
Oxidation of Gaseous Elemental Mercury in Acidified Water: Evaluation of Possible Sinking Pathway of Atmospheric Gaseous Mercury in Acid Cloud, Fog, and Rain Droplets


Abstract
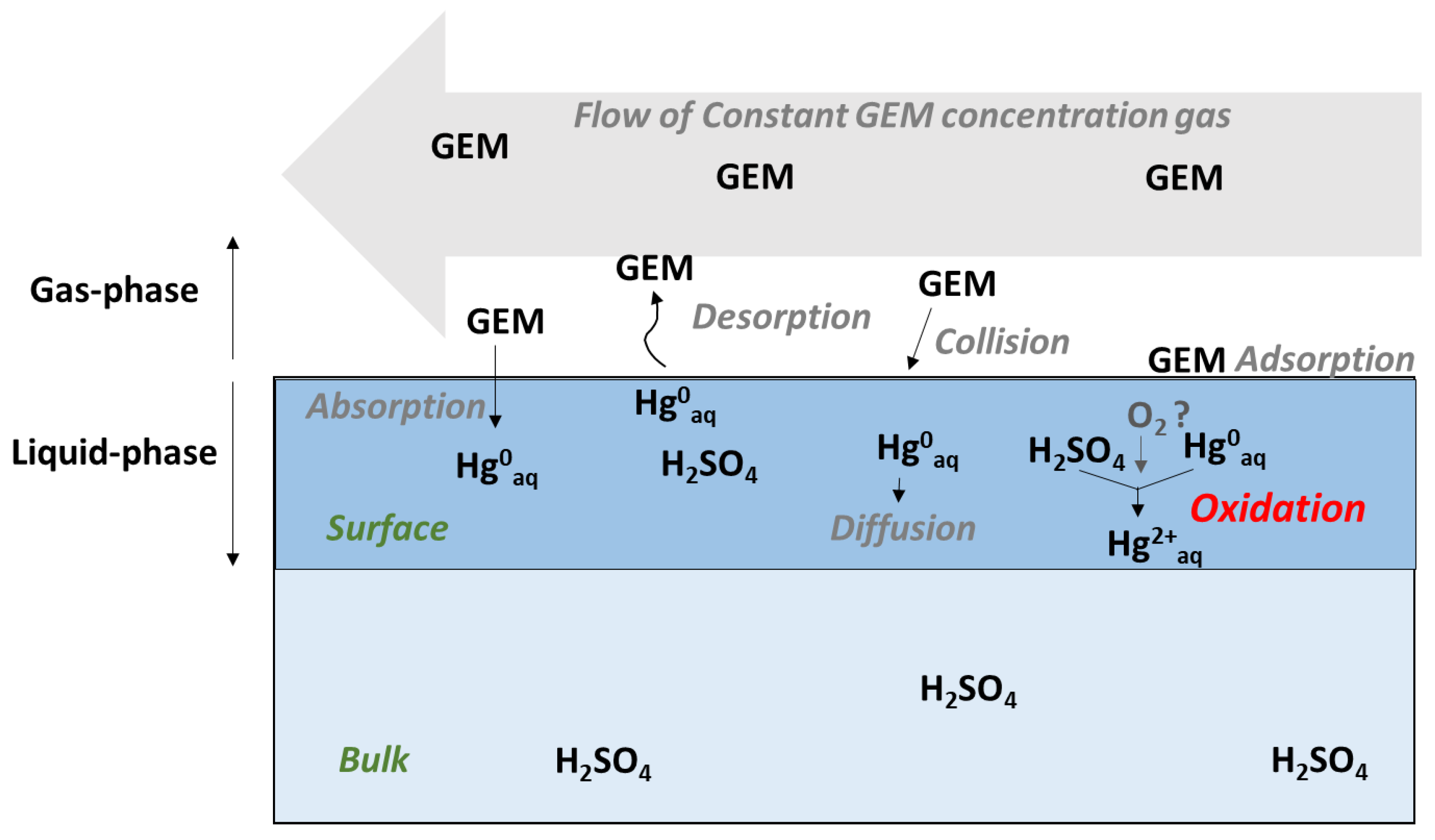
:1. Introduction
2. Materials and Method
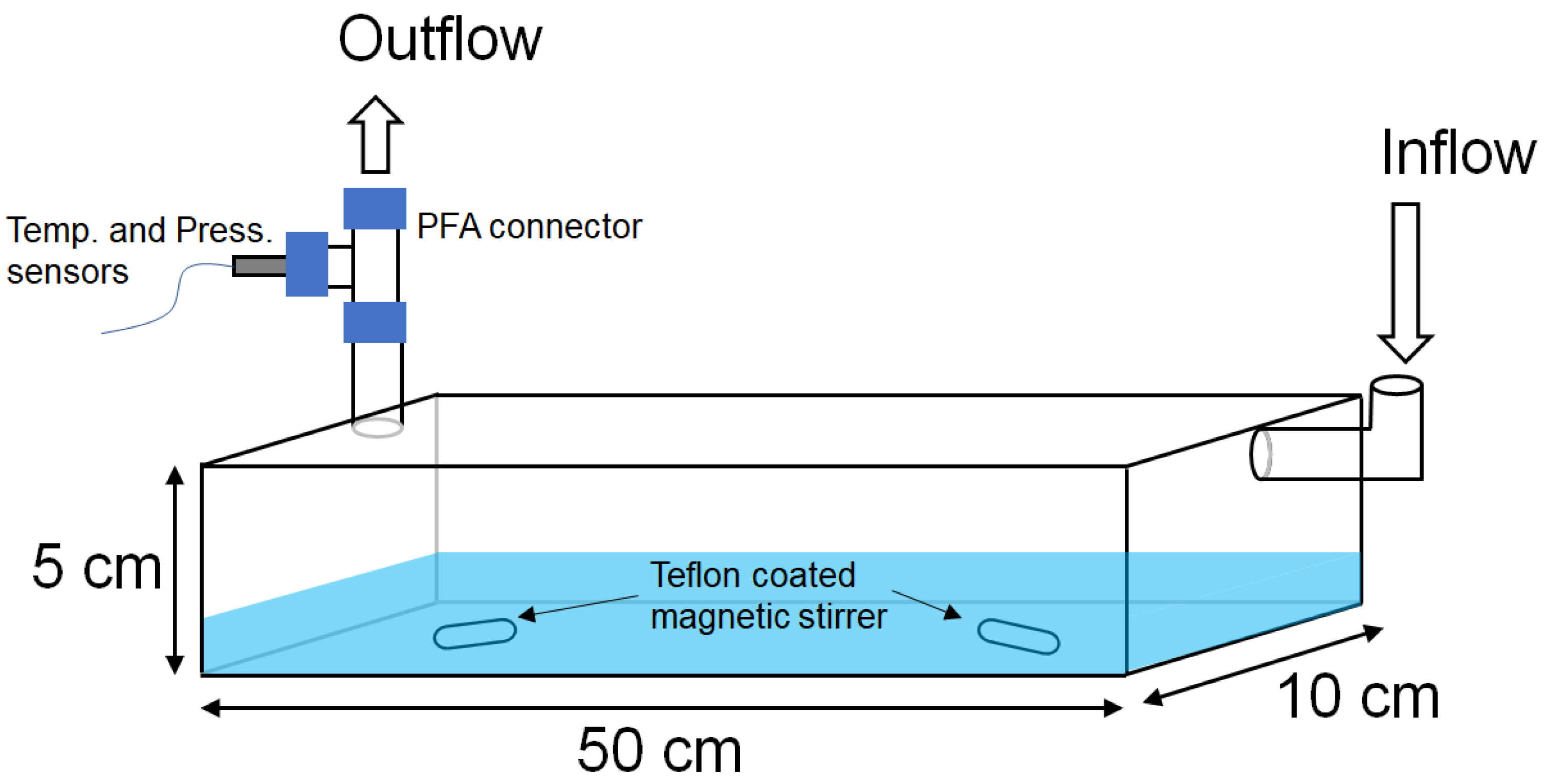
2.1. Uptake Experiment
2.2. Extraction for Dissolved Gaseous and Oxidized Mercury
3. Results and Discussion
3.1. Detection Limits and Sample Blank Values
3.2. Extraction Test
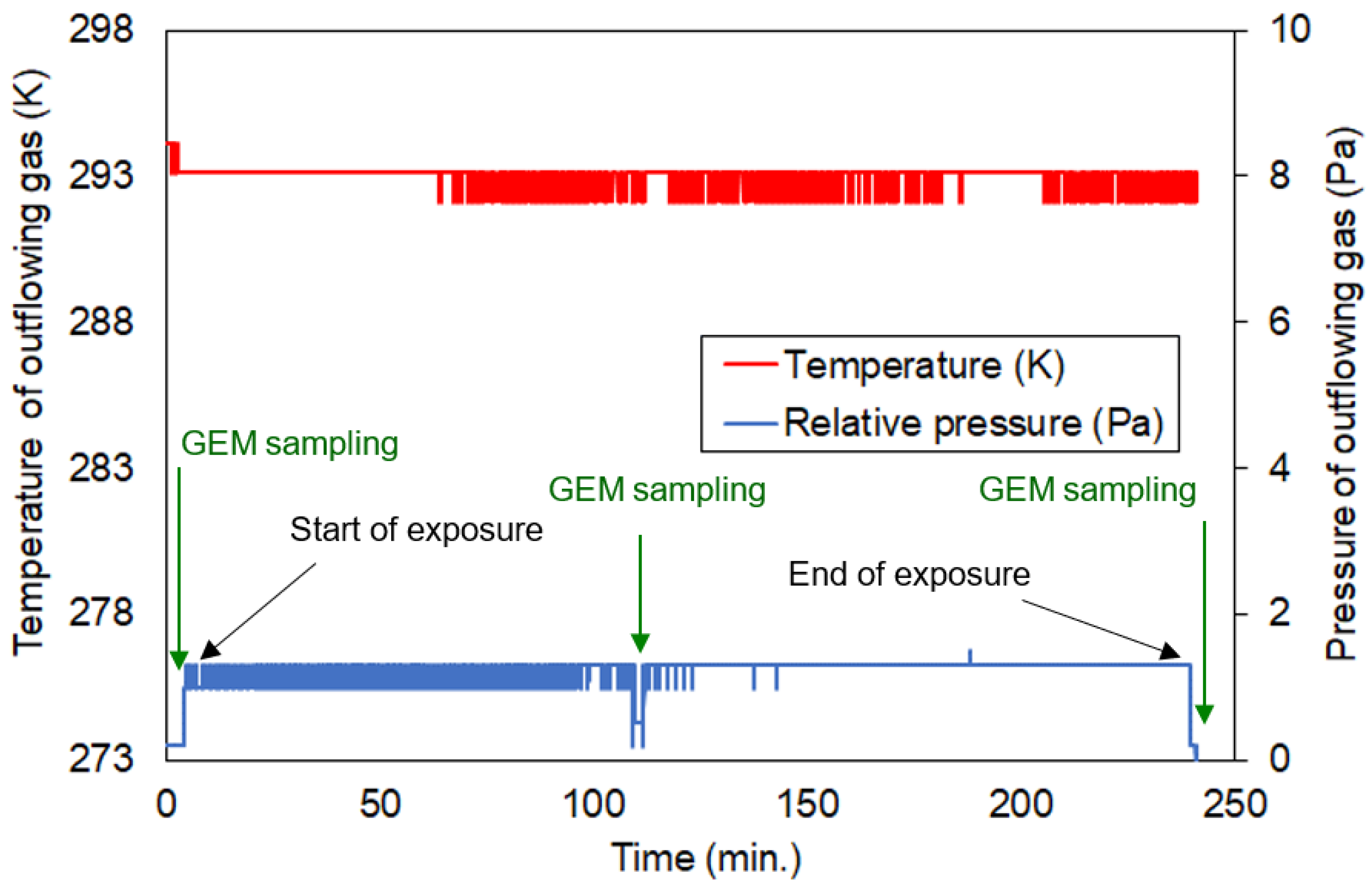
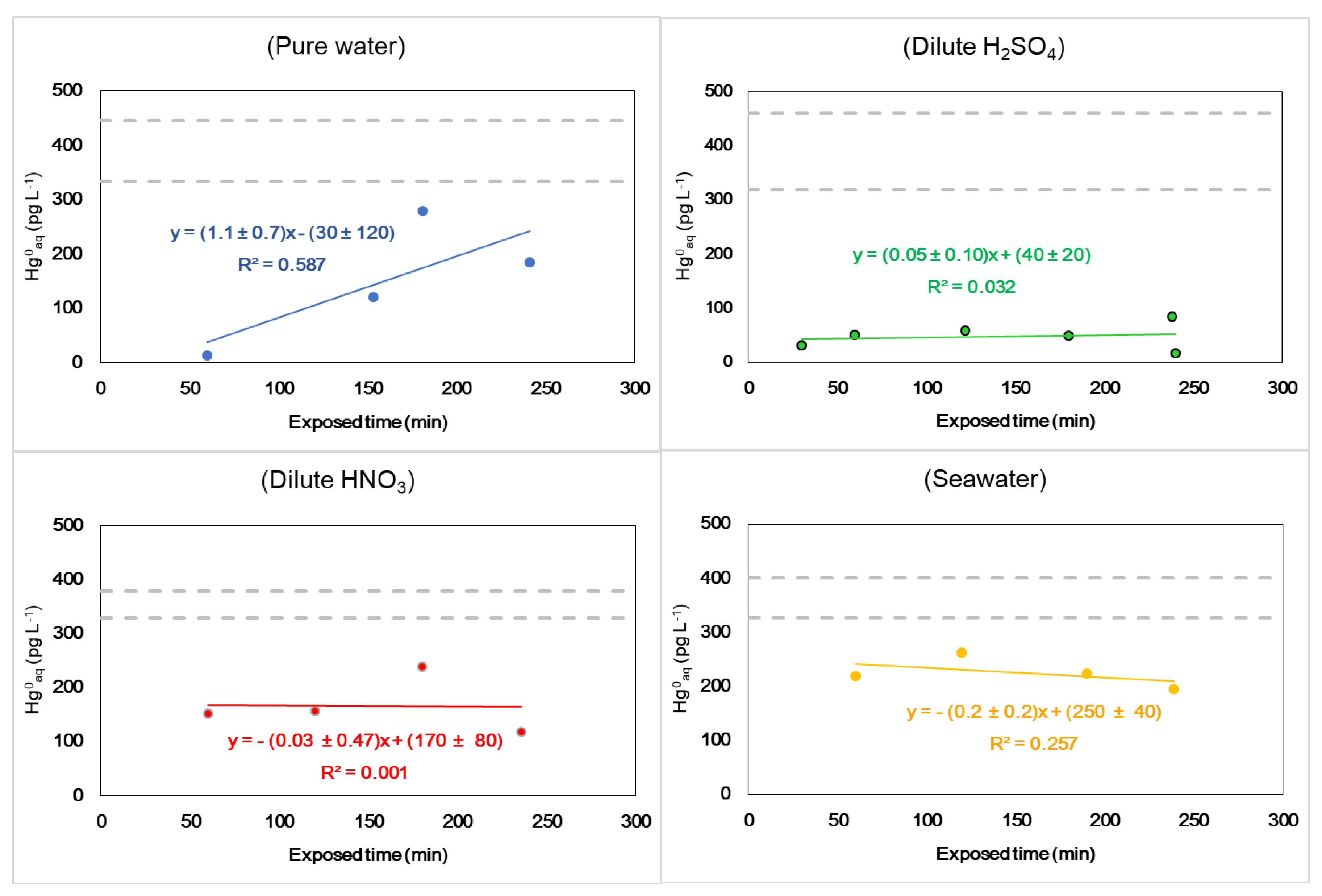
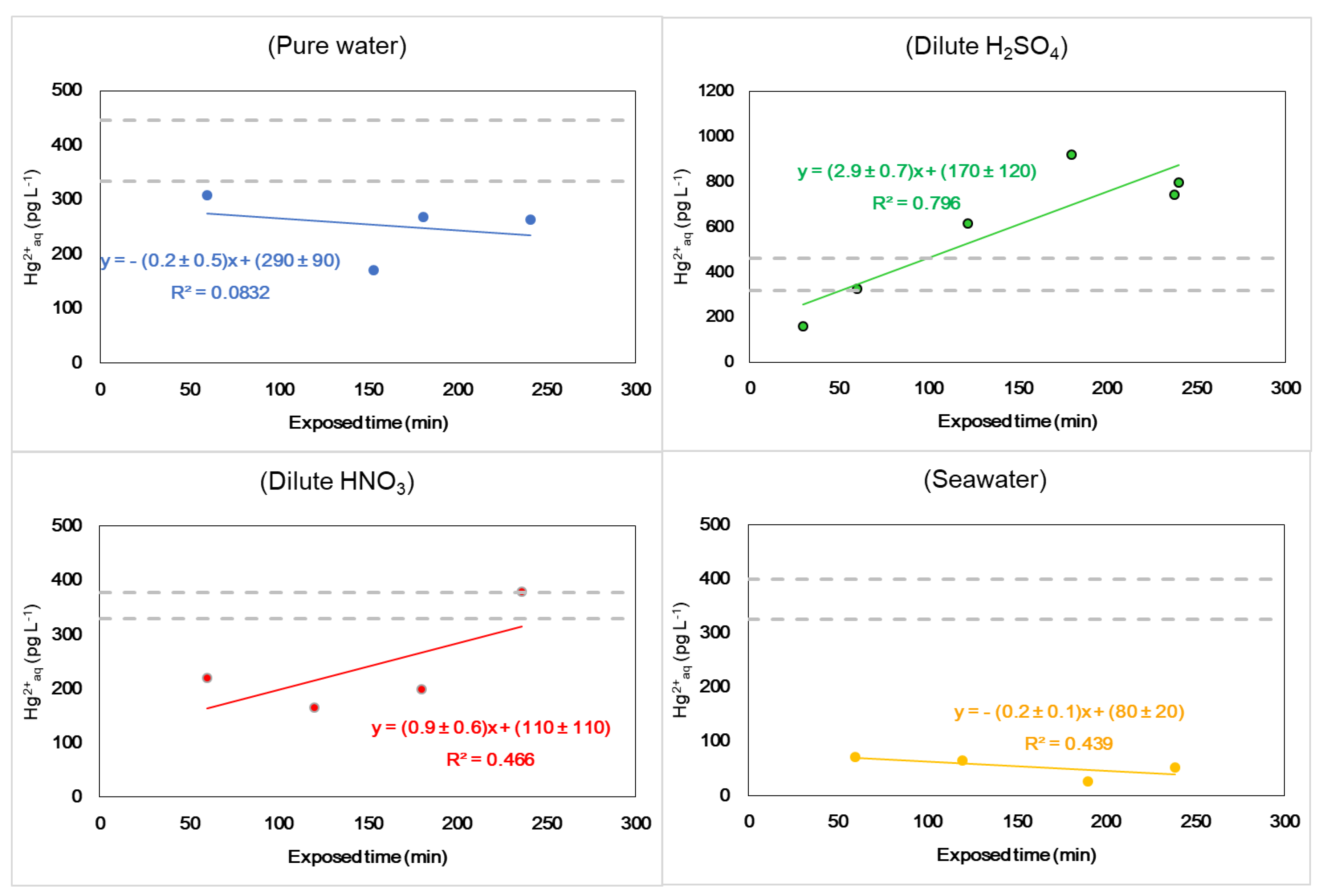
3.3. Uptake Experiment
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UN Environment Program. Global Mercury Assessment 2018; UN Environment Programme, Chemicals and Health Branch: Geneva, Switzerland, 2018. [Google Scholar]
- UN Environment Program. Global Mercury Assessment 2013; UN Environment Programme, Chemicals and Health Branch: Geneva, Switzerland, 2013. [Google Scholar]
- Schroeder, W.H.; Munthe, J. Atmospheric mercury—An overview. Atmos. Environ. 1998, 32, 809–822. [Google Scholar] [CrossRef]
