
Altered miRNA Expression Profiles in the Serum of Beagle Dogs Experimentally Infected with Toxocara canis

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Animals and Collection of Samples
2.3. RNA Extraction and RNA-seq Analysis
2.4. KEGG Pathway Analysis of Differently Expressed miRNAs
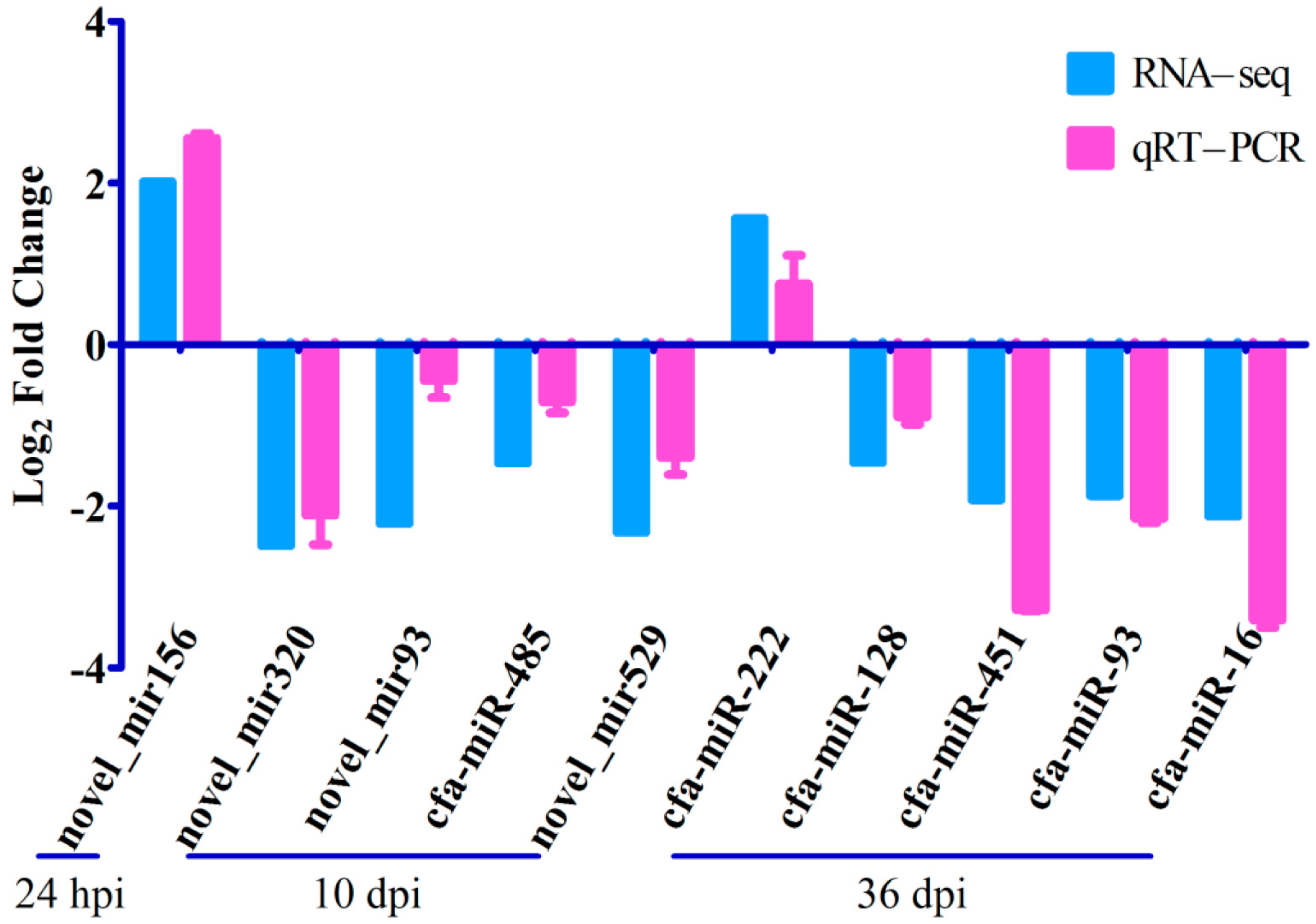
2.5. Quantitative RT-PCR Validation of RNA-seq Results
3. Results
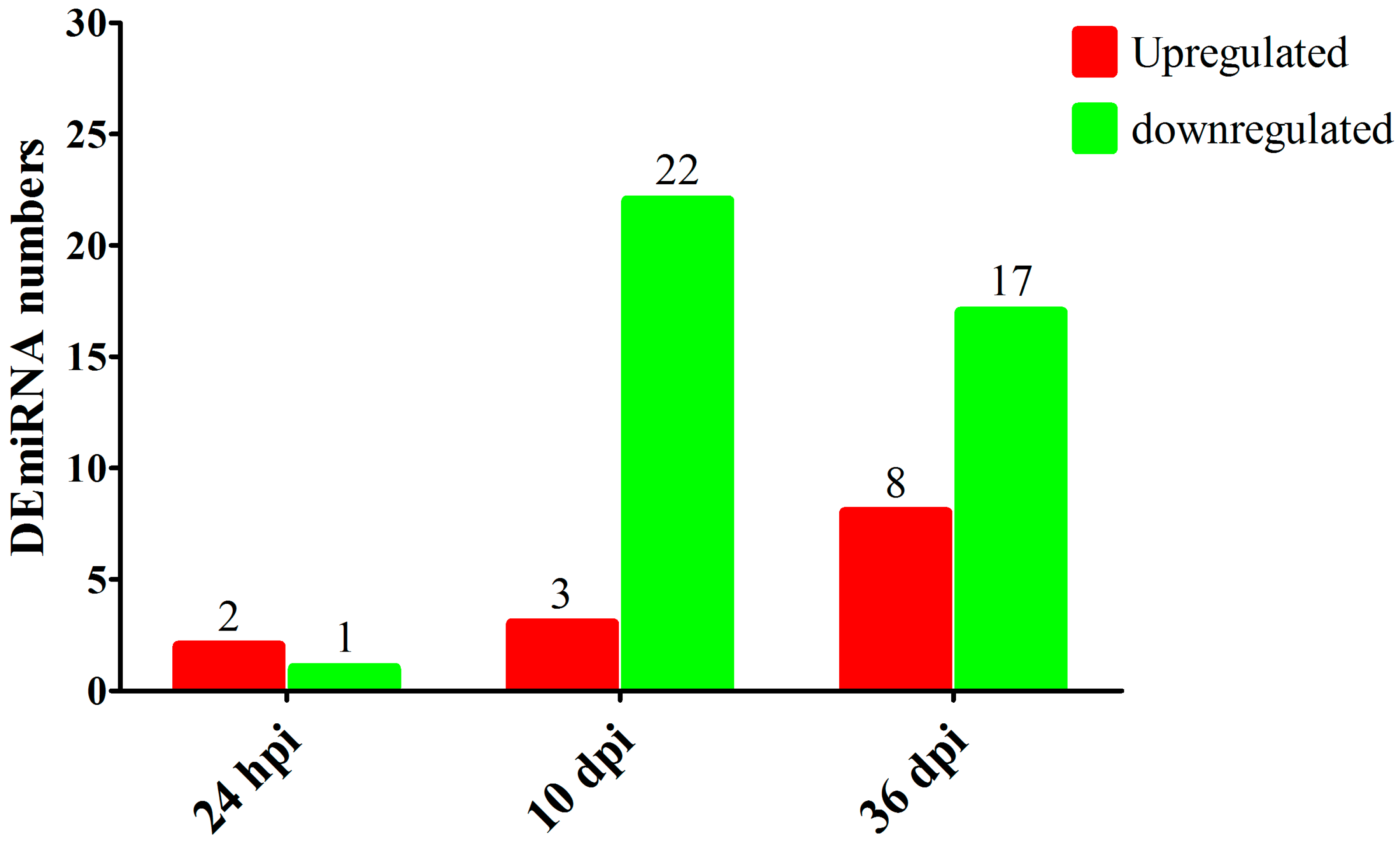
3.1. Overview of RNA-seq Results
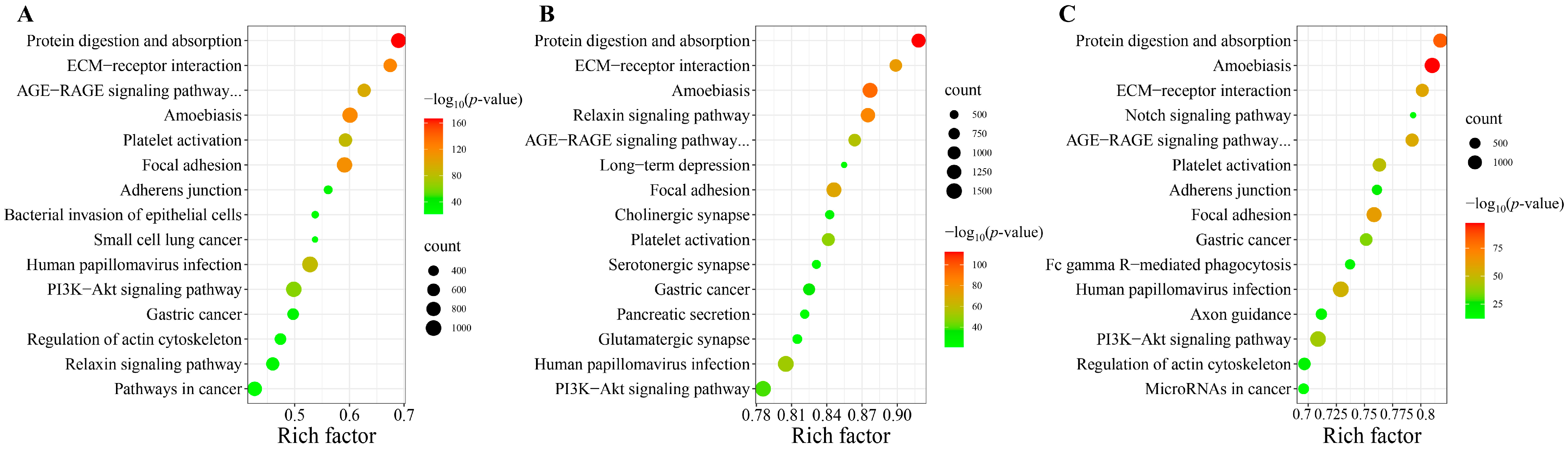
3.2. KEGG Pathway Analysis of Differently Expressed miRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, G.; Holland, C.V.; Wang, T.; Hofmann, A.; Fan, C.K.; Maizels, R.M.; Hotez, P.J.; Gasser, R.B. Human toxocariasis. Lancet Infect. Dis. 2018, 18, e14–e24. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Q.; Liu, G.H.; Zheng, W.B.; Hong, S.J.; Sugiyama, H.; Zhu, X.Q.; Elsheikha, H.M. Toxocariasis: A silent threat with a progressive public health impact. Infect. Dis. Poverty 2018, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostami, A.; Riahi, S.M.; Hofmann, A.; Ma, G.; Wang, T.; Behniafar, H.; Taghipour, A.; Fakhri, Y.; Spotin, A.; Chang, B.C.H.; et al. Global prevalence of Toxocara infection in dogs. Adv. Parasitol. 2020, 109, 561–583. [Google Scholar] [PubMed]
- Rostami, A.; Riahi, S.M.; Holland, C.V.; Taghipour, A.; Khalili-Fomeshi, M.; Fakhri, Y.; Omrani, V.F.; Hotez, P.J.; Gasser, R.B. Seroprevalence estimates for toxocariasis in people worldwide: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2019, 13, e0007809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostami, A.; Ma, G.; Wang, T.; Koehler, A.V.; Hofmann, A.; Chang, B.C.H.; Macpherson, C.N.; Gasser, R.B. Human toxocariasis—A look at a neglected disease through an epidemiological ‘prism’. Infect. Genet. Evol. 2019, 74, 104002. [Google Scholar] [CrossRef]
- Wu, T.; Bowman, D.D. Visceral larval migrans of Toxocara canis and Toxocara cati in non-canid and non-felid hosts. Adv. Parasitol. 2020, 109, 63–88. [Google Scholar]
