
HPA Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
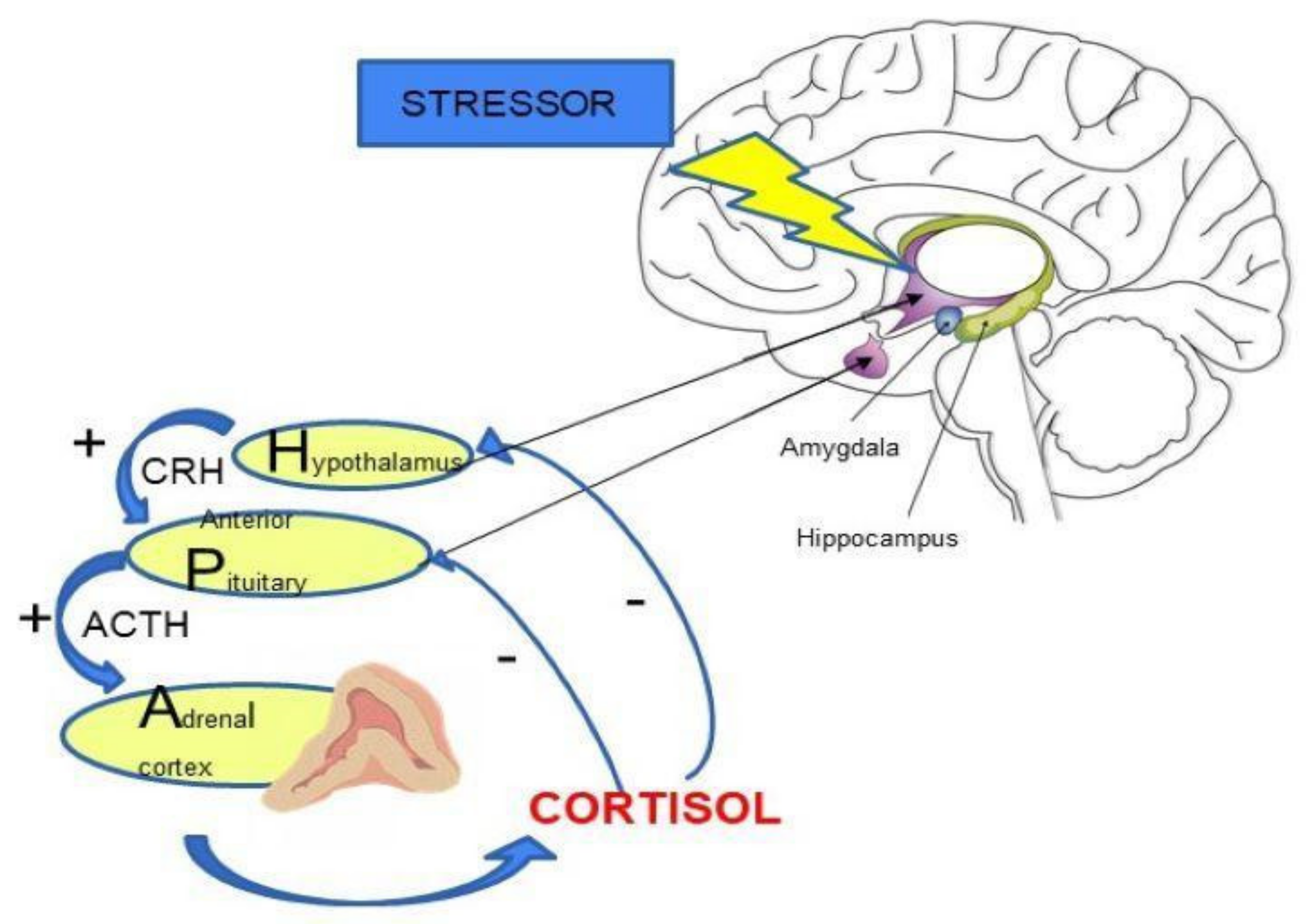
2. HPA Axis Regulation
3. The Role of Cortisol in Depression and Schizophrenia
4. HPA Axis and Gut Microbiota
- Humoral pathways (via microbiota metabolites and gut hormones);
- Activation of immune responses via the vagus nerve [72];
- Production of short-chain fatty acids (SCFAs) in the gut, which affects the balance of microglia and leads to the release of intestinal peptides, influencing the activity of the brain-gut axis [73].
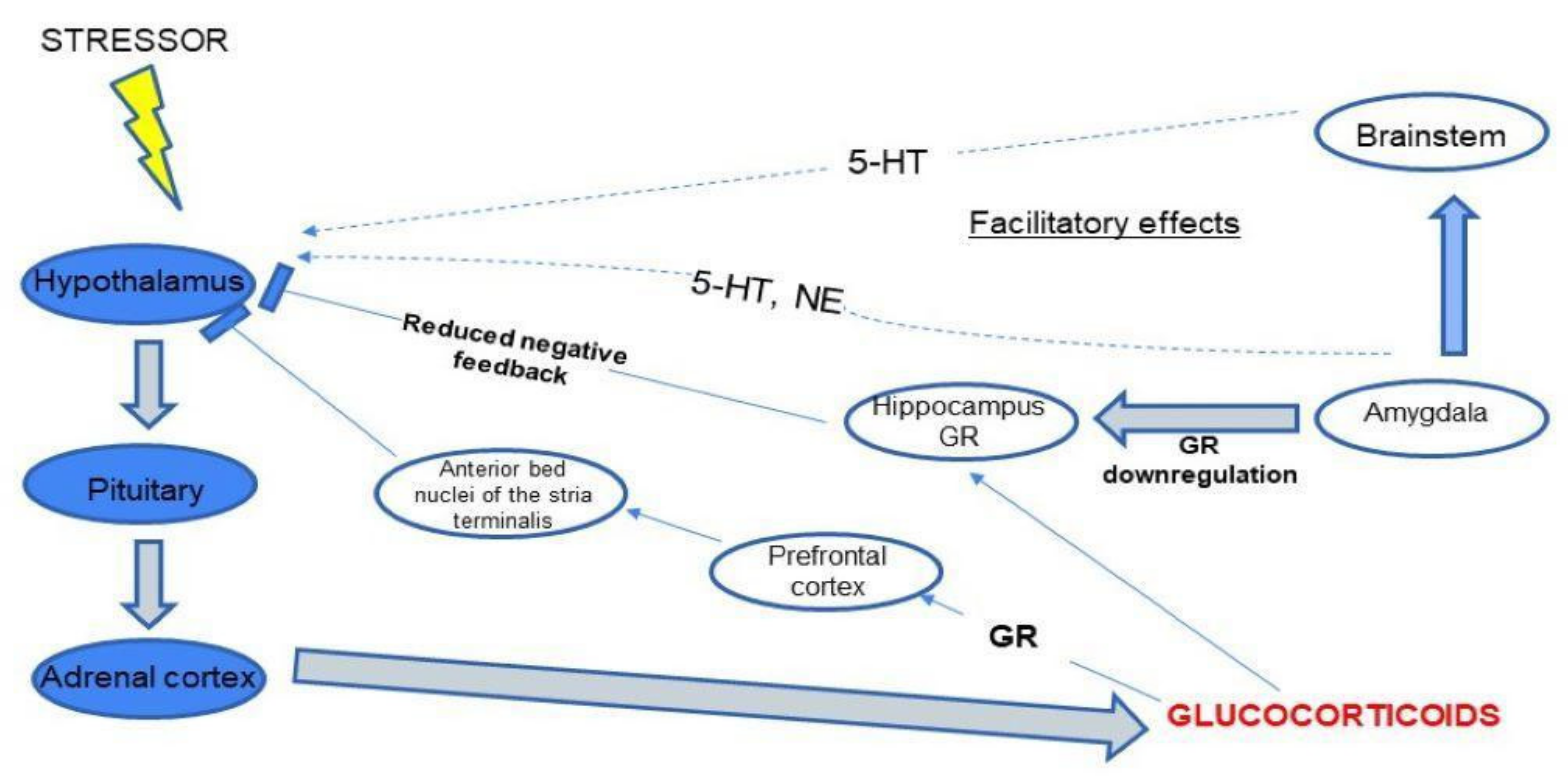
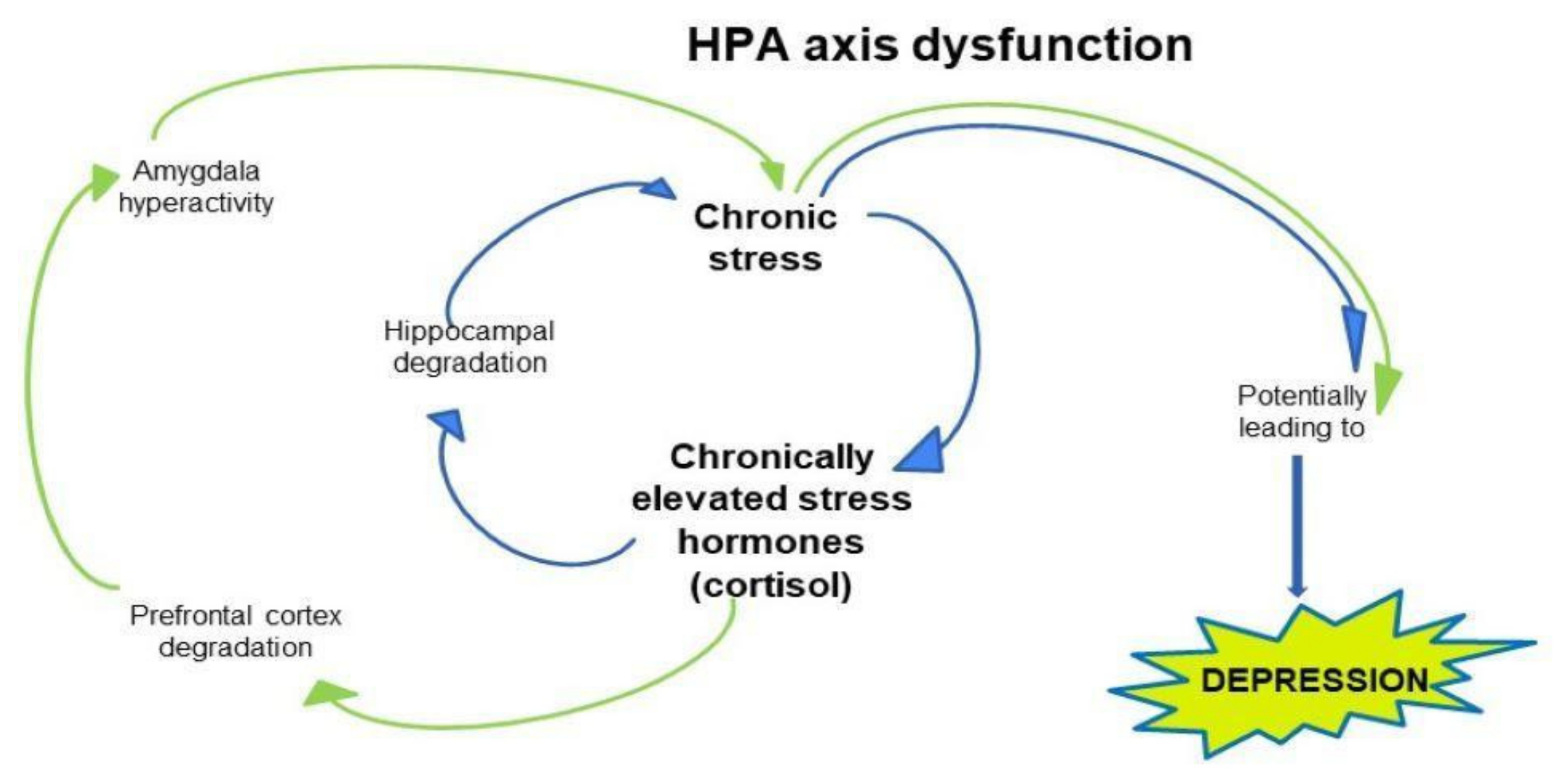
5. The Neurobiology of Depression: Possible Pathophysiological Mechanism Including HPA Axis
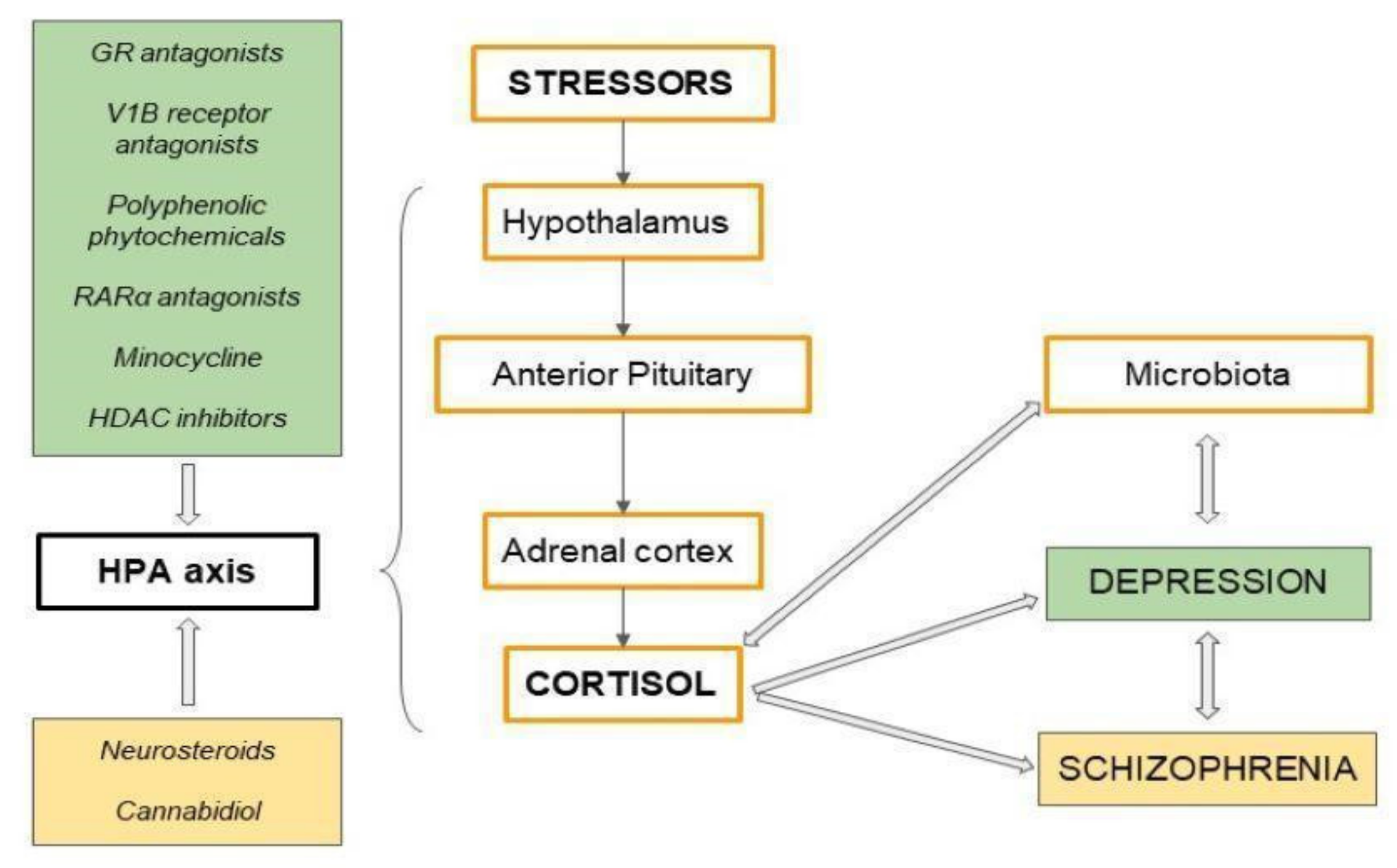
6. Potential Antidepressants—Modulating Overactivity of the HPA Axis
7. The Neurobiology of Schizophrenia: Possible Pathophysiological Mechanism Including HPA Axis
8. New Possible Drugs for Schizophrenia, Targeting the HPA Axis by Psychoactive Substances
9. Correlation—Schizophrenia and Depression
10. Activation of the HPA Axis Due to Environmental/Oxidative Stress as a Risk Factor for Depression and Schizophrenia
11. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kostowski, W. Contemporary directions of research on pathomechanism of stress and depression and their importance in shaping views on the effects of antidepressants. Novel studies on stress and depression and their influence on opinions about mechanisms of action of antidepressants. Psychiatry 2004, 1, 63–71. [Google Scholar]
- Richelson, E. Pharmacology of antidepressants. Mayo Clin. Proc. 2001, 76, 511–527. [Google Scholar] [CrossRef] [Green Version]
- Holsboer, F. The Corticosteroid Receptor Hypothesis of Depression. Neuropsychopharmacology 2000, 23, 477–501. [Google Scholar] [CrossRef] [Green Version]
- Zarkovic, M.; Ignjatovic, S.; Dajak, M.; Ciric, J.; Beleslin, B.; Savić, S.; Stojkovic, M.; Bulat, P.; Trbojevic, B. Cortisol response to ACTH stimulation correlates with blood interleukin 6 concentration in healthy humans. Eur. J. Endocrinol. 2008, 159, 649–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, J.; van Os, J.; Morrison, A.P.; Ross, C.A. Childhood trauma, psychosis and schizophrenia: A literature review with theoretical and clinical implications. Acta Psychiatr. Scand. 2005, 112, 330–350. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.-L.; Li, X.-S.; Chen, G.-Y.; Du, Y.; Wei, Z.-X.; Chen, X.; Zheng, G.-E.; Deng, W.; Cheng, Y. Serum Oxidative Stress Marker Levels in Unmedicated and Medicated Patients with Schizophrenia. J. Mol. Neurosci. 2018, 66, 428–436. [Google Scholar] [CrossRef]
- Jacobson, L. Hypothalamic–Pituitary–Adrenocortical Axis Regulation. Endocrinol. Metab. Clin. N. Am. 2005, 34, 271–292. [Google Scholar] [CrossRef]
- Dallman, M.F.; Akana, S.F.; Cascio, C.S.; Darlington, D.N.; Jacobson, L.; Levin, N. Regulation of ACTH Secretion: Variations on a Theme of B. Rec. Prog. Horm. Res. 1987, 43, 113–173. [Google Scholar] [CrossRef]
- Joseph, D.N.; Whirledge, S. Stress and the HPA Axis: Balancing Homeostasis and Fertility. Int. J. Mol. Sci. 2017, 18, 2224. [Google Scholar] [CrossRef]
- Heaney, J. Hypothalamic-Pituitary-Adrenal Axis; Springer: Berlin/Heidelberg, Germany, 2013; ISBN 978-1-4419-1004-2. [Google Scholar] [CrossRef]
- Engeland, W.C.; Arnhold, M.M. Neural Circuitry in the Regulation of Adrenal Corticosterone Rhythmicity. Endocrine 2005, 28, 325–332. [Google Scholar] [CrossRef]
- Balbo, M.; Leproult, R.; Van Cauter, E. Impact of Sleep and Its Disturbances on Hypothalamo-Pituitary-Adrenal Axis Activity. Int. J. Endocrinol. 2010, 2010, 759234. [Google Scholar] [CrossRef] [Green Version]
- Hansson, P.B.; Murison, R.; Lund, A.; Hammar, Å. Cognitive functioning and cortisol profiles in first episode major depression. Scand. J. Psychol. 2015, 56, 379–383. [Google Scholar] [CrossRef]
- Charles, S.T.; Mogle, J.; Piazza, J.R.; Karlamangla, A.; Almeida, D.M. Going the distance: The diurnal range of cortisol and its association with cognitive and physiological functioning. Psychoneuroendocrinology 2019, 112, 104516. [Google Scholar] [CrossRef]
- Stawski, R.S.; Almeida, D.M.; Lachman, M.E.; Tun, P.A.; Rosnick, C.B.; Seeman, T. Associations Between Cognitive Function and Naturally Occurring Daily Cortisol During Middle Adulthood: Timing Is Everything. J. Gerontol. Ser. B 2011, 66B, i71–i81. [Google Scholar] [CrossRef] [Green Version]
- Marin, M.-F.; Lord, C.; Andrews, J.; Juster, R.-P.; Sindi, S.; Arsenault-Lapierre, G.; Fiocco, A.J.; Lupien, S.J. Chronic stress, cognitive functioning and mental health. Neurobiol. Learn. Mem. 2011, 96, 583–595. [Google Scholar] [CrossRef]
- Wyrwoll, C.S.; Holmes, M.C.; Seckl, J.R. 11β-Hydroxysteroid dehydrogenases and the brain: From zero to hero, a decade of progress. Front. Neuroendocr. 2011, 32, 265–286. [Google Scholar] [CrossRef] [Green Version]
- Evanson, N.K.; Herman, J.P.; Sakai, R.R.; Krause, E.G. Nongenomic Actions of Adrenal Steroids in the Central Nervous System. J. Neuroendocr. 2010, 22, 846–861. [Google Scholar] [CrossRef]
- Hill, M.; Tasker, J. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience 2012, 204, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.N.; McLaughlin, R.; Morrish, A.C.; Viau, V.; Floresco, S.; Hillard, C.J.; Gorzalka, B.B. Suppression of Amygdalar Endocannabinoid Signaling by Stress Contributes to Activation of the Hypothalamic–Pituitary–Adrenal Axis. Neuropsychopharmacology 2009, 34, 2733–2745. [Google Scholar] [CrossRef] [PubMed]
- Pace, T.W.W.; Spencer, R.L. Disruption of mineralocorticoid receptor function increases corticosterone responding to a mild, but not moderate, psychological stressor. Am. J. Physiol. Metab. 2005, 288, E1082–E1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisboa, S.; Gomes, F.; Terzian, A.; Aguiar, D.; Moreira, F.; Resstel, L.; Guimarães, F. The Endocannabinoid System and Anxiety. Vitam. Horm. 2017, 103, 193–279. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.; Cabañero, D.; Martín-García, E. The endocannabinoid system in modulating fear, anxiety, and stress. Dialog-Clin. Neurosci. 2020, 22, 229–239. [Google Scholar] [CrossRef]
- Du, X.; Pang, T.Y. Is Dysregulation of the HPA-Axis a Core Pathophysiology Mediating Co-Morbid Depression in Neurodegenerative Diseases? Front. Psychiatry 2015, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.; Gomez, R.; Williams, G.; Lembke, A.; Lazzeroni, L.; Murphy, G.M.; Schatzberg, A.F. HPA axis in major depression: Cortisol, clinical symptomatology and genetic variation predict cognition. Mol. Psychiatry 2016, 22, 527–536. [Google Scholar] [CrossRef]
- Nandam, L.S.; Brazel, M.; Zhou, M.; Jhaveri, D.J. Cortisol and Major Depressive Disorder—Translating Findings from Humans to Animal Models and Back. Front. Psychiatry 2020, 10. [Google Scholar] [CrossRef]
- Carroll, B.J.; Curtis, G.C.; Mendels, J. Neuroendocrine Regulation in depression. II. Discrimination of depressed from nondepressed patients. Arch. Gen. Psychiatry 1976, 33, 1051–1058. [Google Scholar] [CrossRef]
