10], and in younger school-aged children, its incidence can be as high as 20–50 cases per 1000 children [11]. The approximate incidence of FASDs in the general population was roughly estimated at about 1–2 cases per 1000 individuals [12], while the approximate incidence of FAS is 9–10 cases per 1000 individuals [13]. According to the United States Centers for Disease Control and Prevention, the incidence of FASD in the U.S. is relatively high (1.5–2.0 cases/1000 births) [14], and two studies in Europe showed that the incidence of FASDs is 23–47 cases per 1000 individuals [15] and 40 cases per 1000 individuals [16]. Importantly, the occurrence of FASDs was linked to the socioeconomic status of the mother, with maternal low education level and low income associated with a higher incidence of FASDs [11].
Clinical studies showed alterations in cognitive functions, such as the intelligence quotient score, of children with prenatal alcohol exposure [17,18,19]. In animal studies, the offspring of pregnant rats exposed to alcohol also showed cognitive impairment in fear conditioning and spatial memory tasks [14,20,21]. FASDs are commonly associated with a range of cognitive deficits that include memory impairments, specifically intrusion errors and confabulation, difficulties with strategic manipulation of information to improve recall, difficulties with the initial encoding of information, mixed expressive–receptive language disabilities, visual–motor integration and visual–perceptual deficits, learning disabilities, poor response inhibition, impulsiveness, perseverative behavior, impaired executive functioning, and deficiency in adaptive skills [22]. Prenatal alcohol exposure during the first trimester is associated with deficits in learning, and short- and long-term memory, specifically in the verbal domain [23]. Further, a prospective study in the UK found that alcohol consumption during the second and third trimesters of pregnancy was strongly related to an increased risk of mental health problems, especially inattention and hyperactivity, in girls at 47 months, and in both boys and girls at 81 months [24]. In New Zealand, higher rates of depression were reported among children exposed to alcohol in the prenatal period than those among unexposed children [25]. However, some studies have found only weak associations between alcohol consumption and neurobiological deficits. For example, in a Danish national study, exposure to binge drinking was only weakly associated with impaired behavioral and emotional development measured at age 7 [26]. Further, a study on children in rural Burkina Faso demonstrated that maternal alcohol consumption was associated with poorer neuropsychological performance in children aged 6–8 years, but it could not establish a causal relationship between the two [27]. Such inconsistencies may be a result of differences in study populations, definitions of alcohol consumption, and assessment methods. Despite these differences, studies so far indicate that alcohol consumption during pregnancy can, overall, alter neurodevelopmental processes in the offspring during embryonic development, ultimately leading to cognitive impairment and affecting neurobehavioral performance [28,29].
Alcohol that crosses the placental barrier to reach the fetus [30] is not effectively metabolized because the liver does not mature until the late stage of development; this results in an increase in the alcohol concentration in fetal circulation [31,32]. With regard to its mechanism of action, alcohol binds the gamma-aminobutyric acid (GABA) receptor, which is a major inhibitory neurotransmitter in the brain [33,34]. Through this interaction, alcohol acts as a GABA receptor agonist in central nervous system (CNS) tissues such as the amygdala, hippocampus, cerebellum, and cortex, thereby affecting brain development in the fetus [35,36]. The other reported mechanisms by which prenatal alcohol exposure causes cognitive impairment include the disruption of synaptic plasticity and long-term potentiation (LTP) [14], and the disruption of neurogenesis [37], mediated by the effects of alcohol on protein kinases [14,38,39], and mitochondrial dysfunction [5,6]. However, there are still some gaps in the knowledge regarding the mechanisms underlying the cognitive effects of alcohol on the fetus. Moreover, to the best of our knowledge, there are very few recent reviews that focused on the molecular mechanisms underlying the cognitive effects of prenatal alcohol exposure in offspring. Therefore, in this review, we discuss the recent studies on this topic in order to provide a comprehensive understanding of the mechanisms by which prenatal alcohol exposure induces cognitive impairment in offspring, and lay a basis for potential strategies that can be used to ameliorate these effects.
2. Behavioral Studies on the Effects of Prenatal Alcohol Exposure
Behavioral tests are used to evaluate cognitive function and memory formation in both animals and humans [40]. Some common tests used to assess memory function in animal models include the Y maze, novel object recognition, Morris water maze (MWM), radial arm maze, and contextual fear conditioning tasks [14,41,42]. Contextual fear conditioning tests involve a training session in which animals receive an electric shock that is preceded by a tone. In the test session, the period of time for which the animal remains frozen after the delivery of the shock is compared with that of animals who are exposed to the tone without the shock [14]. Studies using this technique showed that animals exposed to alcohol during pregnancy exhibited a shorter freezing time than that of control animals that had not been exposed to alcohol; this indicates that prenatal alcohol exposure causes cognitive impairment in offspring [14,43].
The Y-maze test is commonly used to evaluate spatial memory [44]. The maze is composed of three arms connected to each other via a chamber at the center. In this test, animals are allowed to explore two arms in an exposure period that ends with a certain cue. The third arm (novel) remains closed during the exposure session and is opened only during the test session. Behavior is evaluated by counting the number of entries and the total time spent in the novel arm in the test animals and comparing the findings with those of nontreated or nondiseased animals. Animals exposed to alcohol in the prenatal period made more spatial reference memory errors in the Y- and T-maze tests than the control animals did; further, spatial working memory errors were more common in males than in females [45,46].
The MWM consists of a large round tub of water with a platform that is hidden from view with the addition of powdered milk to the water [47]. In MWM tests, animals are allowed to swim, find the hidden platform, and stand on it during the training period. During the experiment, the animal is placed in the maze and allowed to find the hidden platform. The time spent on the platform and the distance covered by the animals while locating the hidden platform are tracked and recorded. The time spent reaching the platform is considered to be an indicator of learning and memory ability [47,48]. Studies conducted MWM tests and showed that the offspring of animals exposed to ethanol during pregnancy had impaired cognitive function compared to that of animals who had not been exposed to ethanol [49,50]. The virtual water maze, which is analogous to the MWM in animals, is used to test spatial memory in humans. In this test, children try to learn the location of a hidden platform in a virtual water pool through a sequence of learning trials. In the navigation task, their ability to locate the hidden platform is tested. Similar to the results obtained with rodents exposed to alcohol during pregnancy, the performance of children who had been exposed to alcohol in the prenatal period was poor and indicative of cognitive impairment [51].
Studies using the radial arm maze showed memory impairment in prenatal alcohol-exposed animals compared with control nonexposed animals [52]. In this test, animals are trained in a maze consisting of eight equally spaced arms connected at the center, and a cup with or without food is placed at the end of each arm. After the training session, animals are tested for their ability to identify the arm containing food, and the number of errors is counted and compared with the results of control nontreated or nondiseased animals. Although several studies showed a higher error rate among animals exposed to alcohol in the prenatal period than that of nonexposed controls [53,54], differences in cognitive function between these groups were not observed in other studies that employed this test. These discrepancies may be accounted for by differences in the time point at which the animals were evaluated, as some studies conducted these tests on postnatal day 90 (P90) and other conducted the tests between P26 and P60 [54].
The findings from the experimental animal models discussed above have also been reported in human subjects. For example, one systematic review reported impaired verbal and visual-spatial episodic memory performance in school-aged children and adolescents with prenatal alcohol exposure [55]. Further, another systematic review found that prenatal exposure to alcohol has long-term cognitive, behavioral, social, and emotional developmental consequences in adolescents that vary according to the amount and timing of exposure in utero [56].
To conclude, the findings of behavioral studies in both animal models and humans prove that prenatal exposure affects learning and memory functions in the offspring.
3. Effect of Prenatal Alcohol Exposure on Body and Brain Weight
Body weight is regulated by genetic, metabolic, and environmental factors. However, under certain circumstances, changes in homeostasis and behavior can cause weight gain or loss. Body weight is usually a reflection of health status, and alterations in body weight are a marker of brain function [57]. For example, an increase in body weight due to a high-fat diet can result in the development of Type 2 diabetes, which eventually causes cognitive impairment [58,59] and a reduction in blood cerebral flow [60,61,62]. In addition, an increase in BMI and obesity can cause a reduction in cerebral blood flow as a result of the increase in body weight, which can cause alterations in brain function [63]. Studies also revealed that neurological diseases, such as Alzheimer’s and Parkinson’s diseases, can cause body weight reduction [63,64]. In addition, neurological studies indicated that brain size is a marker of brain and cognitive function [65,66]. For instance, brain size, particularly in terms of the size of grey and white matter, is reduced in cancer patients following chemotherapy [67] and is thought to represent one of the mechanisms underlying cognitive dysfunction following chemotherapy [68]. Accordingly, studies on the effects of prenatal alcohol exposure on offspring revealed a reduction in body weight, and brain size and weight, particularly in the amygdalae and hippocampus, which may affect brain function and lead to cognitive impairment [43,69,70]. Thus, the cognitive effects of prenatal alcohol exposure may involve a reduction in body as well as brain weight.
4. Effect of Prenatal Alcohol Exposure on Neurogenesis
Neurogenesis, the process by which new neurons are generated, occurs in the brain in the embryonic period and continues into adulthood [71]. However, in adulthood, neurogenesis is restricted to specific regions of the brain such as the hippocampus and dentate gyrus [72,73,74]. During neurogenesis, neural stem cells proliferate and differentiate into mature neurons that integrate with other neurons to form the neuronal circuitry [75,76]. Neurons communicate with other neurons via electrochemical interactions at the synapse [77]. Neurogenesis and synaptogenesis in the hippocampus are vital for acquired learning and memory formation [78]. Previous studies revealed that a reduction in hippocampal neurogenesis results in memory impairment [37,79,80]. In addition, neurogenesis is reduced in the hippocampus of the offspring of rats exposed to alcohol in the prenatal period [81]. Thus, it is speculated that impaired neurogenesis is a potential mechanism of prenatal alcohol exposure-induced cognitive dysfunction in offspring. In fact, one study that used human brain organoids as an experimental model found that alcohol exposure impaired neurogenesis in these organoids [82]. This is supported by the findings of another study that used human cortical organoids and reported that alcohol exposure had temporally dependent effects on proliferation, cell cycle, and apoptosis [83].