- Fakhri, Y.; Gasser, R.B.; Rostami, A.; Fan, C.K.; Ghasemi, S.M.; Javanian, M.; Bayani, M.; Armoon, B.; Moradi, B. Toxocara eggs in public places worldwide—A systematic review and meta-analysis. Environ. Pollut. 2018, 242, 1467–1475. [Google Scholar] [CrossRef]
- Carlin, E.P.; Tyungu, D.L. Toxocara: Protecting pets and improving the lives of people. Adv. Parasitol. 2020, 109, 3–16. [Google Scholar]
- Zheng, W.B.; Zou, Y.; Zhu, X.Q.; Liu, G.H. Toxocara “omics” and the promises it holds for medicine and veterinary medicine. Adv. Parasitol. 2020, 109, 89–108. [Google Scholar]
- Zhu, X.Q.; Korhonen, P.K.; Cai, H.; Young, N.D.; Nejsum, P.; von Samson-Himmelstjerna, G.; Boag, P.R.; Tan, P.; Li, Q.; Min, J.; et al. Genetic blueprint of the zoonotic pathogen Toxocara canis. Nat. Commun. 2015, 6, 6145. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.B.; Zou, Y.; He, J.J.; Elsheikha, H.M.; Liu, G.H.; Hu, M.H.; Wang, S.L.; Zhu, X.Q. Global profiling of lncRNAs-miRNAs-mRNAs reveals differential expression of coding genes and non-coding RNAs in the lung of Beagle dogs at different stages of Toxocara canis infection. Int. J. Parasitol. 2021, 51, 49–61. [Google Scholar] [CrossRef]
- Zheng, W.B.; Zou, Y.; Elsheikha, H.M.; Liu, G.H.; Hu, M.H.; Wang, S.L.; Zhu, X.Q. Serum metabolomic alterations in Beagle dogs experimentally infected with Toxocara canis. Parasit. Vectors 2019, 12, 447. [Google Scholar] [CrossRef] [Green Version]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef]
- Trabucchi, M.; Mategot, R. Subcellular heterogeneity of the microRNA machinery. Trends Genet. 2019, 35, 15–28. [Google Scholar] [CrossRef]
- Jia, Y.; Wei, Y. Modulators of microRNA function in the immune system. Int. J. Mol. Sci. 2020, 21, 2357. [Google Scholar] [CrossRef]
- Zou, Y.; Zheng, W.B.; He, J.J.; Elsheikha, H.M.; Zhu, X.Q.; Lu, Y.X. Toxocara canis differentially affects hepatic microRNA expression in beagle dogs at different stages of infection. Front. Vet. Sci. 2020, 7, 587273. [Google Scholar] [CrossRef]
