
An Ensemble Feature Selection Approach for Analysis and Modeling of Transcriptome Data in Alzheimer’s Disease

Abstract
:1. Introduction
2. Materials and Methods
2.1. Feature Selection Techniques
2.2. Important Genes Prioritization
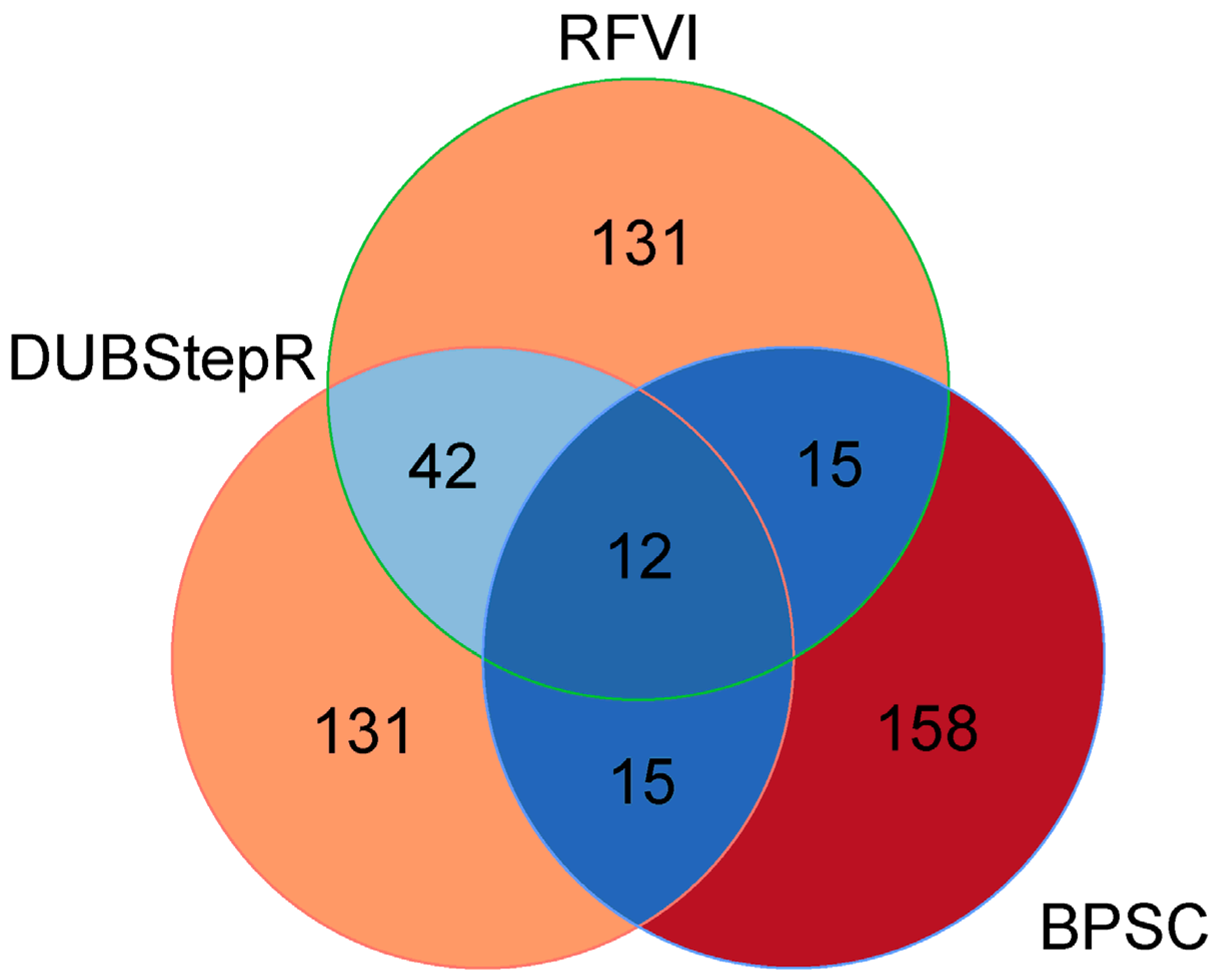
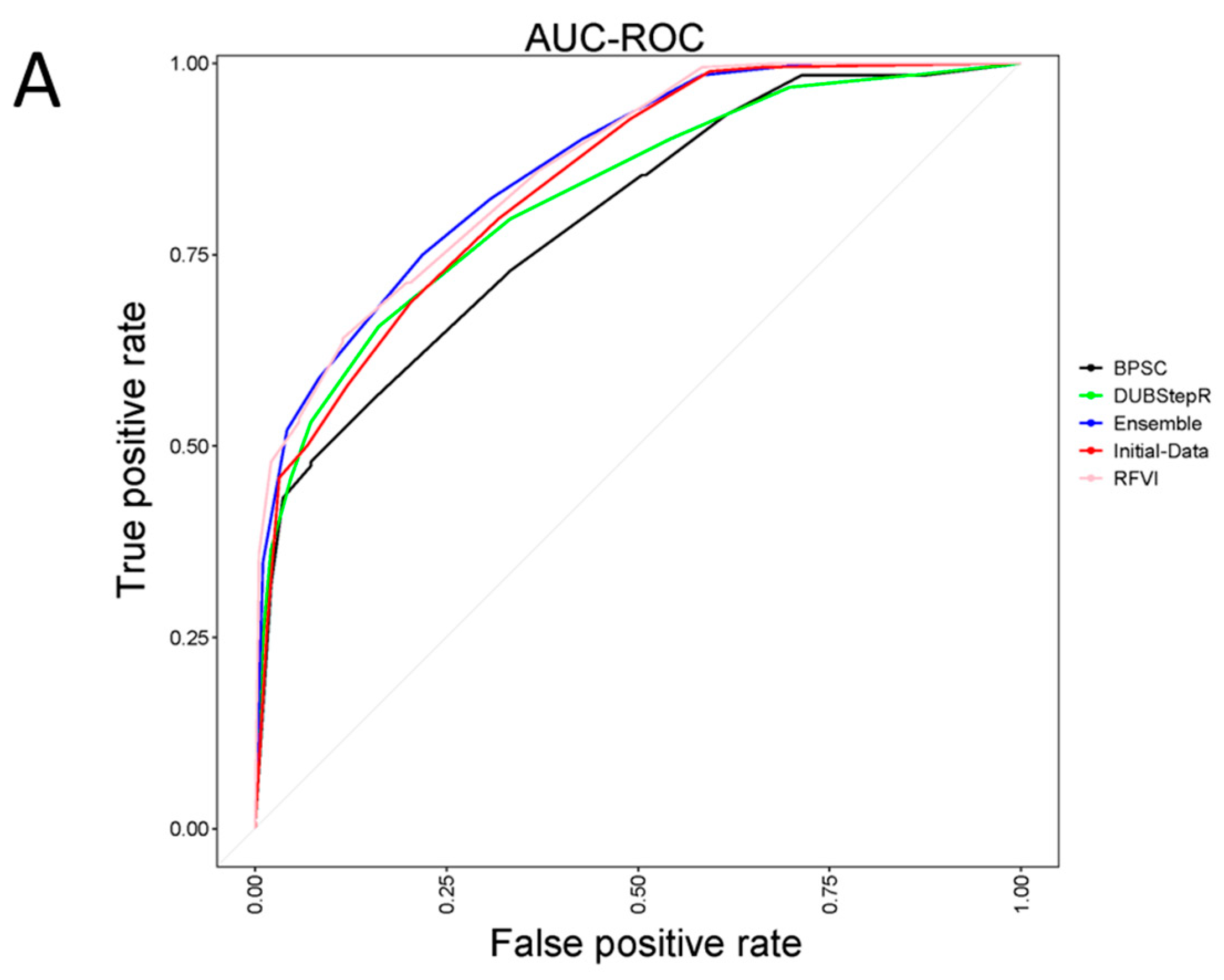
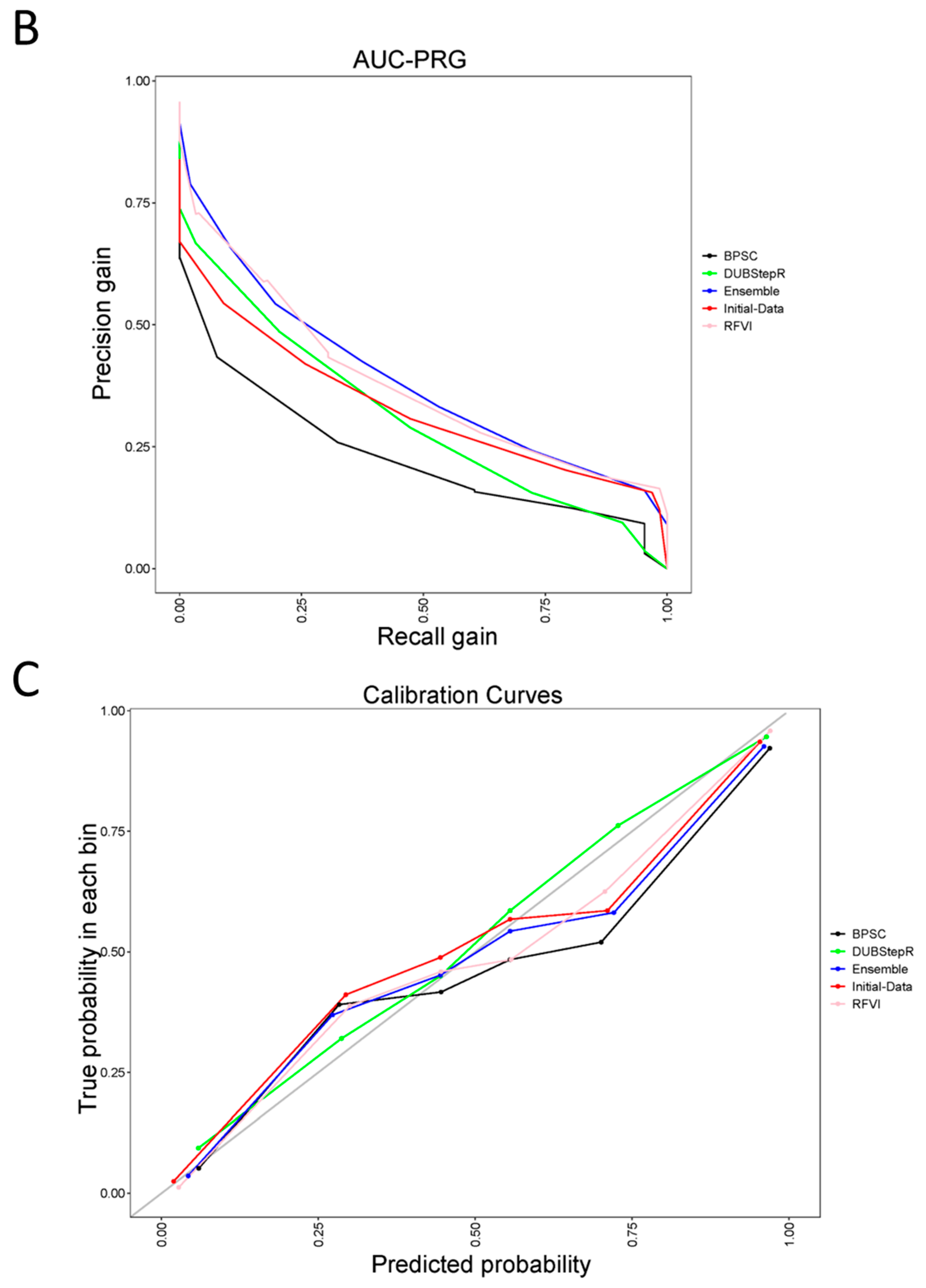
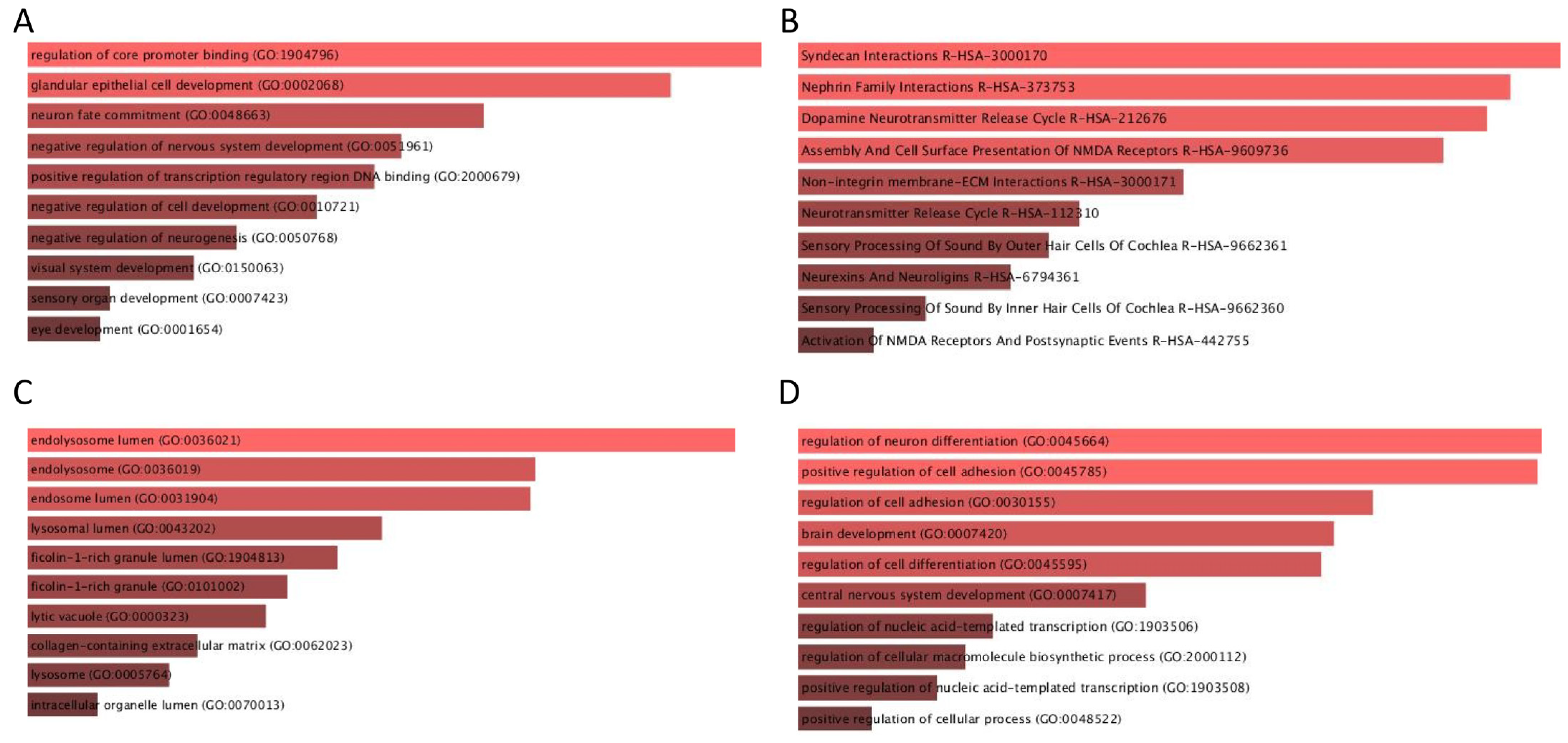
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, D.; Valencia, A. Big data analytics for personalized medicine. Curr. Opin. Biotechnol. 2019, 58, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef]
- Tang, X.; Huang, Y.; Lei, J.; Luo, H.; Zhu, X. The single-cell sequencing: New developments and medical applications. Cell Biosci. 2019, 9, 53. [Google Scholar] [CrossRef]
- Choi, Y.H.; Kim, J.K. Dissecting cellular heterogeneity using single-cell RNA sequencing. Mol. Cells 2019, 42, 189. [Google Scholar]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Wang, R.; Peng, G.; Tam, P.P.; Jing, N. Integration of computational analysis and spatial transcriptomics in single-cell study. Genom. Proteom. Bioinform. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Dokeroglu, T.; Deniz, A.; Kiziloz, H.E. A comprehensive survey on recent metaheuristics for feature selection. Neurocomputing 2022, 494, 269–296. [Google Scholar] [CrossRef]
- Xue, B.; Zhang, M.; Browne, W.N.; Yao, X. A Survey on Evolutionary Computation Approaches to Feature Selection. IEEE Trans. Evol. Comput. 2016, 20, 606–626. [Google Scholar] [CrossRef]