5. Effect of Prenatal Alcohol Exposure on Synaptic Plasticity
Learning and memory processes occur in the hippocampus, which is a part of the limbic system of the brain that is responsible for memory formation. In these processes, the synaptic structure is altered, and its shape is modified, so that more receptors are present in the synaptic cleft; this is known as synaptic plasticity. Synaptic plasticity is also applied at the cellular level of synaptic neurons to describe the communication that occurs during memory formation. The synaptic response is based on the release of neurotransmitters from presynaptic neurons and the response of receptors on the postsynaptic neurons, and any alterations in these processes lead to a reduction or even abolition of LTP. Measurements of LTP and long-term depression (LTD) can be used to determine the strength and weaknesses of synapses in a certain neuronal pathway. Electrophysiological studies using in vitro and in vivo models have revealed changes in synaptic activity in the hippocampus following behavioral tasks, thereby confirming the link between synaptic change and memory formation. Further, studies showed a reduction in the LTP of the offspring of pregnant rats exposed to alcohol [14], indicating that prenatal exposure to alcohol can lead to a persistent reduction in LTP in offspring that subsequently results in cognitive impairment. This is supported by observations in human cortical organoid experiments that demonstrated that alcohol exposure affected glutamatergic synaptic development, and neural network formation and activity [83]. Human studies have not measured LTP in offspring with prenatal alcohol exposure, but one study reported that prenatal alcohol exposure is associated with specific EEG correlates in infants and children [84]. Further, another study on children with FASD found a reduction in theta frequency band power on EEG recordings taken during a memory-guided task [85]. These results could reflect changes in synaptic plasticity that occur as a result of prenatal alcohol exposure, but they need to be confirmed.
6. Effect of Prenatal Alcohol Exposure on Mitochondrial Function
The mitochondrion is the power house of the cell and is involved in many cellular processes, such as oxidative phosphorylation, ATP production, β-fatty acid oxidation, the Krebs cycle, and the regulation of apoptosis [86]. In particular, mitochondrial DNA (mtDNA) is important for enzymes and signaling pathways required for energy production within the mitochondria [42,87,88]. Alcohol exposure during pregnancy can alter mitochondrial morphology and function in the offspring [5,6]. Further, studies also showed that ethanol exposure can alter mtDNA via an increase in 8-hydroxydeoxyguanosine incorporation and mtDNA single-strand breaks, and a reduction in mtDNA content, which lead to mitochondria dysfunction [89,90,91]. Additionally, fetal ethanol exposure can increase lipid peroxidation, DNA damage, and reactive oxygen species generation, and reduce ATP synthesis and storage [92]. As mitochondrial cellular respiration plays a critical role in regulating cellular and cognitive functions, alterations in the process of cellular respiration can impact cognitive function. Interestingly, prenatal alcohol exposure impairs cognitive function by reducing the activity of mitochondrial complex I and complex IV [93,94,95]. Therefore, these mitochondrial complexes warrant further study from the viewpoint of understanding the mechanisms of cognitive impairment induced by prenatal alcohol exposure and identifying targets for treatment.
7. Activation of the GABA Receptor in the CNS of the Fetus in Response to Prenatal Alcohol Exposure
GABA receptors are expressed from Day 12 of the embryonic period and play an essential role in brain development and the general regulation of brain functions, including learning and memory formation [36,36,96]. GABA receptors are highly expressed in the CNS, predominantly in the amygdala, hippocampus, cortex, cerebellum, and hypothalamus [97]. They exist in two distinct forms, namely, GABAA and GABAB [98,99]. GABAA receptors are ligand-gated chloride ion channels, whereas GABAB receptors are G-protein-coupled receptors [100]. They are composed of five subunits, and different types of GABA receptors [101] are distinguished by the composition of these five subunits. The most common type of GABA receptors are composed of two α subunits, two β subunits, and one γ subunit. GABA receptors are involved in synaptic transmission and facilitate learning and memory processes, and GABA receptor activation appears to inhibit neuronal firing and the release of other neurotransmitters, such as glutamate and dopamine, in the CNS.
In experimental animal models, GABA receptor hyperactivation inhibits the release of glutamate and dopamine, thereby reducing the levels of other neurotransmitters linked to behavior modulation through its ability to regulate cognitive function. Further, studies showed that alterations in the expression of GABA receptors in animals impairs memory function [102,103], and prenatal alcohol exposure can chronically alter brain development and function in the offspring [104]. One study on cortical plate samples from fetal and infant brains demonstrated insufficient and delayed production of GABAergic interneurons in the ganglion during the two first trimesters of pregnancy and their delayed incorporation into the laminar structures of the frontal cortex, as well as mispositioning of GABAergic and calretininergic interneurons throughout fetal life [105]. Thus, alterations in GABA expression may be a potential mechanism underlying the cognitive dysfunction caused by prenatal alcohol exposure. This means that GABA receptors may be a viable target for the treatment of cognitive problems caused by prenatal alcohol exposure.
8. Effect of Prenatal Alcohol Exposure on Protein Expression and Activity
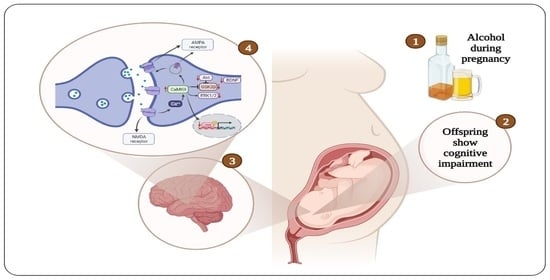
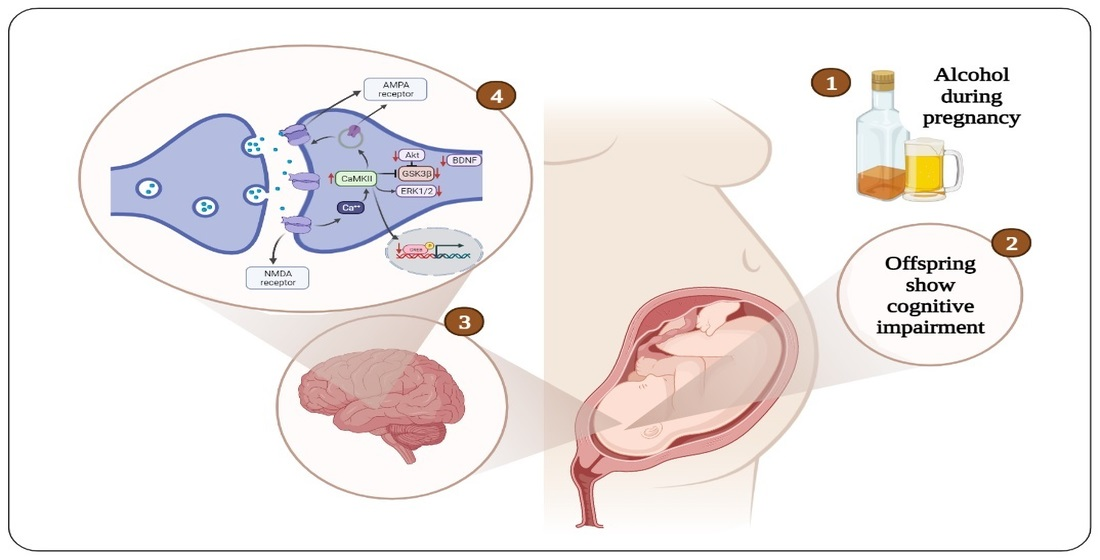
Proteins play a vital role in regulating many physiological functions at the cellular level, including cell structure, metabolism, and differentiation, and synaptic plasticity and learning and memory [106,107,108]. With regard to neuronal stimulation and memory formation, proteins facilitate signal transduction from receptors to activate different protein kinases [109,110]. These signaling pathways regulate the gene expression and protein synthesis required to modify ion channel properties and ion channel density in the synapses [111,112,113]. The expression of several proteins in the hippocampus is altered during hippocampal-dependent tasks, and these proteins include brain-derived neurotrophic factor (BDNF), calcium calmodulin dependent kinase II (CaMKII), extracellular regulated kinase ½ (ERK1/2), cAMP-response element binding protein (CREB), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor, and N-methyl-d-aspartate (NMDA) receptor [114,115,116,117]. In addition, the phosphorylation of protein kinase B (PKB or Akt) and glycogen synthase kinase 3 beta (GSK3β) is altered. These proteins play an essential role in neuronal signaling, signal transduction, and neurotransmitter synthesis and release, thereby facilitating learning and memory processes [112,118] (Figure 1).