- Ising, M.; Künzel, H.E.; Binder, E.B.; Nickel, T.; Modell, S.; Holsboer, F. The combined dexamethasone/CRH test as a potential surrogate marker in depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 1085–1093. [Google Scholar] [CrossRef]
- Dam, H.; Mellerup, E.T.; Rafaelsen, O.J. The dexamethasone suppression test in depression. J. Affect. Disord. 1985, 8, 95–103. [Google Scholar] [CrossRef]
- Stetler, C.; Miller, G.E. Depression and Hypothalamic-Pituitary-Adrenal Activation: A Quantitative Summary of Four Decades of Research. Psychosom. Med. 2011, 73, 114–126. [Google Scholar] [CrossRef]
- Bielajew, C.; Konkle, A.; Merali, Z. The effects of chronic mild stress on male Sprague–Dawley and Long Evans rats: I. Biochemical and physiological analyses. Behav. Brain Res. 2002, 136, 583–592. [Google Scholar] [CrossRef]
- De Andrade, J.; Céspedes, I.; Abrão, R.; dos Santos, T.; Diniz, L.; Britto, L.; Spadari-Bratfisch, R.; Ortolani, D.; Melo-Thomas, L.; Silva, R.C.; et al. Chronic unpredictable mild stress alters an anxiety-related defensive response, Fos immunoreactivity and hippocampal adult neurogenesis. Behav. Brain Res. 2013, 250, 81–90. [Google Scholar] [CrossRef]
- Schatzberg, A.F.; Rothschild, A.J.; Langlais, P.J.; Bird, E.D.; Cole, J.O. A corticosteroid/dopamine hypothesis for psychotic depression and related states. J. Psychiatr. Res. 1985, 19, 57–64. [Google Scholar] [CrossRef]
- Qin, D.; Rizak, J.; Chu, X.; Li, Z.; Yang, S.; Lü, L.; Yang, L.; Yang, Q.; Yang, B.; Pan, L.; et al. A spontaneous depressive pattern in adult female rhesus macaques. Sci. Rep. 2015, 5, 11267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, M.D.; Tiefenbacher, S.; Lutz, C.K.; Novak, M.; Meyer, J.S. Analysis of endogenous cortisol concentrations in the hair of rhesus macaques. Gen. Comp. Endocrinol. 2006, 147, 255–261. [Google Scholar] [CrossRef]
- Feng, X.; Wang, L.; Yang, S.; Qin, D.; Wang, J.; Li, C.; Lv, L.; Ma, Y.; Hu, X. Maternal separation produces lasting changes in cortisol and behavior in rhesus monkeys. Proc. Natl. Acad. Sci. USA 2011, 108, 14312–14317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertollo, A.G.; Grolli, E.R.; Plissari, E.M.; Gasparin, A.V.; Quevedo, J.; Réus, G.Z.; Bagatini, M.D.; Ignácio, Z.M. Stress and serum cortisol levels in major depressive disorder: A cross-sectional study. AIMS Neurosci. 2020, 7, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Hodes, G.; Kana, V.; Menard, C.; Merad, M.; Russo, S.J. Neuroimmune mechanisms of depression. Nat. Neurosci. 2015, 18, 1386–1393. [Google Scholar] [CrossRef]
- Ignácio, Z.M.; Da Silva, R.S.; Plissari, M.E.; Quevedo, J.; Réus, G.Z. Physical Exercise and Neuroinflammation in Major Depressive Disorder. Mol. Neurobiol. 2019, 56, 8323–8335. [Google Scholar] [CrossRef]
- Slavich, G.M.; Irwin, M.R. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychol. Bull. 2014, 140, 774–815. [Google Scholar] [CrossRef]
- Booij, S.H.; Wigman, J.T.; Jacobs, N.; Thiery, E.; Derom, C.; Wichers, M.; Oravecz, Z. Cortisol dynamics in depression: Application of a continuous-time process model. Psychoneuroendocrinology 2020, 115, 104598. [Google Scholar] [CrossRef]
- Sterner, E.Y.; Kalynchuk, L.E. Behavioral and neurobiological consequences of prolonged glucocorticoid exposure in rats: Relevance to depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Fournier, N.M.; Kalynchuk, L.E. Effect of different doses of corticosterone on depression-like behavior and HPA axis responses to a novel stressor. Behav. Brain Res. 2006, 168, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Magariños, A.M.; Orchinik, M.; McEwen, B.S. Morphological changes in the hippocampal CA3 region induced by non-invasive glucocorticoid administration: A paradox. Brain Res. 1998, 809, 314–318. [Google Scholar] [CrossRef]
- Wellman, C.L. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J. Neurobiol. 2001, 49, 245–253. [Google Scholar] [CrossRef]
- Mitra, R.; Sapolsky, R.M. Acute corticosterone treatment is sufficient to induce anxiety and amygdaloid dendritic hypertrophy. Proc. Natl. Acad. Sci. USA 2008, 105, 5573–5578. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Nakagawa, S.; An, Y.; Ito, K.; Kitaichi, Y.; Kusumi, I. The exercise-glucocorticoid paradox: How exercise is beneficial to cognition, mood, and the brain while increasing glucocorticoid levels. Front. Neuroendocr. 2017, 44, 83–102. [Google Scholar] [CrossRef]
- Brown, H.E.; Pearson, N.; Braithwaite, R.E.; Brown, W.J.; Biddle, S. Physical Activity Interventions and Depression in Children and Adolescents. Sports Med. 2013, 43, 195–206. [Google Scholar] [CrossRef]
- Bs, S.B.H.; Zelinski, E.M. Extended Practice and Aerobic Exercise Interventions Benefit Untrained Cognitive Outcomes in Older Adults: A Meta-Analysis. J. Am. Geriatr. Soc. 2011, 60, 136–141. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Takahashi, T.; Nakagawa, S.; Inoue, T.; Kusumi, I. Reinforcement learning in depression: A review of computational research. Neurosci. Biobehav. Rev. 2015, 55, 247–267. [Google Scholar] [CrossRef]
- Pearson-Fuhrhop, K.M.; Dunn, E.; Mortero, S.; Devan, W.J.; Falcone, G.J.; Lee, P.; Holmes, A.; Hollinshead, M.O.; Roffman, J.; Smoller, J.W.; et al. Dopamine Genetic Risk Score Predicts Depressive Symptoms in Healthy Adults and Adults with Depression. PLoS ONE 2014, 9, e93772. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhao, S.; Han, X.; Cui, M.; Fan, S. Optimization of a reshaping rivet to reduce the protrusion height and increase the strength of clinched joints. J. Mater. Process. Technol. 2016, 234, 1–9. [Google Scholar] [CrossRef]
- Yang, F.; Cao, X.; Sun, X.; Wen, H.; Qiu, J.; Xiao, H. Hair Cortisol Is Associated with Social Support and Symptoms in Schizophrenia. Front. Psychiatry 2020, 11, 572656. [Google Scholar] [CrossRef]
- Schifani, C.; Pruessner, J.; Tseng, H.; Rao, N.; Tagore, A.; Wilson, A.A.; Houle, S.; Rusjan, P.; Mizrahi, R. Stress-induced cortical dopamine response is altered in subjects at clinical high risk for psychosis using cannabis. Addict. Biol. 2019, 25, e12812. [Google Scholar] [CrossRef] [PubMed]
- Boiko, A.S.; Mednova, A.I.; Kornetova, E.G.; A Bokhan, N.; Semke, A.V.; Loonen, A.J.; Ivanova, A.S. Cortisol and DHEAS Related to Metabolic Syndrome in Patients with Schizophrenia. Neuropsychiatr. Dis. Treat. 2020, 16, 1051–1058. [Google Scholar] [CrossRef] [Green Version]
- Glassman, M.; Wehring, H.J.; Pocivavsek, A.; Sullivan, K.M.; Rowland, L.M.; McMahon, R.P.; Chiappelli, J.; Liu, F.; Kelly, D.L. Peripheral Cortisol and Inflammatory Response to a Psychosocial Stressor in People with Schizophrenia. J. Neuropsychiatry 2018, 2. [Google Scholar] [CrossRef] [PubMed]
- Ciufolini, S.; Dazzan, P.; Kempton, M.; Pariante, C.M.; Mondelli, V. HPA axis response to social stress is attenuated in schizophrenia but normal in depression: Evidence from a meta-analysis of existing studies. Neurosci. Biobehav. Rev. 2014, 47, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Pruessner, M.; Béchard-Evans, L.; Boekestyn, L.; Iyer, S.N.; Pruessner, J.C.; Malla, A.K. Attenuated cortisol response to acute psychosocial stress in individuals at ultra-high risk for psychosis. Schizophr. Res. 2013, 146, 79–86. [Google Scholar] [CrossRef]