- Mahendran, N.; Durai Raj Vincent, P.M.; Srinivasan, K.; Chang, C.Y. Machine learning based computational gene selection models: A survey, performance evaluation, open issues, and future research directions. Front. Genet. 2020, 11, 603808. [Google Scholar] [CrossRef]
- Rengasamy, D.; Rothwell, B.C.; Figueredo, G.P. Towards a more reliable interpretation of machine learning outputs for safety-critical systems using feature importance fusion. Appl. Sci. 2021, 11, 11854. [Google Scholar] [CrossRef]
- Chen, C.W.; Tsai, Y.H.; Chang, F.R.; Lin, W.C. Ensemble feature selection in medical datasets: Combining filter, wrapper, and embedded feature selection results. Expert Syst. 2020, 37, e12553. [Google Scholar] [CrossRef]
- Aziz, R.; Verma, C.K.; Srivastava, N. Dimension reduction methods for microarray data: A review. AIMS Bioeng. 2017, 4, 179–197. [Google Scholar] [CrossRef]
- Saeys, Y.; Inza, I.; Larrañaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Mera-Gaona, M.; López, D.M.; Vargas-Canas, R.; Neumann, U. Framework for the ensemble of feature selection methods. Appl. Sci. 2021, 11, 8122. [Google Scholar] [CrossRef]
- Alhenawi, E.A.; Al-Sayyed, R.; Hudaib, A.; Mirjalili, S. Feature selection methods on gene expression microarray data for cancer classification: A systematic review. Comput. Biol. Med. 2022, 140, 105051. [Google Scholar] [CrossRef]