- Cai, P.; Mu, Y.; Olveda, R.M.; Ross, A.G.; Olveda, D.U.; McManus, D.P. Circulating miRNAs as footprints for liver fibrosis grading in schistosomiasis. eBioMedicine 2018, 37, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Guo, A. Profiling circulating microRNAs in serum of Fasciola gigantica-infected buffalo. Mol. Biochem. Parasitol. 2019, 232, 111201. [Google Scholar] [CrossRef]
- Ma, G.; Luo, Y.; Zhu, H.; Luo, Y.; Korhonen, P.K.; Young, N.D.; Gasser, R.B.; Zhou, R. MicroRNAs of Toxocara canis and their predicted functional roles. Parasit. Vectors 2016, 9, 229. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, T.D.; Nikapitiya, C.; Lee, J.; De Zoysa, M. Molecular insight into regulation of miRNAs in the spleen of zebrafish (Danio rerio) upon pathogenic Streptococcus parauberis infection. Fish Shellfish Immunol. 2020, 106, 898–909. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [PubMed]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhuo, H.Z.; Wu, J.Y.; Lin, L.Y.; Huang, Z.L.; Lu, J.X.; Cheng, K.L. MiR-92b inhibits proliferation and invasion of lung cancer by targeting EZH2. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3166–3173. [Google Scholar]
- Wang, L.P.; Geng, J.N.; Sun, B.; Sun, C.B.; Shi, Y.; Yu, X.Y.; Ren, K. MiR-92b-3p is induced by advanced glycation end products and involved in the pathogenesis of diabetic nephropathy. Evid.-Based Complement. Alternat. Med. 2020, 2020, 6050874. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Sang, M.; Meng, L.; Gu, L.; Liu, S.; Li, J.; Geng, C. miR-92b promotes autophagy and suppresses viability and invasion in breast cancer by targeting EZH2. Int. J. Oncol. 2018, 53, 1505–1515. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Tian, J.; Li, J. MiR-92b-3p ameliorates inflammation and autophagy by targeting TRAF3 and suppressing MKK3-p38 pathway in caerulein-induced AR42J cells. Int. Immunopharmacol. 2020, 88, 106691. [Google Scholar] [CrossRef]
- Liu, Y.; Song, J.W.; Lin, J.Y.; Miao, R.; Zhong, J.C. Roles of microRNA-122 in cardiovascular fibrosis and related diseases. Cardiovasc. Toxicol. 2020, 20, 463–473. [Google Scholar] [CrossRef]