- Ji, E.; Weickert, C.S.; Purves-Tyson, T.; White, C.; Handelsman, D.J.; Desai, R.; O’’Donnell, M.; Liu, D.; Galletly, C.; Lenroot, R.; et al. Cortisol-dehydroepiandrosterone ratios are inversely associated with hippocampal and prefrontal brain volume in schizophrenia. Psychoneuroendocrinology 2020, 123, 104916. [Google Scholar] [CrossRef]
- Jansen, L.M.C.; Wied, C.C.G.-D.; Kahn, R.S. Selective impairments in the stress response in schizophrenic patients. Psychopharmacology 2000, 149, 319–325. [Google Scholar] [CrossRef]
- Jin, R.O.; Mason, S.; Mellon, S.H.; Epel, E.S.; Reus, V.I.; Mahan, L.; Rosser, R.L.; Hough, C.M.; Burke, H.M.; Mueller, S.G.; et al. Cortisol/DHEA ratio and hippocampal volume: A pilot study in major depression and healthy controls. Psychoneuroendocrinology 2016, 72, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, D.; Kimura, I.; Wakabayashi, M.; Tsumoto, H.; Ozawa, K.; Hara, T.; Takei, Y.; Hirasawa, A.; Ishihama, Y.; Tsujimoto, G. Short-chain fatty acid receptor GPR41-mediated activation of sympathetic neurons involves synapsin 2b phosphorylation. FEBS Lett. 2012, 586, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Hong, T.; Van Pijkeren, J.P.; Hemarajata, P.; Trinh, D.V.; Hu, W.; Britton, R.A.; Kalkum, M.; Versalovic, J. Histamine Derived from Probiotic Lactobacillus reuteri Suppresses TNF via Modulation of PKA and ERK Signaling. PLoS ONE 2012, 7, e31951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzi, A.; Fröhlich, E.E.; Holzer, P. Gut Microbiota and the Neuroendocrine System. Neurotherapeutics 2018, 15, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Doolin, K.; Farrell, C.; Tozzi, L.; Harkin, A.; Frodl, T.; O’’Keane, V.; Doolin, K.; Farrell, C.; Tozzi, L.; Harkin, A.; et al. Diurnal Hypothalamic-Pituitary-Adrenal Axis Measures and Inflammatory Marker Correlates in Major Depressive Disorder. Int. J. Mol. Sci. 2017, 18, 2226. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Hart, R.A.; Charlesworth, R.; Sharpley, C. Gut microbiome and depression: What we know and what we need to know. Rev. Neurosci. 2018, 29, 629–643. [Google Scholar] [CrossRef]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain–gut–enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.A.; Sun, E.W.; Martin, A.; Keating, D.J. The ever-changing roles of serotonin. Int. J. Biochem. Cell Biol. 2020, 125, 105776. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Nardi, D.; Zhang, M.; Yang, H.; Ombrellino, M.; Tracey, K.J. Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton. Neurosci. 2000, 85, 141–147. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Major, A.; Rendon, D.; Lugo, M.; Jackson, V.; Shi, Z.; Mori-Akiyama, Y.; Versalovic, J. Histamine H2 Receptor-Mediated Suppression of Intestinal Inflammation by Probiotic Lactobacillus reuteri. mBio 2015, 6, e01358-15. [Google Scholar] [CrossRef] [Green Version]
- Huo, R.; Zeng, B.; Zeng, L.; Cheng, K.; Li, B.; Luo, Y.; Wang, H.; Zhou, C.; Fang, L.; Li, W.; et al. Microbiota Modulate Anxiety-Like Behavior and Endocrine Abnormalities in Hypothalamic-Pituitary-Adrenal Axis. Front. Cell. Infect. Microbiol. 2017, 7, 489. [Google Scholar] [CrossRef] [Green Version]
- Labad, J.; Soria, V.; Salvat-Pujol, N.; Segalàs, C.; Real, E.; Urretavizcaya, M.; de Arriba-Arnau, A.; Ferrer, A.; Crespo, J.M.; Jiménez-Murcia, S.; et al. Hypothalamic-pituitary-adrenal axis activity in the comorbidity between obsessive-compulsive disorder and major depression. Psychoneuroendocrinology 2018, 93, 20–28. [Google Scholar] [CrossRef]
- Kelly, J.; Borre, Y.; Brien, C.O.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Van Bodegom, M.; Homberg, J.R.; Henckens, M.J.A.G. Modulation of the Hypothalamic-Pituitary-Adrenal Axis by Early Life Stress Exposure. Front. Cell. Neurosci. 2017, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzi, A.; Reichmann, F.; Meinitzer, A.; Mayerhofer, R.; Jain, P.; Hassan, A.; Fröhlich, E.E.; Wagner, K.; Painsipp, E.; Rinner, B.; et al. Synergistic effects of NOD1 or NOD2 and TLR4 activation on mouse sickness behavior in relation to immune and brain activity markers. Brain Behav. Immun. 2014, 44, 106–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Asano, Y.; Hiramoto, T.; Nishino, R.; Aiba, Y.; Kimura, T.; Yoshihara, K.; Koga, Y.; Sudo, N. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am. J. Physiol. Liver Physiol. 2012, 303, G1288–G1295. [Google Scholar] [CrossRef] [Green Version]
- Moya-Pérez, A.; Perez-Villalba, A.; Benitez-Paez, A.; Campillo, I.; Sanz, Y. Bifidobacterium CECT 7765 modulates early stress-induced immune, neuroendocrine and behavioral alterations in mice. Brain Behav. Immun. 2017, 65, 43–56. [Google Scholar] [CrossRef]
- Trzeciak, P.; Herbet, M. Role of the Intestinal Microbiome, Intestinal Barrier and Psychobiotics in Depression. Nutrients 2021, 13, 927. [Google Scholar] [CrossRef] [PubMed]
- Roceri, M.; Hendriks, W.; Racagni, G.; Ellenbroek, A.B.; Riva, M.A. Early maternal deprivation reduces the expression of BDNF and NMDA receptor subunits in rat hippocampus. Mol. Psychiatry 2002, 7, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Maric, N.; Adzic, M. Pharmacological modulation of HPA axis in depression—New avenues for potential therapeutic benefits. Psychiatr. Danub. 2013, 25, 299–305. [Google Scholar] [PubMed]
- Bailey, M.T.; Dowd, S.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gareau, M.G.; Wine, E.; Rodrigues, D.M.; Cho, J.H.; Whary, M.T.; Philpott, D.J.; MacQueen, G.M.; Sherman, P.M. Bacterial infection causes stress-induced memory dysfunction in mice. Gut 2010, 60, 307–317. [Google Scholar] [CrossRef]
- Palazidou, E. The neurobiology of depression. Br. Med. Bull. 2012, 101, 127–145. [Google Scholar] [CrossRef]
- Fuster, J.M. The Prefrontal Cortex—An Update: Time Is of the Essence. Neuron 2001, 30, 319–333. [Google Scholar] [CrossRef] [Green Version]
- Koolschijn, P.C.M.; van Haren, N.E.; Lensvelt-Mulders, G.J.; Pol, H.H.; Kahn, R.S. Brain volume abnormalities in major depressive disorder: A meta-analysis of magnetic resonance imaging studies. Hum. Brain Mapp. 2009, 30, 3719–3735. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Siemer, M.; Gotlib, I.H. Amygdala volume in major depressive disorder: A meta-analysis of magnetic resonance imaging studies. Mol. Psychiatry 2008, 13, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Murialdo, G.; Barreca, A.; Nobili, F.; Rollero, A.; Timossi, G.; Gianelli, M.V.; Copello, F.; Rodriguez, G.; Polleri, A. Relationships between cortisol, dehydroepiandrosterone sulphate and insulin-like growth factor-I system in dementia. J. Endocrinol. Investig. 2001, 24, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Yuan, Y.; Hou, Z.; Jiang, W.; Bai, F.; Zhang, Z. Abnormal Functional Connectivity of Amygdala in Late-Onset Depression Was Associated with Cognitive Deficits. PLoS ONE 2013, 8, e75058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squire, L.R.; Knowlton, B.J. The New Cognitive Neurosciences; Gazzaniga, M.S., Ed.; MIT Press: Cambridge, MA, USA, 2000; pp. 79–765. [Google Scholar]