- Mahendran, N.; Vincent, P.D.R.; Srinivasan, K.; Chang, C.Y. Improving the classification of alzheimer’s disease using hybrid gene selection pipeline and deep learning. Front. Genet. 2021, 12, 784814. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jiang, X.; Xu, J.; Quan, X.; Wu, M.; Zhang, H. Ensemble consensus-guided unsupervised feature selection to identify Huntington’s disease-associated genes. Genes 2018, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, B.; Sun, W.; Park, J.; Mishra, K.; Schmidt, F.; Xie, R.; Alipour, F.; Singhal, V.; Joanito, I.; Honardoost, M.A.; et al. DUBStepR is a scalable correlation-based feature selection method for accurately clustering single-cell data. Nat. Commun. 2021, 12, 5849. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.N.; Wills, Q.F.; Kalari, K.R.; Niu, N.; Wang, L.; Rantalainen, M.; Pawitan, Y. Beta-Poisson model for single-cell RNA-seq data analyses. Bioinformatics 2016, 32, 2128–2135. [Google Scholar] [CrossRef]
- Drotár, P.; Gazda, M.; Vokorokos, L. Ensemble feature selection using election methods and ranker clustering. Inf. Sci. 2019, 480, 365–380. [Google Scholar] [CrossRef]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.M.; Jones, W.; Stokes, T.H.; Phan, J.H.; Moffitt, R.A.; Fang, H.; Shi, L.; Oberthuer, A.; Fischer, M.; Tong, W.; et al. k-Nearest neighbor models for microarray gene expression analysis and clinical outcome prediction. Pharm. J. 2010, 10, 292–309. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef]