Akt is a downstream protein of phosphoinositide-3-kinase (PI3K), and the Akt/PI3K pathway functions as a prosurvival pathway [119]. Akt phosphorylation plays a key role in regulating cellular processes, such as survival, proliferation, and apoptosis, and the neurological pathways involved in learning and memory formation [120]. Akt phosphorylation at Ser473 leads to GSK3β inhibition through phosphorylation at Ser9, thereby reducing glycogen synthesis and increasing the energy available for memory formation. The phosphorylation of Akt and GSK3β was reduced in the brains of offspring exposed to alcohol during the prenatal period, and as a result, energy consumption, signaling, and trafficking of other receptors, such as glutamate AMPA and NMDA receptors, were impaired. AMPA and NMDA receptors play a key role in regulating memory function. These receptors are composed of tetramers of four subunits assembled in different combinations [121]. AMPA receptors consist of GluR1 to GluR4 subunits, while NMDA receptors contain GluN1, GluN2, and GluN3 [122,123]. Under normal physiological conditions, AMPA receptors are impermeable to calcium, while NMDA receptors are permeable [124]. The calcium impermeability of AMPA receptors is due to the presence of the GluR2 subunit that contains Mg2+ [124,125]. The presence of GluR2 in the synaptic AMPA receptors prevents Ca++ influx, thus reducing excitotoxicity [126,127]. In GluR2-knockout mice, increased levels of GluR1 in AMPA receptors resulted in greater Ca++ influx into neurons, and this led to epileptic seizures and thus apoptosis [127]. Therefore, alterations in the surface expression of these subunits could alter memory formation. The surface expression of GluR2 was increased in the hippocampus of rats exposed to alcohol and nicotine during pregnancy; this indicates that cognitive dysfunction in offspring is caused by GluR2 overexpression at the synaptic surface [14]. In addition, alterations in the subunit composition in NMDA receptors can alter cognitive function, and the GluN1 and GluN3 composition of synaptic NMDA receptors is increased following prenatal alcohol exposure. Thus, changes in the subunit composition of AMPA and NMDA receptors may play a role in the mechanism by which cognitive function is impaired in offspring with prenatal alcohol exposure, and these receptors may be viable targets for treatment.
BDNF belongs to a family of neurotrophins that control many physiological functions, including cell survival, differentiation, neurogenesis, and synaptogenesis [128]. With regard to memory function, BDNF plays a critical role in regulating learning and memory processes and synaptic plasticity in the brain. The underlying mechanism involves BDNF-induced activation of tropomyosin-related kinase B (TrkB) receptors, which belong to the tropomyosin-related kinase family. Previous studies revealed that BDNF mRNA expression was increased in the hippocampus of animals after training in the MWM, radial arm maze, and contextual fear conditioning tests [114,128,129]. However, a reduction in BDNF mRNA expression is associated with memory impairment in dopamine transporter-knockout mice [130]. Accordingly, the intrahippocampal injection of BDNF improved the memory function of animals in MWM tests, while the administration of anti-BDNF antibodies led to memory impairment [131,132]. BDNF also regulates the expression and function of various proteins necessary for synaptic plasticity and cognitive function through the modulation of the TrkB receptor [133]. When BDNF binds to the extracellular region of this receptor, it enhances tyrosine kinase activity, leading to autophosphorylation by ATP [134]. The activation of the TrkB receptor activates other pathways, such as the mitogen-activated protein kinase (MAPK) and PI3K/AKT pathways, and regulates CaMKII [135], all of which play essential roles in the regulation of cognitive function [136]. BDNF expression is reduced following prenatal alcohol exposure, and this reduction in BDNF levels could result in alterations in other signaling pathways that ultimately cause cognitive impairment [14]. These findings indicate that alcohol exposure during pregnancy reduces the cognitive function of offspring via inhibition of Akt, GSK3β, and BDNF activities.
CaMKII is a protein kinase that is involved in calcium signaling in eukaryotic cells [137]. This protein is activated by increase in intracellular calcium, and it phosphorylates various proteins that are involved in mobilization of synaptic vesicles, modulation of ion channels, regulation of gene expression, regulation of learning and memory processes, and LTP [138,139]. CaMKII is present downstream of NMDA receptors, and activation by Ca2+ influx through NMDA receptors causes autophosphorylation of CaMKII [140]. There is considerable evidence that alterations in CaMKII activity in the brain can lead to cognitive impairment [141,142,143]. Further, the overactivation of CaMKII can induce neurotoxicity and, thus, apoptosis [144]. Similarly, a reduction in CaMKII activity induces cognitive dysfunction. In addition, CaMKII activation can phosphorylate the AMPA-GluR1 subunit at the Ser831 residue and lead to the mobilization of the GluR1 subunit to the synaptic membrane [113]. CaMKII also plays a critical role in LTP induction and maintenance [145]. In rats with prenatal alcohol exposure, CaMKII activities were significantly increased compared to the control levels [146]. These findings indicate that CaMKII induces apoptosis in the brain, which could be a potential mechanism of cognitive impairment in offspring exposed to alcohol in the prenatal period.
ERK1/2 signaling is required for normal physiological functions [147]. In the brain, ERK1/2 plays a vital role in regulating neuronal development, function, and synaptic plasticity [148]. ERK1/2 is activated through its phosphorylation, and this can enhance signaling pathways that are involved in mediating neuroprotection during oxidative stress responses [38,149] and facilitate memory consolidation [39]. In addition, several lines of evidence showed that ERK1/2 phosphorylation is required for formation of short-term and long-term memory as well as mediating LTP and LTD [150,151]. Likewise, ERK1/2 can regulate the activities of transcriptional factors, such as CREB and BDNF, which are well-established as vital factors in learning and memory processes [152]. Therefore, alterations in ERK1/2 activity can alter synaptic plasticity and memory formation [153,154]. With regard to the association of ERK1/2 with prenatal alcohol exposure, ERK1/2 phosphorylation is reduced in offspring with alcohol exposure during pregnancy [43,155,156]. Thus, a reduction in ERK1/2 phosphorylation may play an important role in memory impairment in offspring.
CREB is a transcription factor that plays a critical role in regulating several physiological functions [157]. CREB is activated by several signaling pathways, such as the insulin, PI3K/AKT, and MAPK pathways [158,159]. This activation of CREB occurs through phosphorylation at the Ser133 residue [152,160]. Once CREB is activated, it is transported from the cytoplasm to the nucleus, where it binds the CRE promoter region, which is involved in gene transcription, translation, and protein synthesis [152,161]. Recent studies revealed that activation of CREB plays a role in hippocampal-dependent tasks by enhancing synaptic plasticity, LTP, and memory formation. In contrast, the reduced activity of CREB induces LTD by causing synaptic AMPA receptor endocytosis [162,163,164]. Interestingly, studies reported that CREB phosphorylation is reduced in the offspring of mothers exposed to alcohol during pregnancy [165,166,167]; this might represent another mechanism of cognitive impairment caused by prenatal alcohol exposure. Thus, prenatal alcohol exposure may induce cognitive impairment through inhibition of CREB activities, and this may cause a reduction in GluR1-containing AMPA receptors in the synaptic surface and induce LTD.
Thus, various proteins have been implicated in the mechanisms underlying the cognitive effects of prenatal alcohol exposure, namely, Akt, GSK3β, NMDA and AMPA receptors, BDNF, CaMKII, ERK1/2, and CREB. In the future, it would be interesting to explore their potential as treatment targets in offspring affected by cognitive dysfunction caused by alcohol exposure.