- Liu, W.; Ge, T.; Leng, Y.; Pan, Z.; Fan, J.; Yang, W.; Cui, R. The Role of Neural Plasticity in Depression: From Hippocampus to Prefrontal Cortex. Neural Plast. 2017, 2017, 6871089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, E.; Yau, S.S.Y.; Lau, W.M.; Ma, H.; Lee, T.M.C.; Chang, R.C.-C.; So, K.-F. Synaptic Plasticity, but not Hippocampal Neurogenesis, Mediated the Counteractive Effect of Wolfberry on Depression in Rats. Cell Transplant. 2012, 21, 2635–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murialdo, G.; Nobili, F.A.; Rollero, A.; Gianelli, M.V.; Copello, F.; Rodriguez, G.; Polleri, A. Hippocampal perfusion and pituitary-adrenal axis in Alzheimer’s disease. Neuropsychobiology 2000, 42, 51–57. [Google Scholar] [CrossRef]
- Ge, J.-F.; Qi, C.-C.; Zhou, J.-N. Imbalance of leptin pathway and hypothalamus synaptic plasticity markers are associated with stress-induced depression in rats. Behav. Brain Res. 2013, 249, 38–43. [Google Scholar] [CrossRef]
- Salvat-Pujol, N.; Labad, J.; Urretavizcaya, M.; de Arriba-Arnau, A.; Segalàs, C.; Real, E.; Ferrer, A.; Crespo, J.M.; Jiménez-Murcia, S.; Soriano-Mas, C.; et al. Hypothalamic-pituitary-adrenal axis activity and cognition in major depression: The role of remission status. Psychoneuroendocrinology 2016, 76, 38–48. [Google Scholar] [CrossRef]
- Roubos, E.W.; Dahmen, M.; Kozicz, T.; Xu, L. Leptin and the hypothalamo-pituitary–adrenal stress axis. Gen. Comp. Endocrinol. 2012, 177, 28–36. [Google Scholar] [CrossRef]
- Kloiber, S.; Ripke, S.; Kohli, M.A.; Reppermund, S.; Salyakina, D.; Uher, R.; McGuffin, P.; Perlis, R.H.; Hamilton, S.P.; Pütz, B.; et al. Resistance to antidepressant treatment is associated with polymorphisms in the leptin gene, decreased leptin mRNA expression, and decreased leptin serum levels. Eur. Neuropsychopharmacol. 2013, 23, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Fernstrom, J.D. Large neutral amino acids: Dietary effects on brain neurochemistry and function. Amino Acids 2012, 45, 419–430. [Google Scholar] [CrossRef]
- De Jong, R.A.; Nijman, H.W.; Boezen, H.M.; Volmer, M.; Hoor, K.A.T.; Krijnen, J.; van der Zee, A.G.; Hollema, H.; Kema, I.P. Serum Tryptophan and Kynurenine Concentrations as Parameters for Indoleamine 2,3-Dioxygenase Activity in Patients with Endometrial, Ovarian, and Vulvar Cancer. Int. J. Gynecol. Cancer 2011, 21, 1320–1327. [Google Scholar] [CrossRef]
- Sorgdrager, F.; Doornbos, B.; Penninx, B.; de Jonge, P.; Kema, I. The association between the hypothalamic pituitary adrenal axis and tryptophan metabolism in persons with recurrent major depressive disorder and healthy controls. J. Affect. Disord. 2017, 222, 32–39. [Google Scholar] [CrossRef]
- Zoubovsky, S.P.; Hoseus, S.; Tumukuntala, S.; Schulkin, J.O.; Williams, M.T.; Vorhees, C.V.; Muglia, L.J. Chronic psychosocial stress during pregnancy affects maternal behavior and neuroendocrine function and modulates hypothalamic CRH and nuclear steroid receptor expression. Transl. Psychiatry 2020, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Renthal, N.E.; Williams, K.C.; Montalbano, A.P.; Chen, C.-C.; Gao, L.; Mendelson, C.R. Molecular Regulation of Parturition: A Myometrial Perspective. Cold Spring Harb. Perspect. Med. 2015, 5, a023069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, E.P.; Korja, R.; Karlsson, L.; Glynn, L.M.; Sandman, C.A.; Vegetabile, B.; Kataja, E.-L.; Nolvi, S.; Sinervä, E.; Pelto, J.; et al. Across continents and demographics, unpredictable maternal signals are associated with children’s cognitive function. EBioMedicine 2019, 46, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Glynn, L.M.; Baram, T.Z. The influence of unpredictable, fragmented parental signals on the developing brain. Front. Neuroendocr. 2019, 53, 100736. [Google Scholar] [CrossRef]
- Baram, T.Z.; Davis, E.P.; Obenaus, A.; Sandman, C.A.; Small, S.; Solodkin, A.; Stern, H. Fragmentation and Unpredictability of Early-Life Experience in Mental Disorders. Am. J. Psychiatry 2012, 169, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pariante, C.M.; Lightman, S.L. The HPA axis in major depression: Classical theories and new developments. Trends Neurosci. 2008, 31, 464–468. [Google Scholar] [CrossRef]
- Keck, E.M.; Welt, T.; Müller, M.B.; Uhr, M.; Ohl, F.; Wigger, A.; Toschi, N.; Holsboer, F.; Landgraf, R. Reduction of Hypothalamic Vasopressinergic Hyperdrive Contributes to Clinically Relevant Behavioral and Neuroendocrine Effects of Chronic Paroxetine Treatment in a Psychopathological Rat Model. Neuropsychopharmacology 2002, 28, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Shalev, I.; Israel, S.; Uzefovsky, F.; Gritsenko, I.; Kaitz, M.; Ebstein, R. Vasopressin needs an audience: Neuropeptide elicited stress responses are contingent upon perceived social evaluative threats. Horm. Behav. 2011, 60, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Y.; Liang, J.; Xia, Q.R. Novel insights into the pharmacological effects of resveratrol on the management of depression: A short review. Die Pharm. 2017, 72, 499–502. [Google Scholar]
- Menke, A.; Klengel, T.; Binder, E.B. Epigenetics, Depression and Antidepressant Treatment. Curr. Pharm. Des. 2012, 18, 5879–5889. [Google Scholar] [CrossRef]
- Ge, J.-F.; Peng, L.; Cheng, J.-Q.; Pan, C.-X.; Tang, J.; Chen, F.-H.; Li, J. Antidepressant-like effect of resveratrol: Involvement of antioxidant effect and peripheral regulation on HPA axis. Pharmacol. Biochem. Behav. 2013, 114–115, 64–69. [Google Scholar] [CrossRef]
- Yang, X.-H.; Song, S.-Q.; Xu, Y. Resveratrol ameliorates chronic unpredictable mild stress-induced depression-like behavior: Involvement of the HPA axis, inflammatory markers, BDNF, and Wnt/β-catenin pathway in rats. Neuropsychiatr. Dis. Treat. 2017, 13, 2727–2736. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Xie, K.; Yang, X.; Gu, J.; Ge, L.; Wang, X.; Wang, Z. Resveratrol reverses the effects of chronic unpredictable mild stress on behavior, serum corticosterone levels and BDNF expression in rats. Behav. Brain Res. 2014, 264, 9–16. [Google Scholar] [CrossRef]
- Harris, A.; Seckl, J. Glucocorticoids, prenatal stress and the programming of disease. Horm. Behav. 2011, 59, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Li, R.; Cai, L.; Wu, S.-D.; Li, C.-M. Ro41-5253, a selective antagonist of retinoic acid receptor α, ameliorates chronic unpredictable mild stress-induced depressive-like behaviors in rats: Involvement of regulating HPA axis and improving hippocampal neuronal deficits. Brain Res. Bull. 2019, 146, 302–309. [Google Scholar] [CrossRef]
- Shapiro, L.P.; Parsons, R.; Koleske, A.J.; Gourley, S.L. Differential expression of cytoskeletal regulatory factors in the adolescent prefrontal cortex: Implications for cortical development. J. Neurosci. Res. 2016, 95, 1123–1143. [Google Scholar] [CrossRef] [Green Version]
- Boksa, P. Effects of prenatal infection on brain development and behavior: A review of findings from animal models. Brain Behav. Immun. 2010, 24, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Majidi, J.; Kosari-Nasab, M.; Salari, A.-A. Developmental minocycline treatment reverses the effects of neonatal immune activation on anxiety- and depression-like behaviors, hippocampal inflammation, and HPA axis activity in adult mice. Brain Res. Bull. 2016, 120, 1–13. [Google Scholar] [CrossRef]