- Motley, W.W.; Züchner, S.; Scherer, S.S. Isoform-specific loss of dystonin causes hereditary motor and sensory neuropathy. Neurol. Genet. 2020, 6, e496. [Google Scholar] [CrossRef] [PubMed]
- Ström, A.L.; Gal, J.; Shi, P.; Kasarskis, E.J.; Hayward, L.J.; Zhu, H. Retrograde axonal transport and motor neuron disease. J. Neurochem. 2008, 106, 495–505. [Google Scholar] [CrossRef]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef]
- Sadow, T.F.; Rubin, R.T. Effects of hypothalamic peptides on the aging brain. Psychoneuroendocrinology 1992, 17, 293–314. [Google Scholar] [CrossRef]
- Boczek, T.; Kapiloff, M.S. Compartmentalization of local cAMP signaling in neuronal growth and survival. Neural Regen. Res. 2020, 15, 453. [Google Scholar] [PubMed]
- Kaneko, K.; Xu, P.; Cordonier, E.L.; Chen, S.S.; Ng, A.; Xu, Y.; Morozov, A.; Fukuda, M. Neuronal Rap1 regulates energy balance, glucose homeostasis, and leptin actions. Cell Rep. 2016, 16, 3003–3015. [Google Scholar] [CrossRef]
- Sansom, S.N.; Griffiths, D.S.; Faedo, A.; Kleinjan, D.J.; Ruan, Y.; Smith, J.; Van Heyningen, V.; Rubenstein, J.L.; Livesey, F.J. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 2009, 5, e1000511. [Google Scholar] [CrossRef]
- Thakurela, S.; Tiwari, N.; Schick, S.; Garding, A.; Ivanek, R.; Berninger, B.; Tiwari, V.K. Mapping gene regulatory circuitry of Pax6 during neurogenesis. Cell Discov. 2016, 2, 15045. [Google Scholar] [CrossRef]