- Gustin, M.S.; Amos, H.M.; Huang, J.; Miller, M.B.; Heidecorn, K. Measuring and modeling mercury in the atmosphere: A critical review. Atmos. Chem. Phys. 2015, 15, 5697–5713. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, W.H.; Yarwood, G.; Niki, H. Transformation processes involving mercury species in the atmosphere–Results from a literature survey. WaterAirSoil Pollut. 1991, 56, 653–666. [Google Scholar] [CrossRef]
- AMAP/UNEP. Global Mercury Modelling: Update of Modelling Results in the Global Mercury Assessment 2013; The Arctic Monitoring and Assessment Programme: Tromsø, Norway, 2015. [Google Scholar]
- Clever, H.L.; Johnson, S.A.; Derrick, M.E. The solubility of mercury and some sparingly soluble mercury salts in water and aqueous electrolyte solutions. J. Phys. Chem. Ref. Data 1985, 14, 631–680. [Google Scholar] [CrossRef]
- Andersson, M.E.; Gårdfeldt, K.; Wängberg, I.; Strömberg, D. Determination of Henry’s law constant for elemental mercury. Chemosphere 2008, 73, 587–592. [Google Scholar] [CrossRef]
- Kuss, J. Water-air gas exchange of elemental mercury: An experimentally determined mercury diffusion coefficient for Hg0 water-air flux calculations. Limnol. Oceanogr. 2014, 59, 1461–1467. [Google Scholar] [CrossRef]
- Kwon, S.-Y.; Selin, N.E. Uncertainties in atmospheric mercury modelling for policy evaluation. Curr. Pollut. Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Pal, B.; Ariya, P.A. Gas-phase HO-initiated reactions of elemental mercury: Kinetics, product studies, and atmospheric implications. Environ. Sci. Technol. 2004, 38, 5555–5566. [Google Scholar] [CrossRef]
- Calvert, J.G.; Lindberg, S.E. Mechanisms of mercury removal by O3 and OH in the atmosphere. Atmos. Environ. 2005, 39, 3355–3367. [Google Scholar] [CrossRef]
- Si, L.; Ariya, P.A. Recent advances in atmospheric chemistry of mercury. Atmosphere 2018, 9, 76. [Google Scholar] [CrossRef] [Green Version]
- Holmes, C.D.; Jacob, D.J.; Corbitt, E.S.; Mao, J.; Yang, X.; Talbot, R.; Slemr, F. Global atmospheric model for mercury including oxidation by bromine atoms. Atmos. Chem. Phys. 2010, 10, 12037–12057. [Google Scholar] [CrossRef] [Green Version]