- Doosti, M.-H.; Bakhtiari, A.; Zare, P.; Amani, M.; Majidi-Zolbanin, N.; Babri, S.; Salari, A.-A. Impacts of early intervention with fluoxetine following early neonatal immune activation on depression-like behaviors and body weight in mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 43, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.J.; Huang, Y.; Wynne, A.; Hanke, M.; Himler, J.; Bailey, M.T.; Sheridan, J.F.; Godbout, J.P. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J. Neuroinflamm. 2008, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soczynska, J.K.; Mansur, R.B.; Brietzke, E.; Swardfager, W.; Kennedy, S.H.; Woldeyohannes, H.O.; Powell, A.M.; Manierka, M.S.; McIntyre, R.S. Novel therapeutic targets in depression: Minocycline as a candidate treatment. Behav. Brain Res. 2012, 235, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, T.; Yasukawa, R.; Yasuda, H.; Hayashida, M.; Inagaki, T.; Horiguchi, J. Minocycline as Adjunctive Therapy for Schizophrenia: An open-label study. Clin. Neuropharmacol. 2008, 31, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Chaki, S. Vasopressin V1B Receptor Antagonists as Potential Antidepressants. Int. J. Neuropsychopharmacol. 2021, 24, 450–463. [Google Scholar] [CrossRef]
- Iijima, M.; Yoshimizu, T.; Shimazaki, T.; Tokugawa, K.; Fukumoto, K.; Kurosu, S.; Kuwada, T.; Sekiguchi, Y.; Chaki, S. Antidepressant and anxiolytic profiles of newly synthesized arginine vasopressin V1Breceptor antagonists: TASP0233278 and TASP0390325. Br. J. Pharmacol. 2014, 171, 3511–3525. [Google Scholar] [CrossRef] [Green Version]
- Griebel, G.; Holsboer, F. Neuropeptide receptor ligands as drugs for psychiatric diseases: The end of the beginning? Nat. Rev. Drug Discov. 2012, 11, 462–478. [Google Scholar] [CrossRef] [Green Version]
- Cohan, P. Mifepristone Accelerates HPA Axis Recovery in Secondary Adrenal Insufficiency. Case Rep. Endocrinol. 2016, 2016, 4709597. [Google Scholar] [CrossRef] [Green Version]
- Wulsin, A.C.; Herman, J.P.; Solomon, M.B. Mifepristone decreases depression-like behavior and modulates neuroendocrine and central hypothalamic–pituitary–adrenocortical axis responsiveness to stress. Psychoneuroendocrinology 2010, 35, 1100–1112. [Google Scholar] [CrossRef] [Green Version]
- Mailliet, F.; Qi, H.; Rocher, C.; Spedding, M.; Svenningsson, P.; Jay, T.M. Protection of stress-induced impairment of hippocampal/prefrontal LTP through blockade of glucocorticoid receptors: Implication of MEK signaling. Exp. Neurol. 2008, 211, 593–596. [Google Scholar] [CrossRef]
- Howland, R.H. Mifepristone as a Therapeutic Agent in Psychiatry. J. Psychosoc. Nurs. Ment. Health Serv. 2013, 51, 11–14. [Google Scholar] [CrossRef]
- Gallagher, P.; Watson, S.; Smith, M.S.; Ferrier, I.N.; Young, A.H. Effects of adjunctive mifepristone (RU-486) administration on neurocognitive function and symptoms in schizophrenia. Biol. Psychiatry 2005, 57, 155–161. [Google Scholar] [CrossRef]
- Chong, W.; Li, Y.; Liu, B.; Liu, Z.; Zhao, T.; Wonsey, D.R.; Chen, C.; Velmahos, G.C.; Demoya, M.A.; King, D.R.; et al. Anti-inflammatory properties of histone deacetylase inhibitors: A mechanistic study. J. Trauma Inj. Infect. Crit. Care 2012, 72, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.-M.; Ding, Q.-H.; Chen, W.-P.; Luo, R.-B. Vorinostat, a HDAC inhibitor, showed anti-osteoarthritic activities through inhibition of iNOS and MMP expression, p38 and ERK phosphorylation and blocking NF-κB nuclear translocation. Int. Immunopharmacol. 2013, 17, 329–335. [Google Scholar] [CrossRef]
- Covington, H.E.; Maze, I.; LaPlant, Q.C.; Vialou, V.; Ohnishi, Y.N.; Berton, O.; Fass, D.M.; Renthal, W.; Rush, A.; Wu, E.Y.; et al. Antidepressant Actions of Histone Deacetylase Inhibitors. J. Neurosci. 2009, 29, 11451–11460. [Google Scholar] [CrossRef]
- Chen, S.; Sang, N. Histone Deacetylase Inhibitors: The Epigenetic Therapeutics That Repress Hypoxia-Inducible Factors. J. Biomed. Biotechnol. 2010, 2011, 197946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorn, J.V.; Schür, R.R.; Boks, M.P.; Kahn, R.S.; Joels, M.; Vinkers, C.H. Cortisol stress reactivity across psychiatric disorders: A systematic review and meta-analysis. Psychoneuroendocrinology 2017, 77, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ehlert, U.; Gaab, J.; Heinrichs, M. Psychoneuroendocrinological contributions to the etiology of depression, posttraumatic stress disorder, and stress-related bodily disorders: The role of the hypothalamus–pituitary–adrenal axis. Biol. Psychol. 2001, 57, 141–152. [Google Scholar] [CrossRef]
- Sapolsky, R.M. Glucocorticoids and Hippocampal Atrophy in Neuropsychiatric Disorders. Arch. Gen. Psychiatry 2000, 57, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Chaumette, B.; Kebir, O.; Mam-Lam-Fook, C.; Morvan, Y.; Bourgin, J.; Godsil, B.; Plaze, M.; Gaillard, R.; Jay, T.M.; Krebs, M.-O. Salivary cortisol in early psychosis: New findings and meta-analysis. Psychoneuroendocrinology 2016, 63, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, R.; Addington, J.; Rusjan, P.; Suridjan, I.; Ng, A.; Boileau, I.; Pruessner, J.; Remington, G.; Houle, S.; Wilson, A.A. Increased Stress-Induced Dopamine Release in Psychosis. Biol. Psychiatry 2012, 71, 561–567. [Google Scholar] [CrossRef]
- Steiner, M.; Wotjak, C. Role of the endocannabinoid system in regulation of the hypothalamic-pituitary-adrenocortical axis. Prog. Brain Res. 2008, 170, 397–432. [Google Scholar] [CrossRef]
- Dewey, W.L.; Peng, T.-C.; Harris, L.S. The effect of 1-trans-Δ9-tetrahydrocannabinol on the hypothalamo-hypophyseal-adrenal axis of rats. Eur. J. Pharmacol. 1970, 12, 382–384. [Google Scholar] [CrossRef]
- Finn, D.P.; Jhaveri, M.D.; Beckett, S.R.G.; Kendall, D.A.; Marsden, C.A.; Chapman, V. Cannabinoids modulate ultrasound-induced aversive responses in rats. Psychopharmacology 2004, 172, 41–51. [Google Scholar] [CrossRef]
- Gorzalka, B.B.; Hill, M.N. Integration of Endocannabinoid Signaling into the Neural Network Regulating Stress-Induced Activation of the Hypothalamic–Pituitary–Adrenal Axis. Curr. Top Behav. Neurosci. 2009, 1, 289–306. [Google Scholar] [CrossRef]
- McEwen, B.S. Glucocorticoids, depression, and mood disorders: Structural remodeling in the brain. Metabolism 2005, 54, 20–23. [Google Scholar] [CrossRef]
- Herman, J.P.; Figueiredo, H.; Mueller, N.K.; Ulrich-Lai, Y.; Ostrander, M.M.; Choi, D.C.; Cullinan, W.E. Central mechanisms of stress integration: Hierarchical circuitry controlling hypothalamo–pituitary–adrenocortical responsiveness. Front. Neuroendocr. 2003, 24, 151–180. [Google Scholar] [CrossRef]
- Ulrich-Lai, Y.M.; Xie, W.; Meij, J.T.; Dolgas, C.M.; Yu, L.; Herman, J. Limbic and HPA axis function in an animal model of chronic neuropathic pain. Physiol. Behav. 2006, 88, 67–76. [Google Scholar] [CrossRef]
- Barna, I.; Zelena, D.; Arszovszki, A.; Ledent, C. The role of endogenous cannabinoids in the hypothalamo-pituitary-adrenal axis regulation: In vivo and in vitro studies in CB1 receptor knockout mice. Life Sci. 2004, 75, 2959–2970. [Google Scholar] [CrossRef]