- Hsueh, Y.P. The role of the MAGUK protein CASK in neural development and synaptic function. Curr. Med. Chem. 2006, 13, 1915–1927. [Google Scholar] [CrossRef]
- McSweeney, D.; Gabriel, R.; Jin, K.; Pang, Z.P.; Aronow, B.; Pak, C. CASK loss of function differentially regulates neuronal maturation and synaptic function in human induced cortical excitatory neurons. Iscience 2022, 25, 105187. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Montilla-Perez, P.; Hillary, R.; Wen, J.; Benner, C.; Telese, F. The Function of CASK in Transcriptional Regulation in Neurons. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Oberstein, T.J.; Utz, J.; Spitzer, P.; Klafki, H.W.; Wiltfang, J.; Lewczuk, P.; Kornhuber, J.; Maler, J.M. The role of Cathepsin B in the degradation of Aβ and in the production of Aβ peptides starting with Ala2 in cultured astrocytes. Front. Mol. Neurosci. 2021, 13, 615740. [Google Scholar] [CrossRef]
- Hook, V.Y.; Kindy, M.; Reinheckel, T.; Peters, C.; Hook, G. Genetic cathepsin B deficiency reduces β-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem. Biophys. Res. Commun. 2009, 386, 284–288. [Google Scholar] [CrossRef]
- Pišlar, A.; Bolčina, L.; Kos, J. New insights into the role of cysteine cathepsins in neuroinflammation. Biomolecules 2021, 11, 1796. [Google Scholar] [CrossRef]