9. Conclusions
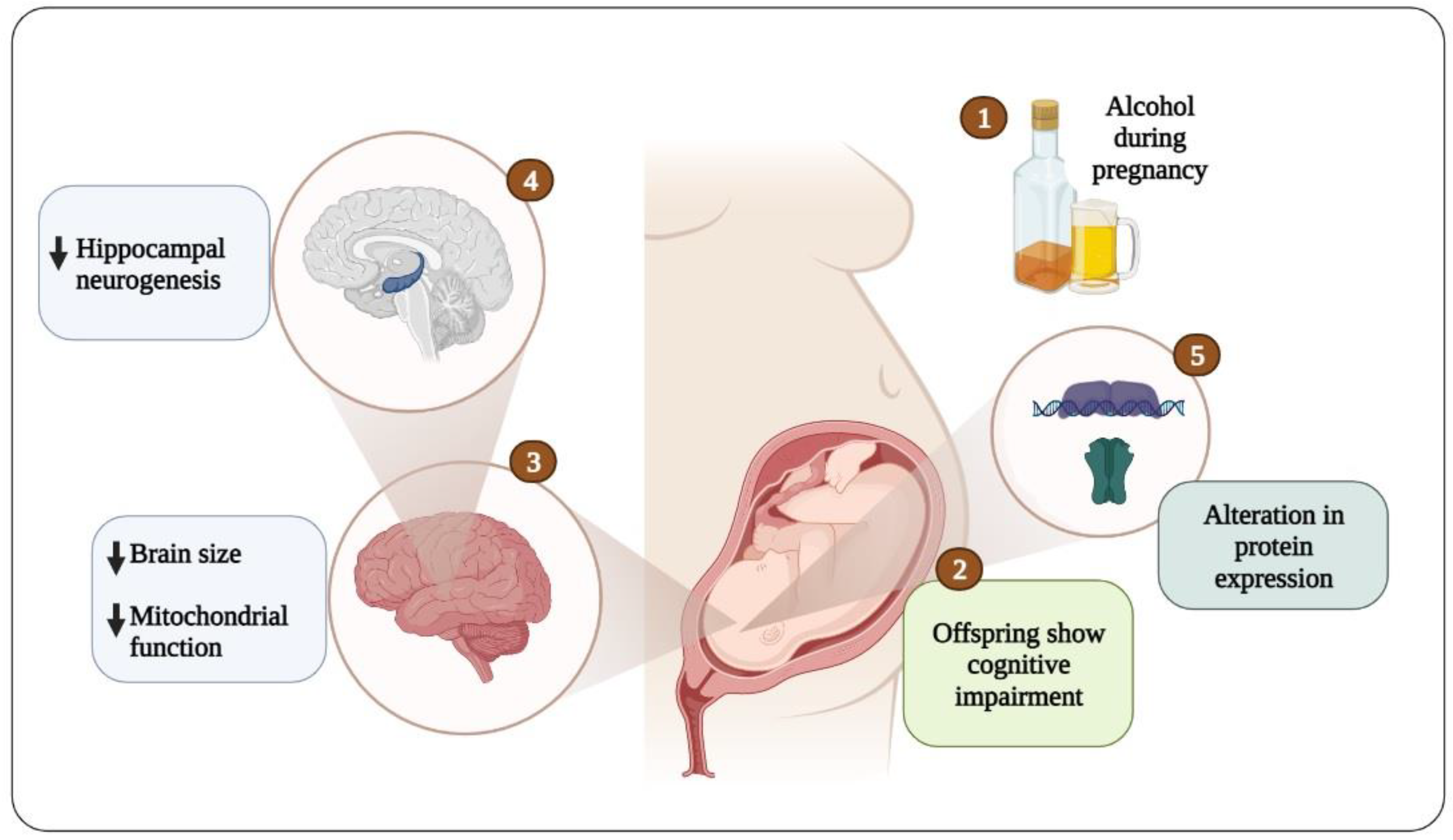
Prenatal alcohol exposure caused cognitive impairment via different mechanisms in humans and experimental animal models, and these include the disruption of synaptic plasticity, LTP, neurogenesis, and mitochondrial dysfunction (Figure 2). Alcohol has the ability to pass through the placental and blood–brain barriers to access the fetal brain and activate GABA receptors, thereby affecting development and leading to irreversible changes in brain function. Prenatal exposure to alcohol reduces the expression and function of proteins, such as phosphorylated Akt, phosphorylated GSK3β, BDNF, phosphorylated ERK1/2, phosphorylated CREB, and phosphorylated CaMKII, and alters the composition of the AMPA and NMDA receptor subunits, and this leads to an increase in glutamate GluR2 surface expression. These changes are associated with mitochondrial dysfunction, and decreased neurogenesis in the hippocampus and cortex, and ultimately lead to cognitive dysfunction in the offspring. Thus, these proteins and their pathways would be interesting topics of investigation in future studies on the treatment of cognitive dysfunction caused by alcohol exposure.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Rehm, J.; Parry, C.D. Alcohol consumption and infectious diseases in South Africa. Lancet 2009, 374, 2053. [Google Scholar] [CrossRef] [PubMed]
- Corrao, G. A meta-analysis of alcohol consumption and the risk of 15 diseases. Prev. Med. 2004, 38, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Fitzpatrick, A.L.; Rapp, S.R.; Nahin, R.L.; Williamson, J.D.; Lopez, O.L.; DeKosky, S.T.; Kuller, L.H.; Mackey, R.H.; Mukamal, K.J.; et al. Alcohol Consumption and Risk of Dementia and Cognitive Decline Among Older Adults With or Without Mild Cognitive Impairment. JAMA Netw. Open 2019, 2, e1910319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachdeva, A.; Chandra, M.; Choudhary, M.; Dayal, P.; Anand, K.S. Alcohol-Related Dementia and Neurocognitive Impairment: A Review Study. Int. J. High Risk Behav. Addict. 2016, 5, e27976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popova, S.; Lange, S.; Probst, C.; Gmel, G.; Rehm, J. Global prevalence of alcohol use and binge drinking during pregnancy, and fetal alcohol spectrum disorder. Biochem. Cell Biol. 2018, 96, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Popova, S.; Lange, S.; Probst, C.; Gmel, G.; Rehm, J. Estimation of national, regional, and global prevalence of alcohol use during pregnancy and fetal alcohol syndrome: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e290–e299. [Google Scholar] [CrossRef] [Green Version]
- Easey, K.E.; Dyer, M.L.; Timpson, N.J.; Munafò, M.R. Prenatal alcohol exposure and offspring mental health: A systematic review. Drug Alcohol Depend. 2019, 197, 344–353. [Google Scholar] [CrossRef]
- O’Leary, C.; Leonard, H.; Bourke, J.; D’Antoine, H.; Bartu, A.; Bower, C. Intellectual disability: Population-based estimates of the proportion attributable to maternal alcohol use disorder during pregnancy. Dev. Med. Child Neurol. 2013, 55, 271–277. [Google Scholar] [CrossRef]
- Charness, M.E.; Riley, E.P.; Sowell, E.R. Drinking During Pregnancy and the Developing Brain: Is Any Amount Safe? Trends Cogn. Sci. 2016, 20, 80–82. [Google Scholar] [CrossRef] [Green Version]
- Nulman, I.; Shulman, T.; Liu, F. Fetal Alcohol Spectrum Disorder. In Handbook of Developmental Neurotoxicology; Sciencedirect: Amsterdam, The Netherlands, 2018; pp. 427–437. [Google Scholar] [CrossRef]
- May, P.A.; Gossage, J.P.; Kalberg, W.O.; Robinson, L.K.; Buckley, D.; Manning, M.; Hoyme, H.E. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev. Disabil. Res. Rev. 2009, 15, 176–192. [Google Scholar] [CrossRef]
- May, P.A.; Baete, A.; Russo, J.; Elliott, A.J.; Blankenship, J.; Kalberg, W.O.; Buckley, D.; Brooks, M.; Hasken, J.; Abdul-Rahman, O.; et al. Prevalence and Characteristics of Fetal Alcohol Spectrum Disorders. Pediatrics 2014, 134, 855–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, S.; Probst, C.; Gmel, G.; Rehm, J.; Burd, L.; Popova, S. Global Prevalence of Fetal Alcohol Spectrum Disorder Among Children and Youth. JAMA Pediatr. 2017, 171, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Bhattacharya, D.; Dunaway, E.P.; Bhattacharya, S.; Bloemer, J.; Buabeid, M.; Escobar, M.; Suppiramaniam, V.; Dhanasekaran, M. Impaired ILK Function Is Associated with Deficits in Hippocampal Based Memory and Synaptic Plasticity in a FASD Rat Model. PLoS ONE 2015, 10, e0135700. [Google Scholar] [CrossRef] [Green Version]
- May, P.; Fiorentino, D.; Coriale, G.; Kalberg, W.; Hoyme, H.E.; Aragón, A.; Buckley, D.; Stellavato, C.; Gossage, J.P.; Robinson, L.; et al. Prevalence of Children with Severe Fetal Alcohol Spectrum Disorders in Communities Near Rome, Italy: New Estimated Rates Are Higher than Previous Estimates. Int. J. Environ. Res. Public Health 2011, 8, 2331–2351. [Google Scholar] [CrossRef]
- Petković, G.; Barišić, I. FAS prevalence in a sample of urban schoolchildren in Croatia. Reprod. Toxicol. 2010, 29, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, L.; Lewis, S.J.; Davey Smith, G.; Sayal, K.; Draper, E.S.; Fraser, R.; Barrow, M.; Alati, R.; Ring, S.; Macleod, J.; et al. Prenatal alcohol exposure and offspring cognition and school performance. A ‘Mendelian randomization’ natural experiment. Int. J. Epidemiol. 2013, 42, 1358–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjölund, S.; Hemmingsson, T.; Allebeck, P. Survey of Swedish Conscripts. Alcohol. Clin. Exp. Res. 2015, 39, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Kodituwakku, P.; Kodituwakku, E. Cognitive and Behavioral Profiles of Children with Fetal Alcohol Spectrum Disorders. Curr. Dev. Disord. Rep. 2014, 1, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Hunt, P.S.; Jacobson, S.E.; Torok, E.J. Deficits in trace fear conditioning in a rat model of fetal alcohol exposure: Dose–response and timing effects. Alcohol 2009, 43, 465–474. [Google Scholar] [CrossRef]
- Sanchez, L.M.; Goss, J.; Wagner, J.; Davies, S.; Savage, D.D.; Hamilton, D.A.; Clark, B.J. Moderate prenatal alcohol exposure impairs performance by adult male rats in an object-place paired-associate task. Behav. Brain Res. 2019, 360, 228–234. [Google Scholar] [CrossRef]
- Gibbard, W.B.; Wass, P.; Clarke, M.E. The neuropsychological implications of prenatal alcohol exposure. Can. Child Adolesc. Psychiatr. Rev. 2003, 12, 72–76. [Google Scholar] [PubMed]
- Willford, J.A.; Richardson, G.A.; Leech, S.L.; Day, N.L. Verbal and Visuospatial Learning and Memory Function in Children With Moderate Prenatal Alcohol Exposure. Alcohol. Clin. Exp. Res. 2004, 28, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayal, K.; Heron, J.; Golding, J.; Alati, R.; Smith, G.D.; Gray, R.; Emond, A. Binge Pattern of Alcohol Consumption During Pregnancy and Childhood Mental Health Outcomes: Longitudinal Population-Based Study. Pediatrics 2009, 123, e289–e296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easey, K.E.; Timpson, N.J.; Munafò, M.R. Association of Prenatal Alcohol Exposure and Offspring Depression: A Negative Control Analysis of Maternal and Partner Consumption. Alcohol. Clin. Exp. Res. 2020, 44, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Niclasen, J.; Nybo Andersen, A.M.; Teasdale, T.W.; Strandberg-Larsen, K. Prenatal exposure to alcohol, and gender differences on child mental health at age seven years. J. Epidemiol. Community Health 2014, 68, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Sanou, A.S.; Diallo, A.H.; Holding, P.; Nankabirwa, V.; Engebretsen, I.M.S.; Ndeezi, G.; Tumwine, J.K.; Meda, N.; Tylleskar, T.; Kashala-Abotnes, E. Maternal alcohol consumption during pregnancy and child’s cognitive performance at 6–8 years of age in rural Burkina Faso: An observational study. PeerJ 2017, 5, e3507. [Google Scholar] [CrossRef] [Green Version]
- Carpita, B.; Migli, L.; Chiarantini, I.; Battaglini, S.; Montalbano, C.; Carmassi, C.; Cremone, I.M.; Dell’Osso, L. Autism Spectrum Disorder and Fetal Alcohol Spectrum Disorder: A Literature Review. Brain Sci. 2022, 12, 792. [Google Scholar] [CrossRef]
- Zhang, M.; Marjonen, H.; Sierra, A.; Nyman, A.; Rogojin, V.; Gröhn, O.; Linden, A.-M.; Hautaniemi, S.; Kaminen-Ahola, N. Early Maternal Alcohol Consumption Alters Hippocampal DNA Methylation, Gene Expression and Volume in a Mouse Model. PLoS ONE 2015, 10, e0124931. [Google Scholar] [CrossRef]