- Soria, V.; González-Rodríguez, A.; Huerta-Ramos, E.; Usall, J.; Cobo, J.; Bioque, M.; Barbero, J.D.; Garcia-Rizo, C.; Tost, M.; Monreal, J.A.; et al. Targeting hypothalamic-pituitary-adrenal axis hormones and sex steroids for improving cognition in major mood disorders and schizophrenia: A systematic review and narrative synthesis. Psychoneuroendocrinology 2018, 93, 8–19. [Google Scholar] [CrossRef]
- Streit, F.; Memic, A.; Hasandedić, L.; Rietschel, L.; Frank, J.; Lang, M.; Witt, S.H.; Forstner, A.J.; Degenhardt, F.; Wüst, S.; et al. Perceived stress and hair cortisol: Differences in bipolar disorder and schizophrenia. Psychoneuroendocrinology 2016, 69, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Pruessner, M.; Cullen, A.; Aas, M.; Walker, E.F. The neural diathesis-stress model of schizophrenia revisited: An update on recent findings considering illness stage and neurobiological and methodological complexities. Neurosci. Biobehav. Rev. 2017, 73, 191–218. [Google Scholar] [CrossRef] [Green Version]
- Heim, C.; Newport, D.J.; Heit, S.; Graham, Y.P.; Wilcox, M.; Bonsall, R.; Miller, A.H.; Nemeroff, C.B. Pituitary-Adrenal and Autonomic Responses to Stress in Women After Sexual and Physical Abuse in Childhood. JAMA 2000, 284, 592–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondelli, V.; Dazzan, P.; Hepgul, N.; Di Forti, M.; Aas, M.; D’Albenzio, A.; Di Nicola, M.; Fisher, H.; Handley, R.; Marques, T.R.; et al. Abnormal cortisol levels during the day and cortisol awakening response in first-episode psychosis: The role of stress and of antipsychotic treatment. Schizophr. Res. 2009, 116, 234–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatzber, A.F.; Lindley, S. Glucocorticoid antagonists in neuropsychotic disorders. Eur. J. Pharmacol. 2008, 583, 358–364. [Google Scholar] [CrossRef]
- Revell, E.R.; Neill, J.C.; Harte, M.; Khan, Z.; Drake, R.J. A systematic review and meta-analysis of cognitive remediation in early schizophrenia. Schizophr. Res. 2015, 168, 213–222. [Google Scholar] [CrossRef]
- Yang, A.C.; Tsai, S.-J. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. Int. J. Mol. Sci. 2017, 18, 1689. [Google Scholar] [CrossRef]
- Weill-Engerer, S.; David, J.-P.; Sazdovitch, V.; Liere, P.; Schumacher, M.; Delacourte, A.; Baulieu, E.-E.; Akwa, Y. In vitro metabolism of dehydroepiandrosterone (DHEA) to 7α-hydroxy-DHEA and Δ5-androstene-3β,17β-diol in specific regions of the aging brain from Alzheimer’’s and non-demented patients. Brain Res. 2003, 969, 117–125. [Google Scholar] [CrossRef]
- Labrie, F.; Bélanger, A.; Simard, J.; Luu-The, V.; Labrie, C. DHEA and Peripheral Androgen and Estrogen Formation: Intracrinology. Ann. N. Y. Acad. Sci. 1995, 774, 16–28. [Google Scholar] [CrossRef]
- Hill, M.; Starka, L. The non-genomic actions of dehydroepiandrosterone and its metabolites. J. Steroid Biochem. Mol. Biol. 2015, 145, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Kreinin, A.; Bawakny, N.; Ritsner, M.S. Adjunctive Pregnenolone Ameliorates the Cognitive Deficits in Recent-Onset Schizophrenia: An 8-Week, Randomized, Double-Blind, Placebo-Controlled Trial. Clin. Schizophr. Relat. Psychoses 2017, 10, 201–210. [Google Scholar] [CrossRef]
- Ritsner, M.S.; Strous, R.D. Neurocognitive deficits in schizophrenia are associated with alterations in blood levels of neurosteroids: A multiple regression analysis of findings from a double-blind, randomized, placebo-controlled, crossover trial with DHEA. J. Psychiatr. Res. 2010, 44, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Kamin, H.S.; Kertes, D.A. Cortisol and DHEA in development and psychopathology. Horm. Behav. 2017, 89, 69–85. [Google Scholar] [CrossRef]
- Ritsner, M.S.; Bawakny, H.; Kreinin, A. Pregnenolone treatment reduces severity of negative symptoms in recent-onset schizophrenia: An 8-week, double-blind, randomized add-on two-center trial. Psychiatry Clin. Neurosci. 2014, 68, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Vallée, M. Neurosteroids and potential therapeutics: Focus on pregnenolone. J. Steroid Biochem. Mol. Biol. 2015, 160, 78–87. [Google Scholar] [CrossRef]
- Ritsner, M.S. Pregnenolone, Dehydroepiandrosterone, and Schizophrenia: Alterations and Clinical Trials. CNS Neurosci. Ther. 2010, 16, 32–44. [Google Scholar] [CrossRef]
- Robson, P.J.; Guy, G.W.; di Marzo, V. Cannabinoids and Schizophrenia: Therapeutic Prospects. Curr. Pharm. Des. 2014, 20, 2194–2204. [Google Scholar] [CrossRef]
- Walter, M.; Denier, N.; Vogel, M.; Lang, U. Effects of Psychoactive Substances in Schizophrenia—Findings of Structural and Functional Neuroimaging. Curr. Top. Med. Chem. 2012, 12, 2426–2433. [Google Scholar] [CrossRef]
- Shafi, A.; Berry, A.J.; Sumnall, H.; Wood, D.M.; Tracy, D.K. New psychoactive substances: A review and updates. Ther. Adv. Psychopharmacol. 2020, 10. [Google Scholar] [CrossRef]
- Banister, S.D.; Connor, M. The Chemistry and Pharmacology of Synthetic Cannabinoid Receptor Agonists as New Psychoactive Substances: Origins. Handb. Exp. Pharmacol. 2018, 252, 165–190. [Google Scholar] [CrossRef]
- Upthegrove, R.; Marwaha, S.; Birchwood, M. Depression and Schizophrenia: Cause, Consequence, or Trans-diagnostic Issue? Schizophr. Bull. 2016, 43, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manetti, L.; Cavagnini, F.; Martino, E.; Ambrogio, A.G. Effects of cocaine on the hypothalamic–pituitary–adrenal axis. J. Endocrinol. Investig. 2014, 37, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetti, V.; Solowij, N.; Yücel, M. The Role of Cannabinoids in Neuroanatomic Alterations in Cannabis Users. Biol. Psychiatry 2015, 79, e17–e31. [Google Scholar] [CrossRef] [Green Version]
- Schubart, C.; Sommer, I.; Fusar-Poli, P.; de Witte, L.; Kahn, R.; Boks, M. Cannabidiol as a potential treatment for psychosis. Eur. Neuropsychopharmacol. 2014, 24, 51–64. [Google Scholar] [CrossRef]
- Schoevers, J.; Leweke, J.E.; Leweke, F.M. Cannabidiol as a treatment option for schizophrenia: Recent evidence and current studies. Curr. Opin. Psychiatry 2020, 33, 185–191. [Google Scholar] [CrossRef]
- McGuire, P.; Robson, P.; Cubała, W.; Vasile, D.; Morrison, P.D.; Barron, R.; Taylor, A.; Wright, S. Cannabidiol (CBD) as an Adjunctive Therapy in Schizophrenia: A Multicenter Randomized Controlled Trial. Am. J. Psychiatry 2018, 175, 225–231. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, D.; Esposito, G.; Cirillo, C.; Cipriano, M.; de Winter, B.; Scuderi, C.; Sarnelli, G.; Cuomo, R.; Steardo, L.; de Man, J.G.; et al. Cannabidiol Reduces Intestinal Inflammation through the Control of Neuroimmune Axis. PLoS ONE 2011, 6, e28159. [Google Scholar] [CrossRef]
- Iseger, T.A.; Bossong, M.G. A systematic review of the antipsychotic properties of cannabidiol in humans. Schizophr. Res. 2015, 162, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Long, E.L.; Malone, D.T.; Taylor, A.D. Cannabidiol Reverses MK-801-Induced Disruption of Prepulse Inhibition in Mice. Neuropsychopharmacology 2005, 31, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leweke, F.M.; Piomelli, D.; Pahlisch, F.; Muhl, D.; Gerth, C.W.; Hoyer, C.; Klosterkötter, J.; Hellmich, M.; Koethe, D. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl. Psychiatry 2012, 2, e94. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yang, Y.; An, F.-R.; Zhang, L.; Ungvari, G.S.; Jackson, T.; Yuan, Z.; Xiang, Y.-T. Prevalence of comorbid depression in schizophrenia: A meta-analysis of observational studies. J. Affect. Disord. 2020, 273, 524–531. [Google Scholar] [CrossRef]