- Siklos, M.; BenAissa, M.; Thatcher, G.R. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, V.V.; Maday, S. Neuronal endosomes to lysosomes: A journey to the soma. J. Cell Biol. 2018, 217, 2977. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Kawaguchi, M.; Suzuki, M.; Jung, C.G.; Asai, K.; Shibamoto, Y.; Lavin, M.F.; Khanna, K.K.; Miura, Y. The ZFHX3 (ATBF1) transcription factor induces PDGFRB, which activates ATM in the cytoplasm to protect cerebellar neurons from oxidative stress. Dis. Model. Mech. 2010, 3, 752–762. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Santana, L.E.A.D.S.; de Paula Canuto, A.M. Filter-based optimization techniques for selection of feature subsets in ensemble systems. Expert Syst. Appl. 2014, 41, 1622–1631. [Google Scholar] [CrossRef]
- Tadist, K.; Najah, S.; Nikolov, N.S.; Mrabti, F.; Zahi, A. Feature selection methods and genomic big data: A systematic review. J. Big Data 2019, 6, 79. [Google Scholar] [CrossRef]
- Uncu, Ö.; Türkşen, I.B. A novel feature selection approach: Combining feature wrappers and filters. Inf. Sci. 2007, 177, 449–466. [Google Scholar] [CrossRef]
- Sarkar, C.; Cooley, S.; Srivastava, J. Robust feature selection technique using rank aggregation. Appl. Artif. Intell. 2014, 28, 243–257. [Google Scholar] [CrossRef]
- Gough, A.; Stern, A.; Maier, J.; Lezon, T.; Shun, T.-Y.; Chennubhotla, C.; Schurdak, M.; Haney, S.; Taylor, D. Biologically Relevant Heterogeneity: Metrics and Practical Insights. SLAS Discov. 2017, 22, 213–237. [Google Scholar] [CrossRef]