- Loureiro, D.; Tout, I.; Narguet, S.; Benazzouz, S.M.; Mansouri, A.; Asselah, T. miRNAs as potential biomarkers for viral hepatitis B and C. Viruses 2020, 12, 1440. [Google Scholar] [CrossRef]
- Ghosh, J.; Bose, M.; Roy, S.; Bhattacharyya, S.N. Leishmania donovani targets Dicer1 to downregulate miR-122, lower serum cholesterol, and facilitate murine liver infection. Cell Host Microbe 2013, 13, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Ingle, H.; Kumar, S.; Raut, A.A.; Mishra, A.; Kulkarni, D.D.; Kameyama, T.; Takaoka, A.; Akira, S.; Kumar, H. The microRNA miR-485 targets host and influenza virus transcripts to regulate antiviral immunity and restrict viral replication. Sci. Signal. 2015, 8, ra126. [Google Scholar] [CrossRef]
- Mou, X.; Liu, S. MiR-485 inhibits metastasis and EMT of lung adenocarcinoma by targeting Flot2. Biochem. Biophys. Res. Commun. 2016, 477, 521–526. [Google Scholar] [CrossRef]
- Pan, W.; Song, X.; Hu, Q.; Zhang, Y. miR-485 inhibits histone deacetylase HDAC5, HIF1alpha and PFKFB3 expression to alleviate epilepsy in cellular and rodent models. Aging 2021, 13, 14416–14432. [Google Scholar] [CrossRef]
- Roush, S.; Slack, F.J. The let-7 family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef]
- Bernstein, D.L.; Gajghate, S.; Reichenbach, N.L.; Winfield, M.; Persidsky, Y.; Heldt, N.A.; Rom, S. let-7g counteracts endothelial dysfunction and ameliorating neurological functions in mouse ischemia/reperfusion stroke model. Brain Behav. Immun. 2020, 87, 543–555. [Google Scholar] [CrossRef]
- Delić, D.; Dkhil, M.; Al-Quraishy, S.; Wunderlich, F. Hepatic miRNA expression reprogrammed by Plasmodium chabaudi malaria. Parasitol. Res. 2011, 108, 1111–1121. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, W.; Du, Y.; Guo, Q.; Mao, Y.; Zhou, X.; Hua, D. Serum miR-16 as a potential biomarker for human cancer diagnosis: Results from a large-scale population. J. Cancer Res. Clin. Oncol. 2019, 145, 787–796. [Google Scholar] [CrossRef]