- Naguy, A. Depression in schizophrenia-A good or bad omen? Asia-Pac. Psychiatry 2018, 10, e12312. [Google Scholar] [CrossRef] [PubMed]
- Ayesa-Arriola, R.; Alcaraz, E.G.; Hernández, B.V.; Pérez-Iglesias, R.; Moríñigo, J.D.L.; Duta, R.; David, A.S.; Tabares-Seisdedos, R.; Crespo-Facorro, B. Suicidal behaviour in first-episode non-affective psychosis: Specific risk periods and stage-related factors. Eur. Neuropsychopharmacol. 2015, 25, 2278–2288. [Google Scholar] [CrossRef]
- Bagarić, D.; Brečić, P.; Ostojić, D.; Jukić, V.; Goleš, A. The relationship between depressive syndrome and suicidal risk in patients with acute schizophrenia. Croat. Med. J. 2013, 54, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Huang, X.; Zhu, T. A systematic analysis of online broadcasts of suicidality in China. Asia-Pac. Psychiatry 2017, 10, e12302. [Google Scholar] [CrossRef] [PubMed]
- Sher, L.; Kahn, R.S. Suicide in Schizophrenia: An Educational Overview. Medicina 2019, 55, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumtaz, F.; Khan, M.I.; Zubair, M.; Dehpour, A.R. Neurobiology and consequences of social isolation stress in animal model—A comprehensive review. Biomed. Pharmacother. 2018, 105, 1205–1222. [Google Scholar] [CrossRef]
- Shargh, N.A.; Rostami, B.; Kosari, B.; Toosi, Z.; Majelan, G.A. Study of Relationship Between Depression and Quality of Life in Patients with Chronic Schizophrenia. Glob. J. Health Sci. 2015, 8, 224–229. [Google Scholar] [CrossRef]
- Majadas, S.; Olivares, J.; Galan, J.; Diez, T. Prevalence of depression and its relationship with other clinical characteristics in a sample of patients with stable schizophrenia. Compr. Psychiatry 2012, 53, 145–151. [Google Scholar] [CrossRef]
- Dai, J.; Du, X.; Yin, G.; Zhang, Y.; Xia, H.; Li, X.; Cassidy, R.; Tong, Q.; Chen, D.; Teixeira, A.L.; et al. Prevalence, demographic and clinical features of comorbid depressive symptoms in drug naïve patients with schizophrenia presenting with first episode psychosis. Schizophr. Res. 2017, 193, 182–187. [Google Scholar] [CrossRef]
- Siris, S.G. Depression in Schizophrenia: Perspective in the Era of “Atypical” Antipsychotic Agents. Am. J. Psychiatry 2000, 157, 1379–1389. [Google Scholar] [CrossRef]
- Krynicki, C.R.; Upthegrove, R.; Deakin, J.; Barnes, T.R.E. The relationship between negative symptoms and depression in schizophrenia: A systematic review. Acta Psychiatr. Scand. 2018, 137, 380–390. [Google Scholar] [CrossRef]
- Ampalam, P.; Deepthi, R.; Vadaparty, P. Schizophrenia—Insight, Depression: A Correlation Study. Indian J. Psychol. Med. 2012, 34, 44–48. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.G.C.; Carroll, A.; Fattah, S.; Clyde, Z.; Coffey, I.; Johnstone, E.C. A randomized, controlled trial of a brief interventional package for schizophrenic out-patients. Acta Psychiatr. Scand. 2001, 103, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Mintz, A.R.; Dobson, K.; Romney, D.M. Insight in schizophrenia: A meta-analysis. Schizophr. Res. 2002, 61, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Cotton, S.; Gleeson, J.; Alvarez-Jimenez, M.; McGorry, P. Quality of life in patients who have remitted from their first episode of psychosis. Schizophr. Res. 2010, 121, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Green, H.F.; Nolan, Y.M. Inflammation and the developing brain: Consequences for hippocampal neurogenesis and behavior. Neurosci. Biobehav. Rev. 2014, 40, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Hueston, C.; Cryan, J.F.; Nolan, Y.M. Stress and adolescent hippocampal neurogenesis: Diet and exercise as cognitive modulators. Transl. Psychiatry 2017, 7, e1081. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Vega, R.; Kong, X.; Deng, W.; Wang, Q.; Ma, X.; Li, M.; Hu, X.; Greenshaw, A.J.; Greiner, R.; et al. Neurocognitive Graphs of First-Episode Schizophrenia and Major Depression Based on Cognitive Features. Neurosci. Bull. 2017, 34, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Malone, D.; Kearn, C.; Chongue, L.; Mackie, K.; Taylor, D. Effect of social isolation on CB1 and D2 receptor and fatty acid amide hydrolase expression in rats. Neuroscience 2008, 152, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherian, K.; Schatzberg, A.F.; Keller, J. HPA axis in psychotic major depression and schizophrenia spectrum disorders: Cortisol, clinical symptomatology, and cognition. Schizophr. Res. 2019, 213, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Coulon, N.; Brailly-Tabard, S.; Walter, M.; Tordjman, S. Altered circadian patterns of salivary cortisol in individuals with schizophrenia: A critical literature review. J. Physiol. 2016, 110, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Patelaros, E.; Zournatzis, E.; Konstantakopoulos, G. Relationship of insight with depression and suicidal ideation in psychotic disorders. Psychiatriki 2015, 25, 265–272. [Google Scholar]
- Ferrer, A.; Labad, J.; Salvat-Pujol, N.; Monreal, J.A.; Urretavizcaya, M.; Crespo, J.M.; Menchón, J.M.; Palao, D.; Soria, V. Hypothalamic-pituitary-adrenal axis-related genes and cognition in major mood disorders and schizophrenia: A systematic review. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 101, 109929. [Google Scholar] [CrossRef]
- Dudzic-Koc, A. Rola stresu w zachorowaniu na schizofrenię. Zesz. Nauk. WSG 2018, 259-27. [Google Scholar]
- Skrzypińska, D.; Słodka, M. Kto może zachorować na schizofrenię? Czyli o modelach podatność–stres. W: Drop E, Maćkiewicz M red. Młoda Psychologia, t. 1. Warszawa Liberi Libri. 2012, 369–385. [Google Scholar]
- Goh, C.; Agius, M. The stress-vulnerability model how does stress impact on mental illness at the level of the brain and what are the consequences? Psychiatr. Danub. 2010, 22, 198–202. [Google Scholar]
- Kinlein, S.A.; Phillips, D.J.; Keller, C.R.; Karatsoreos, I.N. Role of corticosterone in altered neurobehavioral responses to acute stress in a model of compromised hypothalamic-pituitary-adrenal axis function. Psychoneuroendocrinology 2018, 102, 248–255. [Google Scholar] [CrossRef]
- Oitzl, M.S.; Champagne, D.L.; van der Veen, R.; de Kloet, R. Brain development under stress: Hypotheses of glucocorticoid actions revisited. Neurosci. Biobehav. Rev. 2010, 34, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Do, K.Q.; Cabungcal, J.H.; Frank, A.; Steullet, P.; Cuenod, M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr. Opin. Neurobiol. 2009, 19, 220–230. [Google Scholar] [CrossRef]
- Brown, A.S. The environment and susceptibility to schizophrenia. Prog. Neurobiol. 2011, 93, 23–58. [Google Scholar] [CrossRef] [Green Version]