- Guelinckx, I.; Devlieger, R.; Vansant, G. Alcohol during pregnancy and lactation: Recommendations versus real intake. Arch. Public Health 2011, 68, 134–142. [Google Scholar] [CrossRef]
- Kaminen-Ahola, N. Fetal alcohol spectrum disorders: Genetic and epigenetic mechanisms. Prenat. Diagn. 2020, 40, 1185–1192. [Google Scholar] [CrossRef]
- Lee, X.A.; Mostafaie, Y. Acute alcohol exposure in early gestation programmes sex-specific insulin resistance in offspring: A shifting tide in prenatal alcohol exposure models. J. Physiol. 2020, 598, 1807–1808. [Google Scholar] [CrossRef]
- Olsen, R.W.; Liang, J. Role of GABAA receptors in alcohol use disorders suggested by chronic intermittent ethanol (CIE) rodent model. Mol. Brain 2017, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N. Neurotransmitters in alcoholism: A review of neurobiological and genetic studies. Indian J. Hum. Genet. 2014, 20, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, I.A.; Harris, R.A. GABAA receptors and alcohol. Pharmacol. Biochem. Behav. 2008, 90, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2014, 30, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Faber, D.S.; Pereda, A.E. Two Forms of Electrical Transmission Between Neurons. Front. Mol. Neurosci. 2018, 11, 427. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, J.E.; Jaumotte, J.D.; Lakoski, J.M.; Zigmond, M.J. Neuroprotective role of ERK1/2 and ERK5 in a dopaminergic cell line under basal conditions and in response to oxidative stress. J. Neurosci. Res. 2006, 84, 1367–1375. [Google Scholar] [CrossRef]
- Krawczyk, M.C.; Millan, J.; Blake, M.G.; Feld, M.; Boccia, M.M. Relevance of ERK1/2 Post-retrieval Participation on Memory Processes: Insights in Their Particular Role on Reconsolidation and Persistence of Memories. Front. Mol. Neurosci. 2019, 12, 95. [Google Scholar] [CrossRef] [Green Version]
- Stephan, M.; Volkmann, P.; Rossner, M.J. Assessing behavior and cognition in rodents, nonhuman primates, and humans: Where are the limits of translation? Dialogues Clin. Neurosci. 2022, 21, 249–259. [Google Scholar] [CrossRef]
- Alharbi, I.; Alharbi, H.; Almogbel, Y.; Alalwan, A.; Alhowail, A. Effect of Metformin on Doxorubicin-Induced Memory Dysfunction. Brain Sci. 2020, 10, 152. [Google Scholar] [CrossRef] [Green Version]
- Alhowail, A.H.; Almogbel, Y.S.; Abdellatif, A.A.H.; Alsalehi, N.F.; Alghenaim, F.A.; Aldubayan, M.A.; Felemban, S.G. CMF and MET treatment induce cognitive impairment through upregulation of IL-1alpha in rat brain. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4385–4393. [Google Scholar] [CrossRef] [PubMed]
- Kozanian, O.O.; Rohac, D.J.; Bavadian, N.; Corches, A.; Korzus, E.; Huffman, K.J. Long-Lasting Effects of Prenatal Ethanol Exposure on Fear Learning and Development of the Amygdala. Front. Behav. Neurosci. 2018, 12, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamim, S.; Khan, K.M.; Ullah, N.; Chigurupati, S.; Wadood, A.; Ur Rehman, A.; Ali, M.; Salar, U.; Alhowail, A.; Taha, M.; et al. Synthesis and screening of (E)-3-(2-benzylidenehydrazinyl)-5,6-diphenyl-1,2,4-triazine analogs as novel dual inhibitors of alpha-amylase and alpha-glucosidase. Bioorg. Chem. 2020, 101, 103979. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.E.; Berkowitz, L.E.; Hamilton, D.A.; Clark, B.J. The effects of developmental alcohol exposure on the neurobiology of spatial processing. Neurosci. Biobehav. Rev. 2019, 107, 775–794. [Google Scholar] [CrossRef]
- Subbanna, S.; Basavarajappa, B.S. Binge-like Prenatal Ethanol Exposure Causes Impaired Cellular Differentiation in the Embryonic Forebrain and Synaptic and Behavioral Defects in Adult Mice. Brain Sci. 2022, 12, 793. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Chapouthier, G.; Barnhart, C.D.; Yang, D.; Lein, P.J. Using the Morris Water Maze to Assess Spatial Learning and Memory in Weanling Mice. PLoS ONE 2015, 10, e0124521. [Google Scholar] [CrossRef]
- Ieraci, A.; Herrera, D.G. Early Postnatal Ethanol Exposure in Mice Induces Sex-Dependent Memory Impairment and Reduction of Hippocampal NMDA-R2B Expression in Adulthood. Neuroscience 2020, 427, 105–115. [Google Scholar] [CrossRef]
- Christie, B.R.; Swann, S.E.; Fox, C.J.; Froc, D.; Lieblich, S.E.; Redila, V.; Webber, A. Voluntary exercise rescues deficits in spatial memory and long-term potentiation in prenatal ethanol-exposed male rats. Eur. J. Neurosci. 2005, 21, 1719–1726. [Google Scholar] [CrossRef]
- Dodge, N.C.; Thomas, K.G.F.; Meintjes, E.M.; Molteno, C.D.; Jacobson, J.L.; Jacobson, S.W. Reduced Hippocampal Volumes Partially Mediate Effects of Prenatal Alcohol Exposure on Spatial Navigation on a Virtual Water Maze Task in Children. Alcohol. Clin. Exp. Res. 2020, 44, 844–855. [Google Scholar] [CrossRef]
- Hunt, P.S.; Barnet, R.C. An animal model of fetal alcohol spectrum disorder: Trace conditioning as a window to inform memory deficits and intervention tactics. Physiol. Behav. 2015, 148, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, E.; Wolfe, J.; Savage, D.D. The effects of prenatal alcohol exposure on radial arm maze performance in adult rats. Physiol. Behav. 1989, 46, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, D.; Hartman, R.; Boyle, M.; Vogt, S.; Brooks, A.; Tenkova, T.; Young, C.; Olney, J.; Muglia, L. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis. 2004, 17, 403–414. [Google Scholar] [CrossRef] [PubMed]
- du Plooy, C.P.; Malcolm-Smith, S.; Adnams, C.M.; Stein, D.J.; Donald, K.A. The Effects of Prenatal Alcohol Exposure on Episodic Memory Functioning: A Systematic Review: Table 1. Arch. Clin. Neuropsychol. 2016, 31, 710–726. [Google Scholar] [CrossRef] [PubMed]
- Irner, T.B. Substance exposure in utero and developmental consequences in adolescence: A systematic review. Child Neuropsychol. 2012, 18, 521–549. [Google Scholar] [CrossRef] [PubMed]
- Amen, D.G.; Wu, J.; George, N.; Newberg, A. Patterns of Regional Cerebral Blood Flow as a Function of Obesity in Adults. J. Alzheimer’s Dis. 2020, 77, 1331–1337. [Google Scholar] [CrossRef]
- Elabi, O.F.; Cunha, J.P.M.C.M.; Gaceb, A.; Fex, M.; Paul, G. High-fat diet-induced diabetes leads to vascular alterations, pericyte reduction, and perivascular depletion of microglia in a 6-OHDA toxin model of Parkinson disease. J. Neuroinflammation 2021, 18, 175. [Google Scholar] [CrossRef]
- von Frankenberg, A.D.; Marina, A.; Song, X.; Callahan, H.S.; Kratz, M.; Utzschneider, K.M. A high-fat, high-saturated fat diet decreases insulin sensitivity without changing intra-abdominal fat in weight-stable overweight and obese adults. Eur. J. Nutr. 2015, 56, 431–443. [Google Scholar] [CrossRef] [Green Version]
- de Eulate, R.G.; Goñi, I.; Galiano, A.; Vidorreta, M.; Recio, M.; Riverol, M.; Zubieta, J.L.; Fernández-Seara, M.A.; Bondi, M. Reduced Cerebral Blood Flow in Mild Cognitive Impairment Assessed Using Phase-Contrast MRI. J. Alzheimer’s Dis. 2017, 58, 585–595. [Google Scholar] [CrossRef]
- Leeuwis, A.E.; Smith, L.A.; Melbourne, A.; Hughes, A.D.; Richards, M.; Prins, N.D.; Sokolska, M.; Atkinson, D.; Tillin, T.; Jäger, H.R.; et al. Cerebral Blood Flow and Cognitive Functioning in a Community-Based, Multi-Ethnic Cohort: The SABRE Study. Front. Aging Neurosci. 2018, 10, 279. [Google Scholar] [CrossRef] [Green Version]
- Coucha, M.; Abdelsaid, M.; Ward, R.; Abdul, Y.; Ergul, A. Impact of Metabolic Diseases on Cerebral Circulation: Structural and Functional Consequences. Compr. Physiol. 2018, 8, 773–799. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.P.; Laird, E.; Williamson, W.; O’Connor, J.; Newman, L.; Carey, D.; De Looze, C.; Fagan, A.J.; Chappell, M.A.; Meaney, J.F.; et al. Obesity is associated with reduced cerebral blood flow—Modified by physical activity. Neurobiol. Aging 2021, 105, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Kistner, A.; Lhommée, E.; Krack, P. Mechanisms of Body Weight Fluctuations in Parkinson’s Disease. Front. Neurol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Royle, N.A.; Booth, T.; Valdés Hernández, M.C.; Penke, L.; Murray, C.; Gow, A.J.; Maniega, S.M.; Starr, J.; Bastin, M.E.; Deary, I.J.; et al. Estimated maximal and current brain volume predict cognitive ability in old age. Neurobiol. Aging 2013, 34, 2726–2733. [Google Scholar] [CrossRef] [Green Version]