- Toyota, K.; Dastoor, A.P.; Ryzhkov, A. Air-snowpack exchange of bromine, ozone, and mercury in the springtime Arctic simulated by the 1-D model PHANTAS-Part 2: Mercury and its speciation. Atmos. Chem. Phys. 2014, 14, 4135–4167. [Google Scholar] [CrossRef] [Green Version]
- Kolb, C.E.; Cox, R.A.; Abbatt, J.P.D.; Ammann, M.; Davis, E.J.; Donaldson, D.J.; Garrett, B.C.; George, C.; Griffiths, P.T.; Hanson, D.R.; et al. An overview of current issues in the uptake of atmospheric trace gases by aerosols and clouds. Atmos. Chem. Phys. 2010, 10, 10561–10605. [Google Scholar] [CrossRef] [Green Version]
- Munthe, J. The aqueous oxidation of elemental mercury by ozone. Atmos. Environ. 1992, 26A, 1461–1468. [Google Scholar] [CrossRef]
- Baskerville, C.; Miller, F.W. On the reactions between mercury and concentrated sulfuric acid. J. Am. Chem. Soc. 1897, 19, 873–877. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.A.; Kirk, D.W.; Jia, C.Q. Roles of sulfuric acid in elemental mercury removal by activated carbon and sulfur-impregnated activated carbon. Environ. Sci. Technol. 2012, 46, 7905–7912. [Google Scholar] [CrossRef]