- Maas, D.; Valles, A.; Martens, G. Oxidative stress, prefrontal cortex hypomyelination and cognitive symptoms in schizophrenia. Transl. Psychiatry 2017, 7, e1171. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, D.J.S.; Pedroza, A.A.D.S.; Braz, G.R.F.; da Silva-Filho, R.C.; Lima, T.A.; Fernandes, M.P.; Doi, S.Q.; Lagranha, C.J. Mitochondrial bioenergetics and oxidative status disruption in brainstem of weaned rats: Immediate response to maternal protein restriction. Brain Res. 2016, 1642, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Flatow, J.; Buckley, P.; Miller, B.J. Meta-Analysis of Oxidative Stress in Schizophrenia. Biol. Psychiatry 2013, 74, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Cengotitabengoa, M.; Mac-Dowell, K.S.; Leza, J.C.; Micó, J.A.; Fernandez, M.; Echevarría, E.; Sanjuan, J.; Elorza, J.; González-Pinto, A. Cognitive impairment is related to oxidative stress and chemokine levels in first psychotic episodes. Schizophr. Res. 2012, 137, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Serritella, A.V.; Sawa, A.; Sedlak, T.W. Implications for reactive oxygen species in schizophrenia pathogenesis. Schizophr. Res. 2016, 176, 52–71. [Google Scholar] [CrossRef]
- Sawa, A.; Sedlak, T.W. Oxidative stress and inflammation in schizophrenia. Schizophr. Res. 2016, 176, 1–2. [Google Scholar] [CrossRef]
- Gonzalez-Liencres, C.; Tas, C.; Brown, E.C.; Erdin, S.; Onur, E.; Cubukcoglu, Z.; Aydemir, O.; Esen-Danaci, A.; Brüne, M. Oxidative stress in schizophrenia: A case-control study on the effects on social cognition and neurocognition. BMC Psychiatry 2014, 14, 268. [Google Scholar] [CrossRef] [Green Version]
- Rukmini, M.S.; D”souza, B.; D’Souza, V. Superoxide dismutase and catalase activities and their correlation with malondialdehyde in schizophrenic patients. Indian J. Clin. Biochem. 2004, 19, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Othmen, B.L.; Mechri, A.; Fendri, C.; Bost, M.; Chazot, G.; Gaha, L.; Kerkeni, A. Altered antioxidant defense system in clinically stable patients with schizophrenia and their unaffected siblings. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 155–159. [Google Scholar] [CrossRef]
- Gardiner, J.; Barton, D.; Overall, R.; Marc, J. Neurotrophic Support and Oxidative Stress: Converging Effects in the Normal and Diseased Nervous System. Neuroscience 2008, 15, 47–61. [Google Scholar] [CrossRef]
- Walz, J.C.; Magalhães, P.V.; Giglio, L.M.; Cunha, A.B.; Stertz, L.; Fries, G.R.; Andreazza, A.C.; Kapczinski, F. Increased serum neurotrophin-4/5 levels in bipolar disorder. J. Psychiatr. Res. 2009, 43, 721–723. [Google Scholar] [CrossRef]
- Padurariu, M.; Ciobica, A.; Hritcu, L.; Stoica, B.; Bild, W.; Stefanescu, C. Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2010, 469, 6–10. [Google Scholar] [CrossRef]
- Penner-Goeke, S.; Binder, E.B. Epigenetics and depression. Dialog-Clin. Neurosci. 2019, 21, 397–405. [Google Scholar] [CrossRef]
- Provençal, N.; Arloth, J.; Cattaneo, A.; Anacker, C.; Cattane, N.; Wiechmann, T.; Röh, S.; Ködel, M.; Klengel, T.; Czamara, D.; et al. Glucocorticoid exposure during hippocampal neurogenesis primes future stress response by inducing changes in DNA methylation. Proc. Natl. Acad. Sci. USA 2019, 117, 23280–23285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Kennedy, P.J.; Nestler, E.J. Epigenetics of the Depressed Brain: Role of Histone Acetylation and Methylation. Neuropsychopharmacology 2012, 38, 124–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.; Rosenblat, J.D.; Brietzke, E.; Pan, Z.; Lee, Y.; Cao, B.; Zuckerman, H.; Kalantarova, A.; McIntyre, R.S. Stress, epigenetics and depression: A systematic review. Neurosci. Biobehav. Rev. 2019, 102, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, D.; Dhabhar, F.S.; James, S.J.; Hough, C.M.; Jain, F.A.; Bersani, F.S.; Reus, V.; Verhoeven, J.E.; Epel, E.S.; Mahan, L.; et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology 2016, 76, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; A Pasco, J.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013, 11, 200. [Google Scholar] [CrossRef] [Green Version]
- Kiecolt-Glaser, J.K.; Gouin, J.-P.; Weng, N.-P.; Malarkey, W.B.; Beversdorf, D.; Glaser, R. Childhood Adversity Heightens the Impact of Later-Life Caregiving Stress on Telomere Length and Inflammation. Psychosom. Med. 2011, 73, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.E.; Cole, S.W. Clustering of Depression and Inflammation in Adolescents Previously Exposed to Childhood Adversity. Biol. Psychiatry 2012, 72, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Tafet, G.E.; Nemeroff, C.B. The Links Between Stress and Depression: Psychoneuroendocrinological, Genetic, and Environmental Interactions. J. Neuropsychiatry Clin. Neurosci. 2016, 28, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Leonard, B.E. The Concept of Depression as a Dysfunction of the Immune System. Curr. Immunol. Rev. 2010, 6, 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbet, M.; Korga, A.; Gawrońska-Grzywacz, M.; Izdebska, M.; Piątkowska-Chmiel, I.; Poleszak, E.; Wróbel, A.; Matysiak, W.; Jodłowska-Jędrych, B.; Dudka, J. Chronic Variable Stress Is Responsible for Lipid and DNA Oxidative Disorders and Activation of Oxidative Stress Response Genes in the Brain of Rats. Oxidative Med. Cell. Longev. 2017, 2017, 7313090. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.H. Beyond depression: The expanding role of inflammation in psychiatric disorders. World Psychiatry 2020, 19, 108–109. [Google Scholar] [CrossRef] [PubMed]
- Kappelmann, N.; Lewis, G.; Dantzer, R.; Jones, P.B.; Khandaker, G. Antidepressant activity of anti-cytokine treatment: A systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol. Psychiatry 2016, 23, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A Randomized Controlled Trial of the Tumor Necrosis Factor Antagonist Infliximab for Treatment-Resistant Depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, N.I.; Moieni, M. Inflammation affects social experience: Implications for mental health. World Psychiatry 2020, 19, 109–110. [Google Scholar] [CrossRef] [PubMed]
- Muscatell, K.A.; Moieni, M.; Inagaki, T.K.; Dutcher, J.M.; Jevtic, I.; Breen, E.C.; Irwin, M.R.; Eisenberger, N.I. Exposure to an inflammatory challenge enhances neural sensitivity to negative and positive social feedback. Brain Behav. Immun. 2016, 57, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbet, M.; Natorska-Chomicka, D.; Ostrowska-Lesko, M.; Gawrońska-Grzywacz, M.; Izdebska, M.; Piątkowska-Chmiel, I.; Korga, A.; Wróbel, A.; Dudka, J. Edaravone presents antidepressant-like activity in corticosterone model of depression in mice with possible role of Fkbp5, Comt, Adora1 and Slc6a15 genes. Toxicol. Appl. Pharmacol. 2019, 380, 114689. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikulska, J.; Juszczyk, G.; Gawrońska-Grzywacz, M.; Herbet, M. HPA Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation. Brain Sci. 2021, 11, 1298. https://doi.org/10.3390/brainsci11101298
Mikulska J, Juszczyk G, Gawrońska-Grzywacz M, Herbet M. HPA Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation. Brain Sciences. 2021; 11(10):1298. https://doi.org/10.3390/brainsci11101298
Chicago/Turabian StyleMikulska, Joanna, Gabriela Juszczyk, Monika Gawrońska-Grzywacz, and Mariola Herbet. 2021. "HPA Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation" Brain Sciences 11, no. 10: 1298. https://doi.org/10.3390/brainsci11101298