- Filley, C.M.; Fields, R.D. White matter and cognition: Making the connection. J. Neurophysiol. 2016, 116, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- de Ruiter, M.B.; Reneman, L.; Boogerd, W.; Veltman, D.J.; Caan, M.; Douaud, G.; Lavini, C.; Linn, S.C.; Boven, E.; van Dam, F.S.A.M.; et al. Late effects of high-dose adjuvant chemotherapy on white and gray matter in breast cancer survivors: Converging results from multimodal magnetic resonance imaging. Hum. Brain Mapp. 2012, 33, 2971–2983. [Google Scholar] [CrossRef]
- Inagaki, M.; Yoshikawa, E.; Matsuoka, Y.; Sugawara, Y.; Nakano, T.; Akechi, T.; Wada, N.; Imoto, S.; Murakami, K.; Uchitomi, Y. Smaller regional volumes of brain gray and white matter demonstrated in breast cancer survivors exposed to adjuvant chemotherapy. Cancer 2007, 109, 146–156. [Google Scholar] [CrossRef]
- Hashimoto, K.; Cullen, C.L.; Burne, T.H.J.; Lavidis, N.A.; Moritz, K.M. Low Dose Prenatal Ethanol Exposure Induces Anxiety-Like Behaviour and Alters Dendritic Morphology in the Basolateral Amygdala of Rat Offspring. PLoS ONE 2013, 8, e54924. [Google Scholar] [CrossRef] [Green Version]
- Willoughby, K.A.; Sheard, E.D.; Nash, K.; Rovet, J. Effects of prenatal alcohol exposure on hippocampal volume, verbal learning, and verbal and spatial recall in late childhood. J. Int. Neuropsychol. Soc. 2008, 14, 1022–1033. [Google Scholar] [CrossRef]
- Mayeux, R.; Dupret, D.; Revest, J.-M.; Koehl, M.; Ichas, F.; De Giorgi, F.; Costet, P.; Abrous, D.N.; Piazza, P.V. Spatial Relational Memory Requires Hippocampal Adult Neurogenesis. PLoS ONE 2008, 3, e1959. [Google Scholar] [CrossRef] [Green Version]
- Shivraj Sohur, U.; Emsley, J.G.; Mitchell, B.D.; Macklis, J.D. Adult neurogenesis and cellular brain repair with neural progenitors, precursors and stem cells. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1477–1497. [Google Scholar] [CrossRef] [PubMed]
- Piatti, V.C.; Ewell, L.A.; Leutgeb, J.K. Neurogenesis in the dentate gyrus: Carrying the message or dictating the tone. Front. Neurosci. 2013, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Alhowail, A.H.; Aldubayan, M. Recent progress in the elucidation of the mechanisms of chemotherapy-induced cognitive impairment. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 5807–5817. [Google Scholar] [CrossRef]
- Motamed, S.; Del Borgo, M.P.; Zhou, K.; Kulkarni, K.; Crack, P.J.; Merson, T.D.; Aguilar, M.-I.; Finkelstein, D.I.; Forsythe, J.S. Migration and Differentiation of Neural Stem Cells Diverted From the Subventricular Zone by an Injectable Self-Assembling β-Peptide Hydrogel. Front. Bioeng. Biotechnol. 2019, 7, 315. [Google Scholar] [CrossRef]
- Tau, G.Z.; Peterson, B.S. Normal Development of Brain Circuits. Neuropsychopharmacology 2009, 35, 147–168. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.; Wang, Y.; Borgus, J.R.; Venton, B.J. Electrochemistry at the Synapse. Annu. Rev. Anal. Chem. 2019, 12, 297–321. [Google Scholar] [CrossRef]
- Toda, T.; Parylak, S.L.; Linker, S.B.; Gage, F.H. The role of adult hippocampal neurogenesis in brain health and disease. Mol. Psychiatry 2018, 24, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereda, A.E. Electrical synapses and their functional interactions with chemical synapses. Nat. Rev. Neurosci. 2014, 15, 250–263. [Google Scholar] [CrossRef]
- Gil-Mohapel, J.; Boehme, F.; Kainer, L.; Christie, B.R. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: Insights from different rodent models. Brain Res. Rev. 2010, 64, 283–303. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, L.; Yin, F.; Yu, Y.; Wang, Y.; Shepard, M.J.; Zhuang, Z.; Qin, J. Probing impaired neurogenesis in human brain organoids exposed to alcohol. Integr. Biol. 2017, 9, 968–978. [Google Scholar] [CrossRef]
- Adams, J.W.; Negraes, P.D.; Truong, J.; Tran, T.; Szeto, R.A.; Guerra, B.S.; Herai, R.H.; Teodorof-Diedrich, C.; Spector, S.A.; Del Campo, M.; et al. Impact of alcohol exposure on neural development and network formation in human cortical organoids. Mol. Psychiatry 2022. [Google Scholar] [CrossRef] [PubMed]
- D’Angiulli, A.; Grunau, P.; Maggi, S.; Herdman, A. Electroencephalographic correlates of prenatal exposure to alcohol in infants and children: A review of findings and implications for neurocognitive development. Alcohol 2006, 40, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Hemington, K.S.; Reynolds, J.N. Electroencephalographic correlates of working memory deficits in children with Fetal Alcohol Spectrum Disorder using a single-electrode pair recording device. Clin. Neurophysiol. 2014, 125, 2364–2371. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, S.L.; Lanza, I.R.; Nair, K.S. Mitochondrial DNA alterations and reduced mitochondrial function in aging. Mech. Ageing Dev. 2010, 131, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbari, M.; Kirkwood, T.B.L.; Bohr, V.A. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 2019, 54, 100940. [Google Scholar] [CrossRef]
- Chu, J.; Tong, M.; de la Monte, S.M. Chronic ethanol exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta Neuropathol. 2007, 113, 659–673. [Google Scholar] [CrossRef]
- Ramachandran, V.; Perez, A.; Chen, J.; Senthil, D.; Schenker, S.; Henderson, G.I. In Utero Ethanol Exposure Causes Mitochondrial Dysfunction, Which Can Result in Apoptotic Cell Death in Fetal Brain: A Potential Role for 4-Hydroxynonenal. Alcohol. Clin. Exp. Res. 2001, 25, 862–871. [Google Scholar] [CrossRef]
- Da Lee, R.; Mi An, S.; Sun Kim, S.; Seek Rhee, G.; Jun Kwack, S.; Hyun Seok, J.; Yeong Chae, S.; Hoon Park, C.; Woo Choi, Y.; Sik Kim, H.; et al. Neurotoxic Effects of Alcohol and Acetaldehyde During Embryonic Development. J. Toxicol. Environ. Health Part A 2005, 68, 2147–2162. [Google Scholar] [CrossRef]
- Tong, M.; Longato, L.; Nguyen, Q.-G.; Chen, W.C.; Spaisman, A.; de la Monte, S.M. Acetaldehyde-Mediated Neurotoxicity: Relevance to Fetal Alcohol Spectrum Disorders. Oxidative Med. Cell. Longev. 2011, 2011, 213286. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, P.; Li, Y. Impaired development of mitochondria plays a role in the central nervous system defects of fetal alcohol syndrome. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Bukiya, A.N. Fetal Cerebral Artery Mitochondrion as Target of Prenatal Alcohol Exposure. Int. J. Environ. Res. Public Health 2019, 16, 1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, D.; Fujihashi, A.; Majrashi, M.; Bloemer, J.; Bhattacharya, S.; Buabeid, M.; Escobar, M.; Moore, T.; Suppiramaniam, V.; Dhanasekaran, M. Concurrent nicotine exposure to prenatal alcohol consumption alters the hippocampal and cortical neurotoxicity. Heliyon 2020, 6, e03045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behuet, S.; Cremer, J.N.; Cremer, M.; Palomero-Gallagher, N.; Zilles, K.; Amunts, K. Developmental Changes of Glutamate and GABA Receptor Densities in Wistar Rats. Front. Neuroanat. 2019, 13, 100. [Google Scholar] [CrossRef]
- Brickley, S.G.; Mody, I. Extrasynaptic GABAA Receptors: Their Function in the CNS and Implications for Disease. Neuron 2012, 73, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Stephens, D.N.; King, S.L.; Lambert, J.J.; Belelli, D.; Duka, T. GABAAreceptor subtype involvement in addictive behaviour. Genes Brain Behav. 2017, 16, 149–184. [Google Scholar] [CrossRef] [Green Version]
- Sallard, E.; Letourneur, D.; Legendre, P. Electrophysiology of ionotropic GABA receptors. Cell. Mol. Life Sci. 2021, 78, 5341–5370. [Google Scholar] [CrossRef]
- Terunuma, M. Diversity of structure and function of GABAB receptors: A complexity of GABAB-mediated signaling. Proc. Jpn. Acad. Ser. B 2018, 94, 390–411. [Google Scholar] [CrossRef]
- Sigel, E.; Steinmann, M.E. Structure, Function, and Modulation of GABAA Receptors. J. Biol. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef] [Green Version]
- Sakimoto, Y.; Oo, P.M.-T.; Goshima, M.; Kanehisa, I.; Tsukada, Y.; Mitsushima, D. Significance of GABAA Receptor for Cognitive Function and Hippocampal Pathology. Int. J. Mol. Sci. 2021, 22, 12456. [Google Scholar] [CrossRef] [PubMed]