- Masaki, S.; Ohtsuka, R.; Abe, Y.; Muta, K.; Umemura, T. Expression patterns of microRNAs 155 and 451 during normal human erythropoiesis. Biochem. Biophys. Res. Commun. 2007, 364, 509–514. [Google Scholar] [CrossRef]
- Chamnanchanunt, S.; Fucharoen, S.; Umemura, T. Circulating microRNAs in malaria infection: Bench to bedside. Malar. J. 2017, 16, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Korhonen, P.K.; Gasser, R.B.; Young, N.D. Improved genomic resources and new bioinformatic workflow for the carcinogenic parasite Clonorchis sinensis: Biotechnological implications. Biotechnol. Adv. 2018, 36, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Pak, J.H.; Kim, I.K.; Kim, S.M.; Maeng, S.; Song, K.J.; Na, B.K.; Kim, T.S. Induction of cancer-related microRNA expression profiling using excretory-secretory products of Clonorchis sinensis. Parasitol. Res. 2014, 113, 4447–4455. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primers |
|---|---|
| novel_mir156 | GTGAGATGCCAGAGGAAGGTG |
| novel_mir320 | ATCCTAAGGTTGGACGGTCTGG |
| novel_mir93 | CTGGTGAGAACAGCAGGACG |
| cfa-miR-485 | CTAGAAGAGGCTGGCCGTGAT |
| novel_mir529 | CTGGTAGGAAGGGTGGTAGGG |
| cfa-miR-222 | AGCTACATCTGGCTACTGGGT |
| cfa-miR-128 | CACTCACAGTGAACCGGTCTC |
| cfa-miR-451 | CTGGAAACCGTTACCATTACTGAG |
| cfa-miR-93 | CAAAGTGCTGTTCGTGCAGG |
| cfa-miR-16 | GGTCTGGTAGCAGCACGTAA |
| U6 a | CGCTTCGGCAGCACATATAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, W.-B.; Cai, L.; Zou, Y.; Gao, W.-W.; Liu, Q.; Zhu, X.-Q. Altered miRNA Expression Profiles in the Serum of Beagle Dogs Experimentally Infected with Toxocara canis. Animals 2023, 13, 299. https://doi.org/10.3390/ani13020299
Zheng W-B, Cai L, Zou Y, Gao W-W, Liu Q, Zhu X-Q. Altered miRNA Expression Profiles in the Serum of Beagle Dogs Experimentally Infected with Toxocara canis. Animals. 2023; 13(2):299. https://doi.org/10.3390/ani13020299
Chicago/Turabian StyleZheng, Wen-Bin, Lang Cai, Yang Zou, Wen-Wei Gao, Qing Liu, and Xing-Quan Zhu. 2023. "Altered miRNA Expression Profiles in the Serum of Beagle Dogs Experimentally Infected with Toxocara canis" Animals 13, no. 2: 299. https://doi.org/10.3390/ani13020299