- Xiang, R.; Wang, W.; Yang, L.; Wang, S.; Xu, C.; Chen, X. A comparison for dimensionality reduction methods of single-cell RNA-seq data. Front. Genet. 2021, 12, 646936. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filter Data | Accuracy | Kappa | Precision | Recall | F1 | Elapsed Time (m) |
|---|---|---|---|---|---|---|
| Initial Data | 0.77 | 0.55 | 0.73 | 0.86 | 0.79 | 4.25 |
| Variable Data | 0.69 | 0.39 | 0.72 | 0.63 | 0.67 | 1.03 |
| Importance Data | 0.81 | 0.63 | 0.83 | 0.78 | 0.81 | 1.3 |
| Statistical Data | 0.60 | 0.21 | 0.72 | 0.34 | 0.46 | 1.01 |
| EFSM | 0.82 | 0.65 | 0.83 | 0.81 | 0.82 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paplomatas, P.; Krokidis, M.G.; Vlamos, P.; Vrahatis, A.G. An Ensemble Feature Selection Approach for Analysis and Modeling of Transcriptome Data in Alzheimer’s Disease. Appl. Sci. 2023, 13, 2353. https://doi.org/10.3390/app13042353
Paplomatas P, Krokidis MG, Vlamos P, Vrahatis AG. An Ensemble Feature Selection Approach for Analysis and Modeling of Transcriptome Data in Alzheimer’s Disease. Applied Sciences. 2023; 13(4):2353. https://doi.org/10.3390/app13042353
Chicago/Turabian StylePaplomatas, Petros, Marios G. Krokidis, Panagiotis Vlamos, and Aristidis G. Vrahatis. 2023. "An Ensemble Feature Selection Approach for Analysis and Modeling of Transcriptome Data in Alzheimer’s Disease" Applied Sciences 13, no. 4: 2353. https://doi.org/10.3390/app13042353