- Mousavi Majd, A.; Ebrahim Tabar, F.; Afghani, A.; Ashrafpour, S.; Dehghan, S.; Gol, M.; Ashrafpour, M.; Pourabdolhossein, F. Inhibition of GABA A receptor improved special memory impairment in the local model of demyelination in rat hippocampus. Behav. Brain Res. 2018, 336, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.J.; Thomas, K.G.F.; Molteno, C.D.; Jacobson, J.L.; Jacobson, S.W.; Meintjes, E.M. Prenatal alcohol exposure affects brain function during place learning in a virtual environment differently in boys and girls. Brain Behav. 2018, 8, e01103. [Google Scholar] [CrossRef] [Green Version]
- Marguet, F.; Friocourt, G.; Brosolo, M.; Sauvestre, F.; Marcorelles, P.; Lesueur, C.; Marret, S.; Gonzalez, B.J.; Laquerrière, A. Prenatal alcohol exposure is a leading cause of interneuronopathy in humans. Acta Neuropathol. Commun. 2020, 8, 208. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, T.; Gal-Ben-Ari, S.; Dieterich, D.C.; Kreutz, M.R.; Ziv, N.E.; Gundelfinger, E.D.; Rosenblum, K. The roles of protein expression in synaptic plasticity and memory consolidation. Front. Mol. Neurosci. 2014, 7, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhowail, A.H.; Bloemer, J.; Majrashi, M.; Pinky, P.D.; Bhattacharya, S.; Yongli, Z.; Bhattacharya, D.; Eggert, M.; Woodie, L.; Buabeid, M.A.; et al. Doxorubicin-induced neurotoxicity is associated with acute alterations in synaptic plasticity, apoptosis, and lipid peroxidation. Toxicol. Mech. Methods 2019, 29, 457–466. [Google Scholar] [CrossRef]
- Giese, K.P.; Mizuno, K. The roles of protein kinases in learning and memory. Learn. Mem. 2013, 20, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Abel, T.; Nguyen, P.V. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog. Brain Res. 2008, 169, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Gerber, K.J.; Squires, K.E.; Hepler, J.R. Roles for Regulator of G Protein Signaling Proteins in Synaptic Signaling and Plasticity. Mol. Pharmacol. 2016, 89, 273–286. [Google Scholar] [CrossRef]
- Voglis, G.; Tavernarakis, N. The role of synaptic ion channels in synaptic plasticity. EMBO Rep. 2006, 7, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Elgharabawy, R.M.; Alhowail, A.H.; Emara, A.M.; Aldubayan, M.A.; Ahmed, A.S. The impact of chicory (Cichoriumintybus L.) on hemodynamic functions and oxidative stress in cardiac toxicity induced by lead oxide nanoparticles in male rats. Biomed. Pharm. 2021, 137, 111324. [Google Scholar] [CrossRef] [PubMed]
- Alhowail, A.H.; Pinky, P.D.; Eggert, M.; Bloemer, J.; Woodie, L.N.; Buabeid, M.A.; Bhattacharya, S.; Jasper, S.L.; Bhattacharya, D.; Dhanasekaran, M.; et al. Doxorubicin induces dysregulation of AMPA receptor and impairs hippocampal synaptic plasticity leading to learning and memory deficits. Heliyon 2021, 7, e07456. [Google Scholar] [CrossRef]
- Sakata, K.; Martinowich, K.; Woo, N.H.; Schloesser, R.J.; Jimenez, D.V.; Ji, Y.; Shen, L.; Lu, B. Role of activity-dependent BDNF expression in hippocampal–prefrontal cortical regulation of behavioral perseverance. Proc. Natl. Acad. Sci. USA 2013, 110, 15103–15108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bye, C.M.; McDonald, R.J. A Specific Role of Hippocampal NMDA Receptors and Arc Protein in Rapid Encoding of Novel Environmental Representations and a More General Long-Term Consolidation Function. Front. Behav. Neurosci. 2019, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsushima, D.; Ishihara, K.; Sano, A.; Kessels, H.W.; Takahashi, T. Contextual learning requires synaptic AMPA receptor delivery in the hippocampus. Proc. Natl. Acad. Sci. USA 2011, 108, 12503–12508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavis, P.; Dash, P.K.; Johnson, D.; Clark, J.; Orsi, S.A.; Zhang, M.; Zhao, J.; Grill, R.J.; Moore, A.N.; Pati, S. Involvement of the Glycogen Synthase Kinase-3 Signaling Pathway in TBI Pathology and Neurocognitive Outcome. PLoS ONE 2011, 6, e24648. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, S.D.; Hu, Y.-S.; Long, N.; Pigino, G.; Brady, S.T.; Lazarov, O. Molecular Mechanisms of Environmental Enrichment: Impairments in Akt/GSK3β, Neurotrophin-3 and CREB Signaling. PLoS ONE 2013, 8, e64460. [Google Scholar] [CrossRef] [Green Version]
- Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 11088. [Google Scholar] [CrossRef]
- Asselin, E.; Hossini, A.M.; Quast, A.S.; Plötz, M.; Grauel, K.; Exner, T.; Küchler, J.; Stachelscheid, H.; Eberle, J.; Rabien, A.; et al. PI3K/AKT Signaling Pathway Is Essential for Survival of Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0154770. [Google Scholar] [CrossRef]
- Furukawa, H. Structure and function of glutamate receptor amino terminal domains. J. Physiol. 2012, 590, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R.; et al. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Purkey, A.M.; Dell’Acqua, M.L. Phosphorylation-Dependent Regulation of Ca2+-Permeable AMPA Receptors During Hippocampal Synaptic Plasticity. Front. Synaptic Neurosci. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ma, Y.-Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.M.; Takamiya, K.; Xia, J.; Suh, J.-G.; Johnson, R.; Yu, S.; Huganir, R.L. Calcium-Permeable AMPA Receptor Plasticity Is Mediated by Subunit-Specific Interactions with PICK1 and NSF. Neuron 2005, 45, 903–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.; Vissel, B. The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front. Mol. Neurosci. 2012, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmeyer, D.; Kask, K.; Brusa, R.; Kornau, H.C.; Kolhekar, R.; Rozov, A.; Burnashev, N.; Jensen, V.; Hvalby, O.; Sprengel, R.; et al. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat. Neurosci. 1999, 2, 57–64. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Heldt, S.A.; Stanek, L.; Chhatwal, J.P.; Ressler, K.J. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol. Psychiatry 2007, 12, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Arime, Y.; Hall, F.S.; Uhl, G.R.; Sora, I. Impaired spatial working memory and decreased frontal cortex BDNF protein level in dopamine transporter knockout mice. Eur. J. Pharmacol. 2010, 628, 104–107. [Google Scholar] [CrossRef]
- Gaiarsa, J.-L.; Zhang, L.; Fang, Y.; Lian, Y.; Chen, Y.; Wu, T.; Zheng, Y.; Zong, H.; Sun, L.; Zhang, R.; et al. Brain-Derived Neurotrophic Factor Ameliorates Learning Deficits in a Rat Model of Alzheimer’s Disease Induced by Aβ1-42. PLoS ONE 2015, 10, e0122415. [Google Scholar] [CrossRef] [Green Version]
- Cirulli, F.; Berry, A.; Chiarotti, F.; Alleva, E. Intrahippocampal administration of BDNF in adult rats affects short-term behavioral plasticity in the Morris water maze and performance in the elevated plus-maze. Hippocampus 2004, 14, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Nie, Z.; Shu, H.; Kuang, Y.; Chen, X.; Cheng, J.; Yu, S.; Liu, H. The Role of BDNF on Neural Plasticity in Depression. Front. Cell. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, I.; Takarada, S.; Tatsumi, S.; Azegami, A.; Yasuda, M.; Fukuchi, M.; Tabuchi, A.; Kondo, T.; Tabuchi, Y.; Tsuda, M. Extracellular adenosine 5′-triphosphate elicits the expression of brain-derived neurotrophic factor exon IV mRNA in rat astrocytes. Glia 2008, 56, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Tecuatl, C.; Herrrera-López, G.; Martín-Ávila, A.; Yin, B.; Weber, S.; Barrionuevo, G.; Galván, E.J. TrkB-mediated activation of the phosphatidylinositol-3-kinase/Akt cascade reduces the damage inflicted by oxygen-glucose deprivation in area CA3 of the rat hippocampus. Eur. J. Neurosci. 2018, 47, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Alhowai, A.H. Doxorubicin Attenuates BDNF mRNA Expression in Hippocampal Neuronal Cells. Int. J. Pharmacol. 2021, 17, 414–419. [Google Scholar] [CrossRef]
- Swulius, M.T.; Waxham, M.N. Ca2+/Calmodulin-dependent Protein Kinases. Cell. Mol. Life Sci. 2008, 65, 2637–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalcman, G.; Federman, N.; Romano, A. CaMKII Isoforms in Learning and Memory: Localization and Function. Front. Mol. Neurosci. 2018, 11, 445. [Google Scholar] [CrossRef] [Green Version]
- Rossetti, T.; Banerjee, S.; Kim, C.; Leubner, M.; Lamar, C.; Gupta, P.; Lee, B.; Neve, R.; Lisman, J. Memory Erasure Experiments Indicate a Critical Role of CaMKII in Memory Storage. Neuron 2017, 96, 207–216.e202. [Google Scholar] [CrossRef] [Green Version]
- Giese, K.P. The role of CaMKII autophosphorylation for NMDA receptor-dependent synaptic potentiation. Neuropharmacology 2021, 193, 108616. [Google Scholar] [CrossRef]