- Irei, S. Development of fast sampling and high recovery extraction method for stable isotope measurement of gaseous mercury. Appl. Sci. 2020, 10, 6691. [Google Scholar] [CrossRef]
- Sheu, G.-R.; Lin, N.H. Mercury in cloud water collected on Mt. Bamboo in northern Taiwan during the northeast monsoon season. Atmos. Environ. 2011, 45, 4454–4462. [Google Scholar] [CrossRef]
- Kim, M.-K.; Lee, J.-S.; Jung, G.-B.; Ko, B.-G.; Kwon, S.-I.; Kim, M.-Y.; So, K.-H.; Yun, S.-G. Chemical characterization of rainwater over Suwon region during farming and non-farming periods. J. Agric. Chem. Environ. 2013, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Han, G. Chemical composition of rainwater and anthropogenic influences in Chengdu, Southwest China. Atmos. Res. 2011, 99, 190–196. [Google Scholar] [CrossRef]
- Yamamoto, M. Stimulation of elemental mercury oxidation in the presence of chloride ion in aquatic environments. Chemosphere 1996, 32, 1217–1224. [Google Scholar] [CrossRef]
- Marumoto, K.; Imai, S.; Takeda, K.; Sakugawa, H. Development of seawater sampler for collection of dissolved gaseous mercury preventing vaporization loss and influence of the loss on estimating