- Yabuki, Y.; Nakagawasai, O.; Moriguchi, S.; Shioda, N.; Onogi, H.; Tan-No, K.; Tadano, T.; Fukunaga, K. Decreased CaMKII and PKC activities in specific brain regions are associated with cognitive impairment in neonatal ventral hippocampus-lesioned rats. Neuroscience 2013, 234, 103–115. [Google Scholar] [CrossRef]
- Moriguchi, S.; Tagashira, H.; Sasaki, Y.; Yeh, J.Z.; Sakagami, H.; Narahashi, T.; Fukunaga, K. CaMKII activity is essential for improvement of memory-related behaviors by chronic rivastigmine treatment. J. Neurochem. 2014, 128, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Giese, K.P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 2015, 8, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakaria, M.; Park, S.-Y.; Haque, M.E.; Karthivashan, G.; Kim, I.-S.; Ganesan, P.; Choi, D.-K. Neurotoxic Agent-Induced Injury in Neurodegenerative Disease Model: Focus on Involvement of Glutamate Receptors. Front. Mol. Neurosci. 2018, 11, 307. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Garic, A.; Flentke, G.R.; Amberger, E.; Hernandez, M.; Smith, S.M. CaMKII activation is a novel effector of alcohol’s neurotoxicity in neural crest stem/progenitor cells. J. Neurochem. 2011, 118, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Kong, T.; Liu, M.; Ji, B.; Bai, B.; Cheng, B.; Wang, C. Role of the Extracellular Signal-Regulated Kinase 1/2 Signaling Pathway in Ischemia-Reperfusion Injury. Front. Physiol. 2019, 10, 1038. [Google Scholar] [CrossRef] [Green Version]
- El Gaamouch, F.; Buisson, A.; Moustie, O.; Lemieux, M.; Labrecque, S.; Bontempi, B.; De Koninck, P.; Nicole, O. Interaction between CaMKII and GluN2B Controls ERK-Dependent Plasticity. J. Neurosci. 2012, 32, 10767–10779. [Google Scholar] [CrossRef]
- Mohmmad Abdul, H.; Butterfield, D.A. Involvement of PI3K/PKG/ERK1/2 signaling pathways in cortical neurons to trigger protection by cotreatment of acetyl-L-carnitine and α-lipoic acid against HNE-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Free Radic. Biol. Med. 2007, 42, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Schafe, G.E.; Swank, M.W.; Rodrigues, S.M.; Dȩbiec, J.; Doyère, V. Phosphorylation of ERK/MAP kinase is required for long-term potentiation in anatomically restricted regions of the lateral amygdala in vivo. Learn. Mem. 2008, 15, 55–62. [Google Scholar] [CrossRef]
- Trifilieff, P.; Calandreau, L.; Herry, C.; Mons, N.; Micheau, J. Biphasic ERK1/2 activation in both the hippocampus and amygdala may reveal a system consolidation of contextual fear memory. Neurobiol. Learn. Mem. 2007, 88, 424–434. [Google Scholar] [CrossRef]
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 2018, 11, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgognon, J.-M.; Cavanagh, J. The role of cytokines in modulating learning and memory and brain plasticity. Brain Neurosci. Adv. 2020, 4, 2398212820979802. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.H.; Viola, H. ERK1/2: A Key Cellular Component for the Formation, Retrieval, Reconsolidation and Persistence of Memory. Front. Mol. Neurosci. 2018, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Samudio-Ruiz, S.L.; Allan, A.M.; Valenzuela, C.F.; Perrone-Bizzozero, N.I.; Caldwell, K.K. Prenatal ethanol exposure persistently impairs NMDA receptor-dependent activation of extracellular signal-regulated kinase in the mouse dentate gyrus. J. Neurochem. 2009, 109, 1311–1323. [Google Scholar] [CrossRef]
- Swart, P.C.; Russell, V.A.; Dimatelis, J.J. Maternal separation stress reduced prenatal-ethanol-induced increase in exploratory behaviour and extracellular signal-regulated kinase activity. Behav. Brain Res. 2019, 356, 470–482. [Google Scholar] [CrossRef]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The Role of the Transcription Factor CREB in Immune Function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, P.; Filiz, G.; Brown, D.V.; Hollande, F.; Gonzales, M.; D’Abaco, G.; Papalexis, N.; Phillips, W.A.; Malaterre, J.; Ramsay, R.G.; et al. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis 2014, 3, e108. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.-H.; Quirion, R. Insulin-like growth factor-1 (IGF-1) induces the activation/phosphorylation of Akt kinase and cAMP response element-binding protein (CREB) by activating different signaling pathways in PC12 cells. BMC Neurosci. 2006, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, S.; Martin, K.J.; Arthur, J.S.C. CREB phosphorylation at Ser133 regulates transcription via distinct mechanisms downstream of cAMP and MAPK signalling. Biochem. J. 2014, 458, 469–479. [Google Scholar] [CrossRef]
- Steven, A.; Friedrich, M.; Jank, P.; Heimer, N.; Budczies, J.; Denkert, C.; Seliger, B. What turns CREB on? And off? And why does it matter? Cell. Mol. Life Sci. 2020, 77, 4049–4067. [Google Scholar] [CrossRef]
- Hardt, O.; Nader, K.; Wang, Y.-T. GluA2-dependent AMPA receptor endocytosis and the decay of early and late long-term potentiation: Possible mechanisms for forgetting of short- and long-term memories. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henley, J.M.; Wilkinson, K.A. AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin. Neurosci. 2022, 15, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Middei, S.; Houeland, G.; Cavallucci, V.; Ammassari-Teule, M.; D’Amelio, M.; Marie, H. CREB is necessary for synaptic maintenance and learning-induced changes of the ampa receptor GluA1 subunit. Hippocampus 2013, 23, 488–499. [Google Scholar] [CrossRef]
- Cai, H.; Guo, W.; Crossey, E.L.; Zhang, L.; Zucca, S.; George, O.L.; Valenzuela, C.F.; Zhao, X. Alcohol Exposure Decreases CREB Binding Protein Expression and Histone Acetylation in the Developing Cerebellum. PLoS ONE 2011, 6, e19351. [Google Scholar] [CrossRef] [Green Version]
- Montagud-Romero, S.; Cantacorps, L.; Fernández-Gómez, F.J.; Núñez, C.; Miñarro, J.; Rodríguez-Arias, M.; Milanés, M.V.; Valverde, O. Unraveling the molecular mechanisms involved in alcohol intake and withdrawal in adolescent mice exposed to alcohol during early life stages. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 104, 110025. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xu, D.; Cheng, S.; Zhang, L.; Shi, Z.; Qin, J.; Zhang, Z.; Wang, H. Prenatal ethanol exposure enhances the susceptibility to depressive behavior of adult offspring rats fed a high-fat diet by affecting BDNF-associated pathway. Int. J. Mol. Med. 2019, 45, 365–374. [Google Scholar] [CrossRef]
Figure 1.
Diagrammatic representation of the cellular mechanisms of cognitive impairment induced by prenatal alcohol exposure. AMPA = α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid, NMDA = N-methyl-d-aspartate, CaMKII = calcium calmodulin dependent kinase II, Akt = protein kinase B, GSK3β = glycogen synthase kinase 3 beta, ERK1/2 = extracellular regulated kinase ½, BDNF = brain-derived neurotrophic factor, CREB = cAMP-response element binding protein.
Figure 1.
Diagrammatic representation of the cellular mechanisms of cognitive impairment induced by prenatal alcohol exposure. AMPA = α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid, NMDA = N-methyl-d-aspartate, CaMKII = calcium calmodulin dependent kinase II, Akt = protein kinase B, GSK3β = glycogen synthase kinase 3 beta, ERK1/2 = extracellular regulated kinase ½, BDNF = brain-derived neurotrophic factor, CREB = cAMP-response element binding protein.

Figure 2.
Conceptual diagram of the onset of cognitive impairment due to embryonic alcohol exposure.
Figure 2.
Conceptual diagram of the onset of cognitive impairment due to embryonic alcohol exposure.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Alhowail, A. Mechanisms Underlying Cognitive Impairment Induced by Prenatal Alcohol Exposure. Brain Sci. 2022, 12, 1667. https://doi.org/10.3390/brainsci12121667
AMA Style
Alhowail A. Mechanisms Underlying Cognitive Impairment Induced by Prenatal Alcohol Exposure. Brain Sciences. 2022; 12(12):1667. https://doi.org/10.3390/brainsci12121667
Chicago/Turabian StyleAlhowail, Ahmad. 2022. "Mechanisms Underlying Cognitive Impairment Induced by Prenatal Alcohol Exposure" Brain Sciences 12, no. 12: 1667. https://doi.org/10.3390/brainsci12121667
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.