mercury flux. Bunseki Kagaku 2012, 61, 1063–1072. (In Japanese) [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spiked Hg2+ | § Purging Time | n | † Hg0aq | † Hg2+aq | † Hg2+aq Recovery Yield |
|---|---|---|---|---|---|
| (pg) | (min) | (pg L−1) | (pg L−1) | (%) | |
| 150 | 15 | 5 | 23 ± 5 | 328 ± 46 | 64 ± 14 |
| 150 | 30 | 5 | 30 ± 7 | 348 ± 38 | 68 ± 11 |
| 150 | 60 | 4 | 12 ± 2 | 375 ± 29 | 73 ± 8 |
| 150 | 90 | 3 | 42 ± 16 | 343 ± 27 | 67 ± 8 |
| 150 | 120 | 2 | 27 ± 10 | 446 ± 25 | 86 ± 6 |
| 308 | 60 | 3 | 16 ± 5 | 952 ± 26 | 90 ± 3 |
| 308 | 90 | 3 | 40 ± 4 | 1000 ± 20 | 94 ± 2 |
| 308 | 120 | 3 | 32 ± 5 | 1088 ± 24 | 102 ± 2 |
| Sample | Exposure Duration | Mass of Uptake Solution | pH of Uptake Solution | § GEM Concentration | § Temperature | § Relative Pressure | ‡ Hg0aq | ‡ Hg2+aq |
|---|---|---|---|---|---|---|---|---|
| (min) | (g) | (ng m−3) | (K) | (Pa) | (pg L−1) | (pg L−1) | ||
| Pure water | 60 | 499.7 | † 6.1–6.5 | 153 ± 9 | 299.2 ± 0.2 | 0.70 ± 0.02 | 13.7 | 308 |
| Pure water | 153 | 500 | † 6.1–6.5 | 124 ± 3 | 298.4 ± 0.4 | 1.29 ± 0.06 | 121.2 | 170.9 |
| Pure water | 181 | 500.6 | † 6.1–6.5 | 131.2 ± 0.8 | 299.1 ± 0.3 | 1.2 ± 0.1 | 279.5 | 268.7 |
| Pure water | 241 | 499.7 | † 6.1–6.5 | 116 ± 3 | 297.1 ± 0.3 | 1.1 ± 0.2 | 184.4 | 263 |
| dilute H2SO4 | 122 | 500.3 | 3.3 | 130 ± 10 | 295.2 ± 0.2 | 1.02 ± 0.08 | 58.8 | 614.6 |
| dilute H2SO4 | 60 | 502.2 | 3.24 | 105 ± 6 | 293.4 ± 0.4 | 1.6 ± 0.1 | 50.4 | 325.2 |
| dilute H2SO4 | 238 | 500.3 | 3.25 | 110 ± 3 | 297.3 ± 0.3 | 1.32 ± 0.06 | 83.7 | 742.2 |
| dilute H2SO4 | 30 | 500.4 | 3.3 | 102 ± 8 | 292.6 ± 0.5 | 1.30 ± 0.02 | 30.7 | 157.6 |
| dilute H2SO4 | 180 | 500.1 | 3.2 | 112 ± 2 | 294.2 ± 0.5 | 1.29 ± 0.06 | 48 | 918.4 |
| dilute H2SO4 | 240 | 500 | 3.27 | 110 ± 3 | 295.6 ± 0.6 | 1.30 ± 0.02 | 16.4 | 798.1 |
| dilute HNO3 | 120 | 499.6 | 3.59 | 108.1 ± 0.5 | 294.15 ± 0.01 | 1.1 ± 0.2 | 156.9 | 164.6 |
| dilute HNO3 | 180 | 500.5 | 3.64 | 114.6 ± 0.9 | 295.7 ± 0.5 | 1.1 ± 0.1 | 238 | 198.7 |
| dilute HNO3 | 236 | 500.7 | 3.5 | 106 ± 2 | 293.1 ± 0.3 | 1.3 ± 0.1 | 117.7 | 379.8 |
| dilute HNO3 | 60 | 500.2 | 3.6 | 109.4 ± 0.8 | 295.2 ± 0.1 | 1.2 ± 0.1 | 151.8 | 220 |
| seawater | 239 | 500 | 8.11 | 113.3 ± 0.6 | 296.4 ± 0.4 | 1.2 ± 0.1 | 194.8 | 52.6 |
| seawater | 190 | 501.4 | 7.92 | 107 ± 3 | 294.2 ± 0.1 | 1.1 ± 0.1 | 223.5 | * LDL |
| seawater | 120 | 489.9 | 7.97 | 101 ± 3 | 295.0 ± 0.5 | 1.30 ± 0.01 | 262.7 | 65.5 |
| seawater | 60 | 499.8 | 7.95 | 115.4 ± 0.2 | 295.6 ± 0.5 | 1.03 ± 0.09 | 219.2 | 70.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irei, S. Oxidation of Gaseous Elemental Mercury in Acidified Water: Evaluation of Possible Sinking Pathway of Atmospheric Gaseous Mercury in Acid Cloud, Fog, and Rain Droplets. Appl. Sci. 2021, 11, 1196. https://doi.org/10.3390/app11031196
Irei S. Oxidation of Gaseous Elemental Mercury in Acidified Water: Evaluation of Possible Sinking Pathway of Atmospheric Gaseous Mercury in Acid Cloud, Fog, and Rain Droplets. Applied Sciences. 2021; 11(3):1196. https://doi.org/10.3390/app11031196
Chicago/Turabian StyleIrei, Satoshi. 2021. "Oxidation of Gaseous Elemental Mercury in Acidified Water: Evaluation of Possible Sinking Pathway of Atmospheric Gaseous Mercury in Acid Cloud, Fog, and Rain Droplets" Applied Sciences 11, no. 3: 1196. https://doi.org/10.3390/app11031196